")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Metabolic Activation and Covalent Protein Binding of Berberrubine: Insight into the Underlying Mechanism Related to Its Hepatotoxicity
Authors Wang K, Rao J, Zhang T, Gao Q, Zhang J, Guang C, Ding L, Qiu F
Received 6 August 2020
Accepted for publication 26 September 2020
Published 22 October 2020 Volume 2020:14 Pages 4423—4438
DOI https://doi.org/10.2147/DDDT.S274627
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Kai Wang,1,* Jinqiu Rao,1,* Tingting Zhang,1 Qing Gao,2 Jichao Zhang,3 Chenxi Guang,3 Liqin Ding,3 Feng Qiu1,3
1School of Chinese Materia Medica, Tianjin University of Traditional Chinese Medicine, Tianjin 301617, People’s Republic of China; 2School of Integrative Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin 301617, People’s Republic of China; 3State Key Laboratory of Component-based Chinese Medicine,Tianjin University of Traditional Chinese Medicine, Tianjin, 301617, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Feng Qiu
School of Chinese Materia Medica, Tianjin University of Traditional Chinese Medicine, 10 Poyanghu Road, West Area, Tuanbo New Town, Jinghai District, Tianjin 301617, People’s Republic of China
Tel/ Fax +86-022-59596223
Email [email protected]
Introduction: Berberrubine (BRB), an isoquinoline alkaloid, is a major constituent of medicinal plants Coptis chinensis Franch or Phellodendron chinense Schneid. BRB exhibits various pharmacological activities, whereas exposure to BRB may cause toxicity in experimental animals.
Methods: In this study, we thoroughly investigated the liver injury induced by BRB in mice and rats. To explore the underlying mechanism, a study of the metabolic activation of BRB was conducted. Furthermore, covalent modifications of cysteine residues of proteins were observed in liver homogenate samples of animals after exposure to BRB, by application of an exhaustive proteolytic digestion method.
Results: It was demonstrated that BRB-induced hepatotoxicities in a time- and dose-dependent manner, based on the biochemical parameters ALT and AST. H&E stained histopathological examination showed the occurrence of obvious edema in liver of mice after intraperitoneal (i.p.) administration of BRB at a single dose of 100 mg/kg. Slight hepatotoxicity was also observed in rats given the same doses of BRB after six weeks of gavage. As a result, four GSH adducts derived from reactive metabolites of BRB were detected in microsomal incubations with BRB fortified with GSH as a trapping agent. Moreover, four cys-based adducts derived from reaction of electrophilic metabolites of BBR with proteins were found in livers.
Conclusion: These results suggested that the formation of protein adducts originating from metabolic activation of BRB could be a crucial factor of the mechanism of BRB-induced toxicities.
Keywords: berberrubine, hepatotoxicity, metabolic activation, protein modification
Introduction
Concerns about drug-induced physical toxicity, especially organ injury caused by natural compounds in herbal medicine have been increasing, due to the rapidly growing interest in the application of natural products as medical remedies.1–3 The best known example is that aristolochic acids (AAs), found in Aristolochiae, have been reported to cause severe neurotoxicity, even lead to cancer.4,5 As we know, nitroreductase-mediated biotransformation of the main toxic ingredients AAs into the corresponding electrophilic metabolites aristololactams, which further covalently combine with DNA, is considered as a pivotal factor in AAs-induced toxicity process.6–8 Recently, exposure to Dioscorea bulbifera L., a widely used herbal medicine was reported to display hepatotoxicities in human and experimental animals.9,10 The main toxic ingredients were found to be diosbulbin B (DIOB) and 8‑Epidiosbulbin E Acetate (EEA), which both can be metabolized to the cis-enedial intermediate, and the liver injury correlated with the reaction of these electrophilic intermediates with cell protein.11–13 Obviously, metabolic activation often plays a significantly critical role in the pathogenesis of the observed toxicities induced by drugs.14
Berberrubine (BRB), an isoquinoline alkaloid, is isolated from Berberis vulgaris L.15 BRB is a demethylation metabolites of berberine which is a main component occurring in many common medicinal herbs, such as Berberis vulgaris L. (family Berberidaceae),15 Coptis chinensis Franch. (family Ranunculaceae),16 Phellodendron chinense Schneid. (family Rutaceae),17 and Chelidonium majus L. (family Papaveraceae).18 Despite of the low content of BRB in Rhizoma Coptidis and other traditional Chinese medicines, people are susceptible to high exposure of BRB when they take berberine as a clinical medication. BRB is reported to possess various biological activities, including lipid-lowering, antitumorus, bactericidal, antimalaria, and antidysentery functions.19–22 BRB is also widely distributed in many preparations of the traditional Chinese medicine compound, such as Huanglian Jiedu decoction, Huanglian Shangqing pills and Niuhuang Qingxin pills.23–25
Surprisingly, ingestion of BRB was reportedly related to a high incidence of nephrotoxicity in experimental animals. Animal studies demonstrated that oral administration of BRB resulted in abnormal renal function index, along with severe nephrosis by histopathological examination.26 However, BRB lacks systematic toxicological evaluation, and the toxic mechanism remains unknown. According to the chemical structure of BRB as shown in Figure 1, it is likely that both section A and B of BRB can undergo metabolic activation mediated by drug metabolism enzymes. Given to the fact that liver, which is abundant of drug metabolism enzymes, especially of P450 enzyme, is the main organ responsible for metabolism, we purposed to carefully and thoroughly investigate the hepatotoxicity of BRB in this study. In addition, this article described the electrophilic metabolic intermediates resulting from BRB, for the purpose of providing initial foundation for revealing the possible mechanism of toxicity induced by BRB.
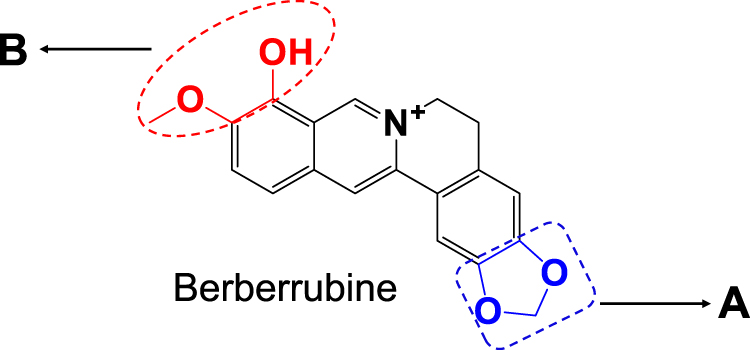 |
Figure 1 Chemical structure of berberrubine: Section (A) the methylenedioxyphenyl structure; section (B) the methylated catechol structure. |
In this study, the acute and sub-chronic hepatoxicities induced by BRB, were comprehensively investigated in mice and rats, respectively. Experiments about time-, dose-dependent toxicities and histopathologic examination were conducted. Furthermore, studies about metabolic activation and protein modification resulting from BRB were also performed to reveal the possible mechanism of BRB-induced hepatotoxicity.
Materials and Methods
Chemicals and Reagents
BRB (purity >98%) was synthesized by our laboratory according to a previously published literature.27 Glutathione (GSH), L-cysteine, and Nicotinamide Adenine Dinucleotide Phosphate (NADPH) were purchased from Sigma-Aldrich (St. Louis, MO). Mouse liver microsomes (MLMs) were purchased from BD Gentest (Woburn, MA). All organic solvents were from Fisher Scientific (Springfield, NJ). All reagents and solvents were of either analytical or high-performance LC grade. Alanine aminotransferase (ALT), aspartate aminotransferase (AST) kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Acute Hepatotoxicity Study in Mice
Male Kunming mice (18–20 g) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The animals had free access to food and water, and were kept in a controlled environment (room temperature and moderate humidity) under 12 h dark/light cycle and maintained on standard rat chow. Animal care and procedures are conformed to “The Guide for The Care and Use of Laboratory Animals by the National Institutes of Health of China” (Documentation 55,2001) and approved by the Committee for Animal Care and Use at Tianjin University of Traditional Chinese Medicine (Permit Number: TCM-LAEC2013005).
Mice were treated intraperitoneally with BRB dissolved in corn oil at doses of 0, 25, 50, or 100 mg/kg. Blood and liver were harvested 2 h after the treatment (dose-dependent experiment) or after 1 h, 2 h, 6 h, 10 h, 24 h and 48 h (time-dependent experiment conducted at the dose of 50 mg/kg) (n=6).
Sub-Chronic Hepatotoxicity Study in Rats
Male Sprague Dawley (SD) rats (160–180 g) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The rats were adapted to laboratory conditions for 4 days, and the rats were randomly divided into 4 groups (n = 8), including control group, low-dose group, middle-dose group and high-dose group. The BRB-treated groups were daily treated with BRB dissolved in 0.7% CMC-Na at doses of 25, 50, or 100 mg/kg/d by gavage administration. The rats in the control group were administered with the 0.7% CMC-Na solvent. The rats were raised for 6 weeks. On the last day of each week, 5mL of blood was collected from the posterior orbital venous plexus of rats. Rat liver tissues were harvested after the last treatment.
Determination of Serum Biochemical Indexes
The blood samples were allowed to clot at room temperature for 2 h and then centrifuged at 8000 rpm for 10 min. The resulting sera were collected for determination of ALT and AST values by quantitative diagnostic kits on VITROS ® 5600 Integrated System (Ortho-Clinical Diagnostics, Rochester, NY) (EnSpire).
Histopathologic Examination
The liver tissues of each animal were divided into two parts. One part was fixed in 10% formalin then embedded in paraffin, and sectioned into 5 μm thickness to be stained with H&E staining and observed under a light microscope (LeicaDM3000, Germany). The remaining organ tissues were stored at −20°C for further treatment.
Reactive Metabolite Trapping in Mouse Liver Microsomes (MLMs)
The microsomal incubation mixture was prepared in a final volume of 0.2 mL phosphate buffer (pH 7.4), containing MLMs (1.0 mg microsomal protein/mL), BRB (0.5 mM), MgCl2 (0.64 mM), and GSH (10 mM). The incubation reaction was initiated by the addition of NADPH (final concentration: 1.0 mM). The control sample contained no NADPH. After 60 minutes of incubation at 37°C, the reactions were terminated by mixing with equal volumes of ice-cold acetonitrile. The resulting mixtures were vortex-mixed and centrifuged at 16,000 rpm for 10 min to remove precipitated protein. A 2μL aliquot of the supernatant was injected to LC-MS/MS for analysis. Each incubation was performed in triplicate.
Protein Adduction and Digestion
The mouse liver tissues (0.2 g) were homogenized in 2.0 mL phosphate buffer (pH 7.4). The resulting protein samples were proteolytically digested by chymotrypsin and pronase E according to the procedures described previously.28 The digested mixtures were centrifuged at 16,000 rpm for 10 min, and the supernatants were subjected to LC-MS/MS for analysis. Each incubation was performed in triplicate.
LC-MS/MS Analysis
The LC-MS system was set up by interfacing a Waters e2695 LC system (Waters, USA) with a Waters Xevo G2-S Q/TOF-MS with electrospray ionization (Waters, USA). The chromatographic separation was achieved on ACQUITY UPLC BEH Shield RP18 column (2.1 × 50 mm, 1.7 µm), using 0.1% (v:v) formic acid in acetonitrile and 0.1% (v:v) formic acid in water as mobile phases A and B, respectively. The gradient started at 10% A for 4 min, ramped to 90% A over 17 min, held at 90% A for 1min, and then returned to 10% Ain 2 min to equilibrate the column. The flow rate was set at 0.4 mL/min. The Q/TOF-MS system with an ESI source was performed in positive ion mode. The optimized parameters were set as follows: capillary potential, 2500 V; sample cone potential, 30V; source temperature, 100 °C; desolvation temperature, 400 °C; cone gas flow, 50L/h; desolvation gas flow, 800 L/h. All data were acquired and processed by Metabolynx XS software under the operating interface of Masslynx V4.1 (Waters, Milford, MA, USA).
Statistical Analysis
The results of the studies are expressed as mean ± standard deviation of the mean. The difference between the control and treated means were analyzed by using one-way analysis of variance (ANOVA) and Student’s t-test. p < 0.05 was selected as the minimum criterion for statistically significant differences.
Results
Evaluation of Acute Hepatotoxicity in Mice After Single Dose of BRB
Hepatotoxicity of BRB in mice was evaluated by monitoring alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities in serum. Intraperitoneal (i.p.) administration of BRB caused elevations of both serum ALT and AST activities in a time-dependent manner at the dose of 50 mg/kg. The hepatotoxicity induced by BRB reached its peak at 1–2 h post-dose, while the ALT and AST values exhibited no statistically significant difference between the control group and the BRB-treated group after 6 h post-administration (Figure 2A and B). The ALT values also exhibited dose dependency as shown in Figure 2C. The AST values of the dose-dependent experiment were as shown in Figure 2D. To further confirm the hepatotoxicity of BRB, H&E staining was performed. As shown in Figure 2E, a wide range of inflammatory cell infiltration was observed, which was pointed out by the black arrows. The histopathologic evaluation (n = 3 each group) showed moderate edema in the livers of mice given BRB at 100 mg/kg (Figure 2E), by comparison with the normal mice liver (Figure 2F). These results suggested that obvious hepatotoxicity was caused in mice 2 h after receiving a single dose of BRB (i.p.).
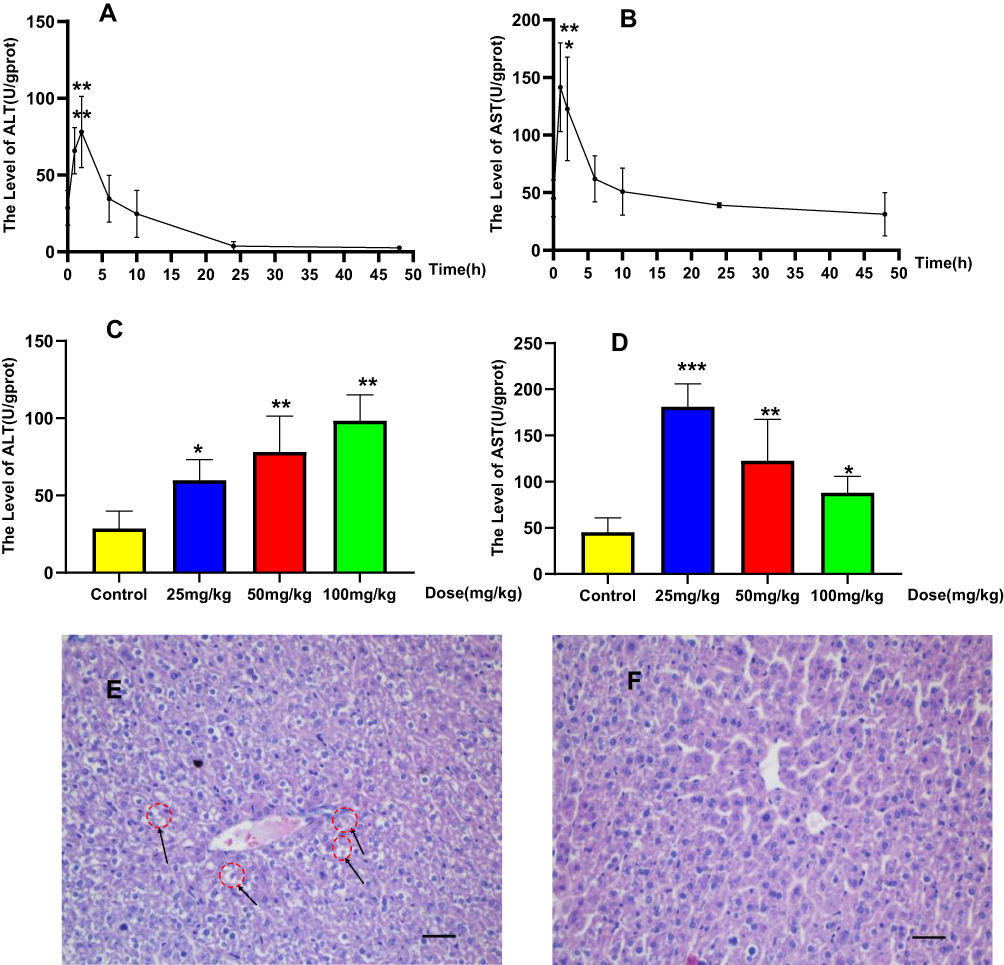 |
Figure 2 Acute hepatotoxicity in mice: Time-dependent manner of ALT (A) or AST (B), mice were treated with BRB (i.p.) at 50mg/kg (n=6); dose-dependent manner of ALT (C) or AST (D), mice were treated with BRB (i.p.) at 0, 25, 50 or 100mg/kg (n=6); representative histopathology of liver from mice in BRB-treated group at 2 h after administration (E) and control group (F), all processed livers were microscopically (Scale bar=50um) examined.*p < 0.05, **p < 0.01, ***p < 0.001 compared with the control group. |
Evaluation of Sub-Chronic Hepatotoxicity Study in Rats
Sub-chronic hepatotoxicity of BRB in rats was evaluated by AST and ALT assays in serum. As shown in Figure 3A, the content of ALT was increased in BRB-treated groups at the first three weeks, compared with that in the control group. The AST level was significantly elevated after administration of BRB at 100 mg/kg for one week (Figure 3B). With the prolongation of administration, both ALT and AST levels were gradually reduced. Obviously, oral administration of BRB at 25, 50 and 100 mg/kg caused elevation of both ALT and AST activities in a dose-dependent manner. To further confirm the hepatotoxicity of BRB, H&E staining was performed and the inflammatory cell infiltration was pointed out by the black arrows in Figure 3C. The histopathologic evaluation (n = 3 each group) showed slight edema in the liver of rats given BRB at 100 mg/kg (Figure 3C), by comparison with the normal rat livers (Figure 3D). These results indicated that slight hepatotoxicity was induced in rats after six weeks of gavage of BRB.
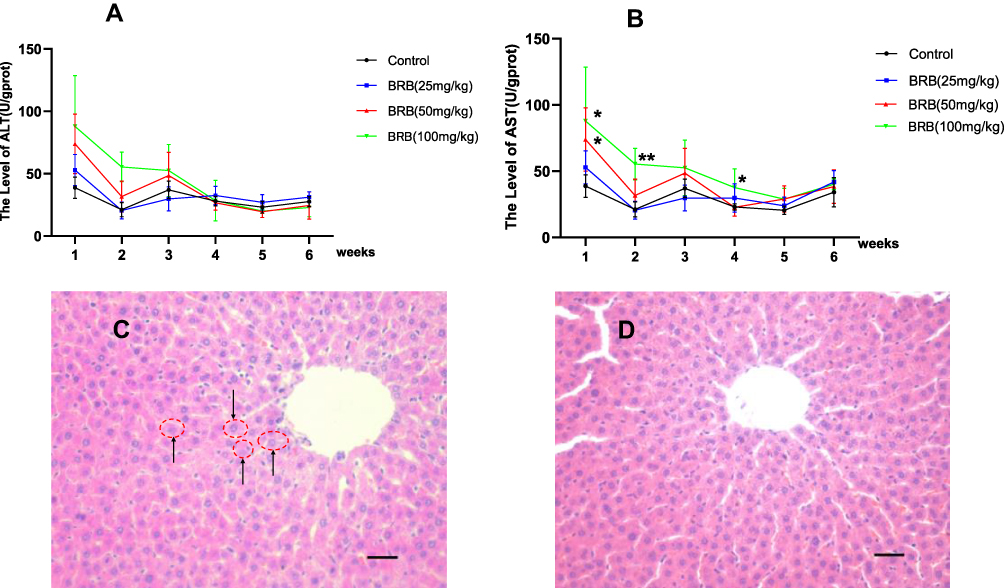 |
Figure 3 Sub-chronic hepatotoxicity in rats: Time-dependent and dose-dependent manner of ALT (A) or AST (B), rats were treated with BRB (i.g.) at 0, 25, 50, or100mg/kg/d (n=8) for 6 weeks; representative histopathology of liver from rats in BRB-treated group at the sixth week after administration (C) and control group (D), all processed livers were microscopically (Scale bar=50um) examined.*p < 0.05, **p < 0.01 compared with the control group. |
Metabolic Activation of BRB
As a next step, we further explored the underlying mechanism related to BRB-induced hepatotoxicity. We proposed that BRB could be metabolized to electrophilic species, quinone intermediates (M1-M6, scheme 1), which played an important role in BRB-induced toxicities. Detection of quinone metabolites was achieved by incubation of BRB in MLMs with glutathione (GSH) as trapping agents, to form chemically stable GSH conjugates, which were analyzed by UPLC-Q/TOF MS. As a result, a total of four GSH-derived conjugates (M1, M2, M4 and M5) were detected in the incubation mixture. M1 (retention time = 10.75 minutes, Figure 4B) showed its protonated molecule ion at m/z 615.1758 in positive ion mode, which matches the elemental composition of C28H31N4O10S+ (calculated protonated molecular mass: 615.1761, Table 1). As shown in Figure 4C, the tandem mass spectrometry (MS/MS) spectrum of M1 showed the indicative characteristic fragment ions associated with the cleavage of the GSH moiety. The product ions at m/z 540.1441 and 486.0742 were derived from the loss of glycine portion (−75 Da) and γ-glutamyl portion (−129 Da) from m/z 615.1761, respectively. Based upon the characteristic fragment ions: m/z 129.1019 (loss of C5H7O3N), 177.0352 (loss of C10H11O2N), 183.1645 (loss of C12H9ON), 213.1599 (loss of C9H15O3N3), 231.0446 (loss of C9H17O4N3), 308.0891 (loss of C10H18O6N3S), 355.0726 (loss of C19H17O4NS), M1 was identified as a GSH conjugate derived from metabolic activation of section A (scheme 1) of BRB structure. No such conjugate was detected in the microsomal incubation system in the absence of NADPH (Figure 4A), indicating that P450 enzyme was involved in the formation of M1.
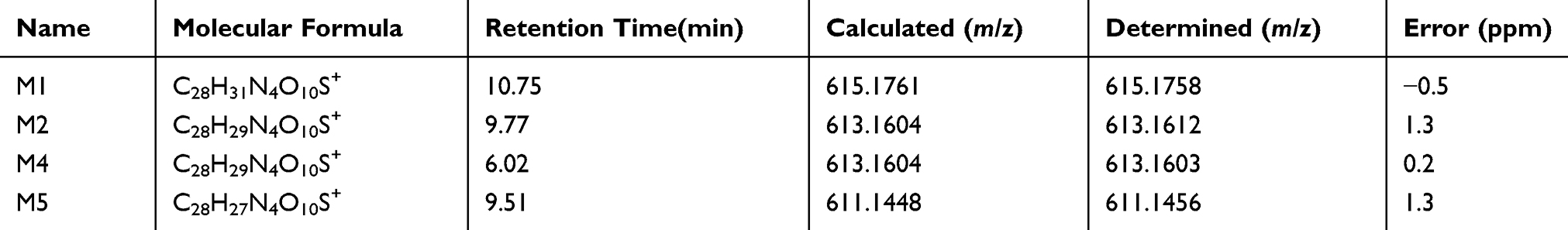 |
Table 1 Mass Spectrometric Profiles of GSH Adducts Derived from Metabolic Activation of BRB |
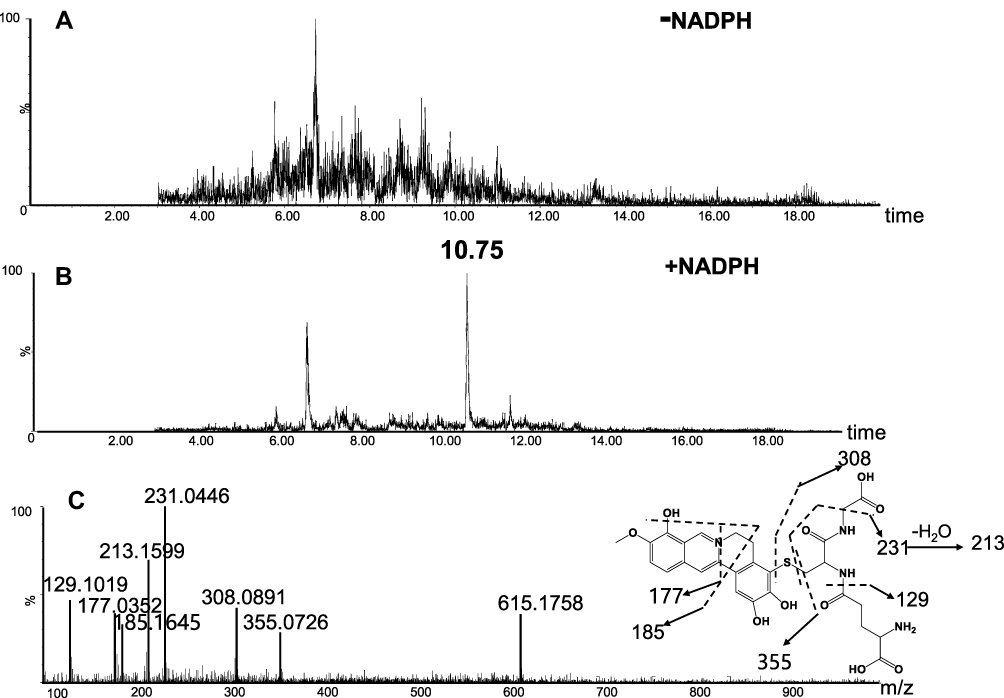 |
Figure 4 Identification of metabolite M1: BRB was incubated with mouse liver microsomes fortified with NADPH and GSH at 37 °C for 1h, followed by LC-MS/MS analysis. Extracted ion chromatogram of M1 obtained from LC-Q-TOF/MS analysis of MLM incubations containing BRB and GSH in the absence (A) or presence (B) of NADPH; MS/MS spectrum of M1 (C). |
M2 (retention time = 9.77 minutes, Figure 5B) and M4 (retention time = 6.02 minutes, Figure 6B) share the same protonated molecular ion at m/z 613.1612 and 613.1603 in positive ion mode, corresponding to the formula of C28H29N4O10S+ (calculated protonated molecular mass: 613.1604, Table 1). The MS/MS spectrum of M2 (Figure 5C) in positive mode elicited the indicative characteristic fragment ions at m/z 129.1030 (loss of C5H7O3N), 231.0453 (loss of C9H17O4N3), 235.0217 (loss of C9H21O4N3), 307.0845 (loss of C10H17O6N3S), 308.0877 (loss of C10H18O6N3S), 340.0641 (loss of C17H14O3N3S), which demonstrated that M2 was produced by metabolic activation of section B (scheme 1) of BRB structure. Interestingly, two unexpected metabolites named as M4 (retention time = 6.02 minutes, Figure 6B) and M5 (retention time = 9.51 minutes, Figure 7B) were observed by the LC-Q/TOF MS. We speculated that BRB was easily bioactivated to dehydro-BRB through dehydrogenation reaction, resulting in formation of more stable conjugated system. The MS/MS spectra acquired for both M4 and M5 showed the major fragmentations produced by loss of 75 Da or 129 Da from their protonated molecular ion, indicating the participation of GSH in the formation of M4 and M5. Based on the MS/MS spectrum of M4 as shown in Figure 6C, M4 was considered as a GSH conjugate resulting from metabolic activation of section A (scheme 1) of dehydro-BRB. M5 exhibited its protonated molecule ion at m/z 611.1456, which matches the elemental composition of C28H27N4O10S+ (calculated protonated molecular mass: 611.1448, Table 1). By comprehensive analysis of the MS/MS spectrum of M5 (Figure 7C), it was speculated to be a GSH conjugate originating from metabolic activation of section B (scheme 1) of dehydro-BRB. The formation of M2, M4 and M5 was also found to be NADPH-dependent (Figures 5A, 6A and 7A). Unfortunately, M3 and M6 derived from two molecules of GSH were not detected in our study.
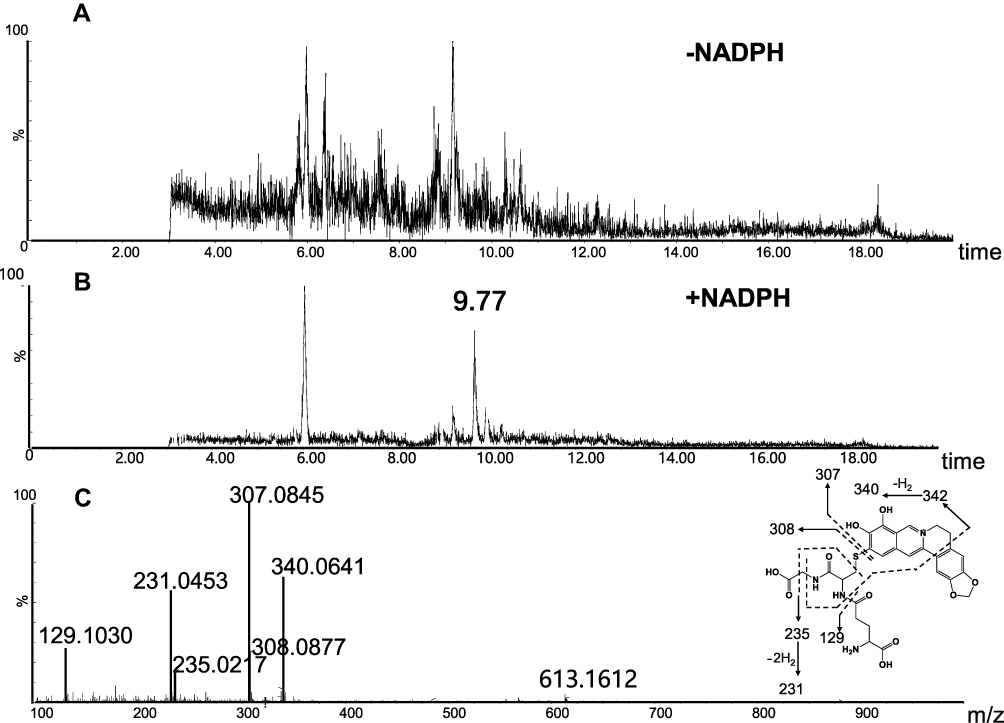 |
Figure 5 Identification of metabolite M2: BRB was incubated with mouse liver microsomes fortified with NADPH and GSH at 37 °C for 1h, followed by LC-MS/MS analysis. Extracted ion chromatogram of M2 obtained from LC-Q-TOF/MS analysis of MLM incubations containing berberrubine and GSH in the absence (A) or presence (B) of NADPH; MS/MS spectrum of M2 (C). |
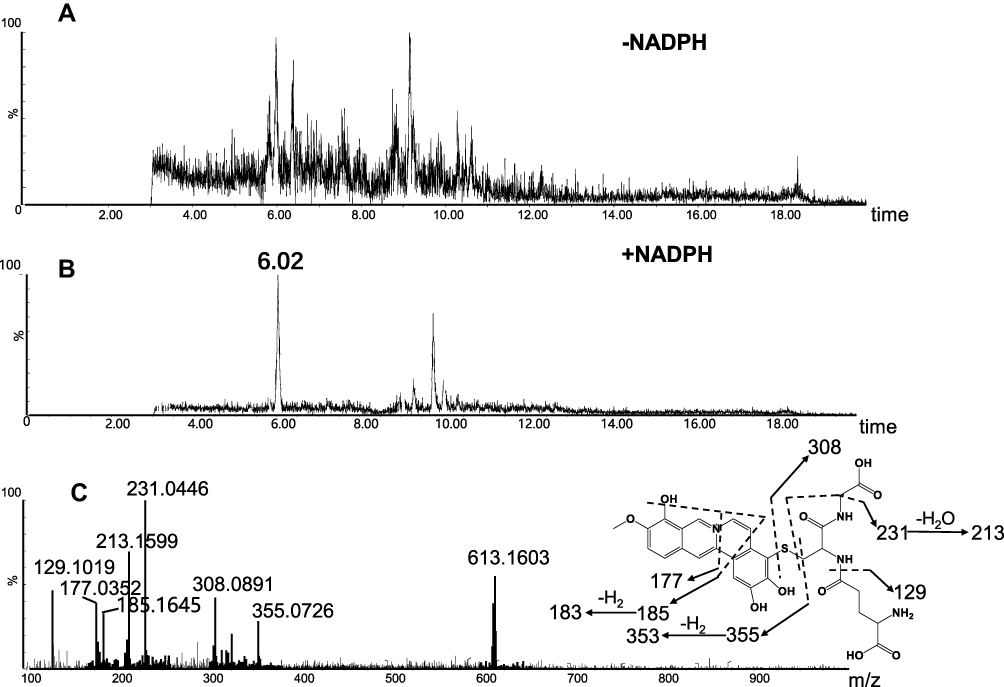 |
Figure 6 Identification of metabolite M4: BRB was incubated with mouse liver microsomes fortified with NADPH and GSH at 37 °C for 1h, followed by LC-MS/MS analysis. Extracted ion chromatogram of M4 obtained from LC-Q-TOF/MS analysis of MLMincubations containing berberrubine and GSH in the absence (A) or presence (B) of NADPH; MS/MS spectrum of M4 (C). |
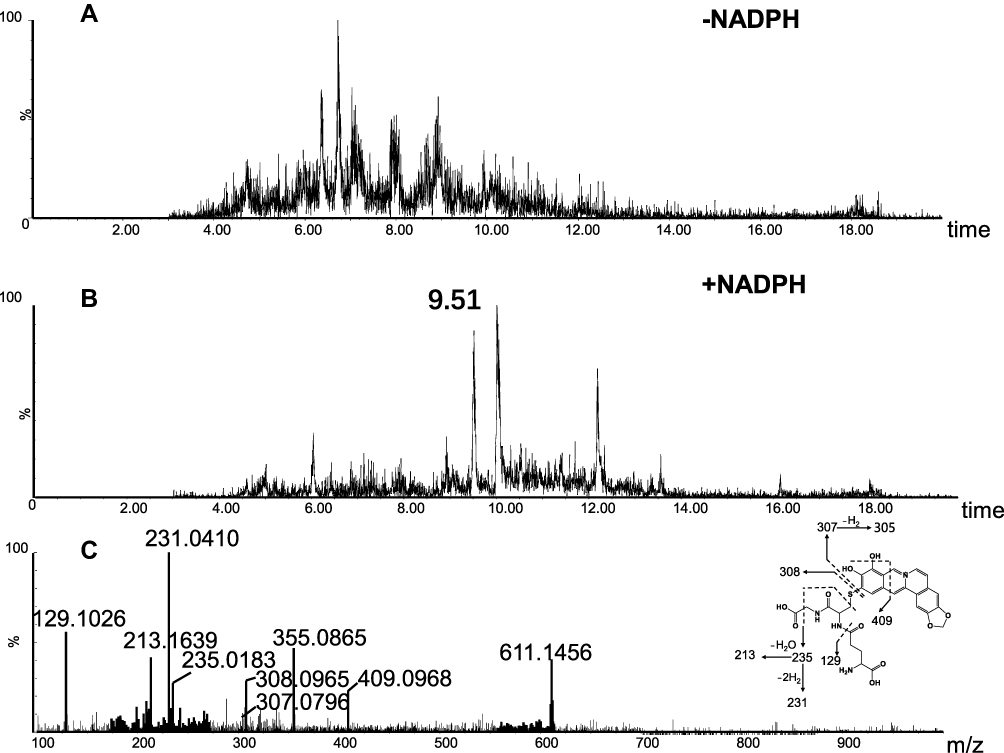 |
Figure 7 Identification of metabolites M5: BRB was incubated with mouse liver microsomes fortified with NADPH and GSH at 37 °C for 1h, followed by LC-MS/MS analysis. Extracted ion chromatogram of M5 obtained from LC-Q-TOF/MS analysis of MLM incubations containing berberrubine and GSH in the absence (A) or presence (B) of NADPH; MS/MS spectrum of M5 (C). |
Cys-Based Protein Adduction by Reactive Metabolite of BRB
The success in the metabolic activation study in vitro encouraged us to further perform protein adduction detection in vivo. As shown in scheme 2, it was speculated that the quinone intermediates of BRB (1–6) reacted with the thiol groups of cysteine residues of cell protein via Michael addition to form protein adductions. However, it may be a challenge to detect these protein adductions for the reason that the structures of proteins are usually complex and their molecular weight is extremely high. Therefore, the resulting thiol-derived protein conjugates were proteolytically digested by chymotrypsin and pronase E. After exhaustive proteolytic digestion, the corresponding cysteine conjugates A1-A6 (scheme 2) produced in the protein samples can be directly analyzed by LC-MS/MS. In this section, BRB-treated mouse liver homogenates were proteolytically digested, followed by LC-MS/MS analysis. As a result, a total of four cys-based conjugates (A1, A3, A5 and A6) were detected in the incubation mixture. A1 (retention time = 11.60 minutes, Figure 8B) had the protonated molecule ion at m/z 429.1142, which matches the molecular weight of the elemental composition C21H21N2O6S+ (calculated protonated molecular mass: 429.1120, Table 2). The MS/MS spectrum of A1 (Figure 8C) in positive mode elicited the indicative characteristic fragment ions at m/z 146.0284 (loss of C8H4O2N), 237.0770 (loss of C15H11O2N), 293.2086 (loss of C18H15O3N), 341.0669 (loss of C18H15O4NS), 385.1009 (loss of C19H15O6NS). So it was inferred that A1 was derived from protein adductions caused by metabolic activation of section A of BRB. A3 (retention time = 7.93 minutes, Figure 9B) showed its protonated molecular ion at m/z 534.0093 in positive ion mode, corresponding to the formula of C23H24N3O8S2+ (calculated protonated molecular mass: 534.1005, Table 2), suggesting that A3 was derived from two molecules of cysteine. By further analysis the MS/MS spectrum (Figure 9C), A3 was identified as a bis-cys–derived BRB conjugate produced by metabolic activation of both section A and B.
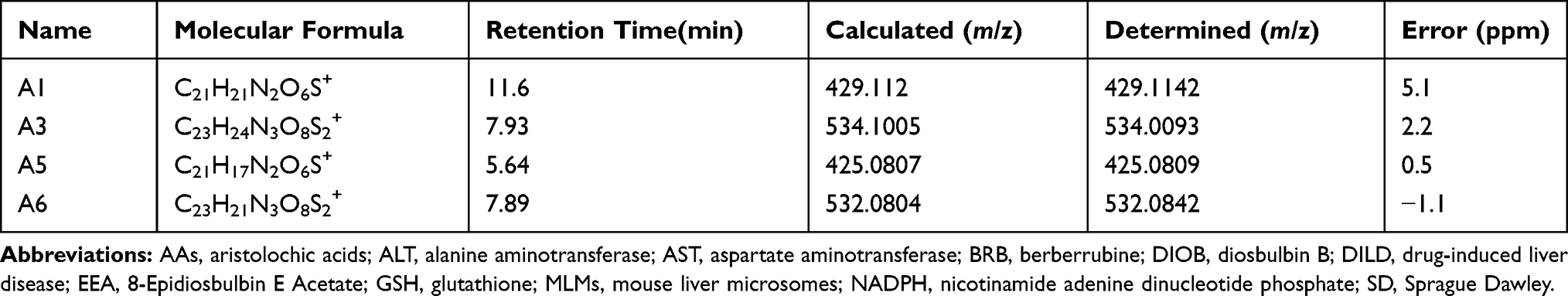 |
Table 2 Mass Spectrometric Profiles of Cys-Based Adducts of BRB in Liver Tissues After Exhaustive Proteolytic Digestion |
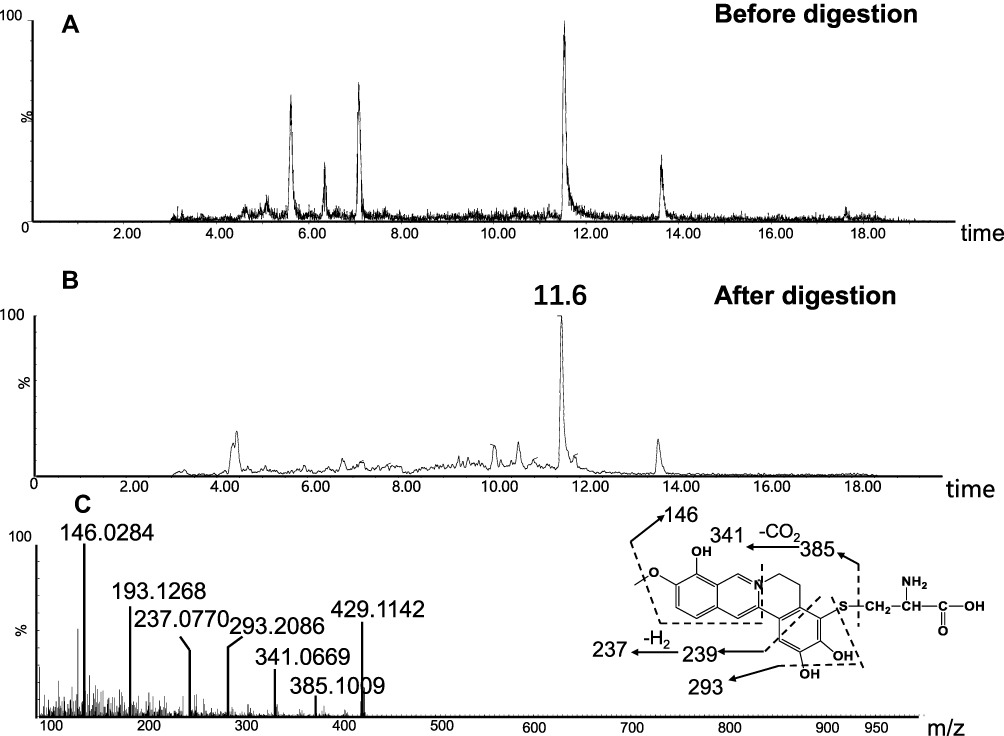 |
Figure 8 Identification of metabolites A1: BRB-treated mouse liver homogenates were proteolytically digested, followed by LC-MS/MS analysis. Extracted ion chromatogram of A1 obtained from LC-Q-TOF/MS before (A) or after exhaustive proteolytic digestion (B); MS/MS spectrum of A1 (C). |
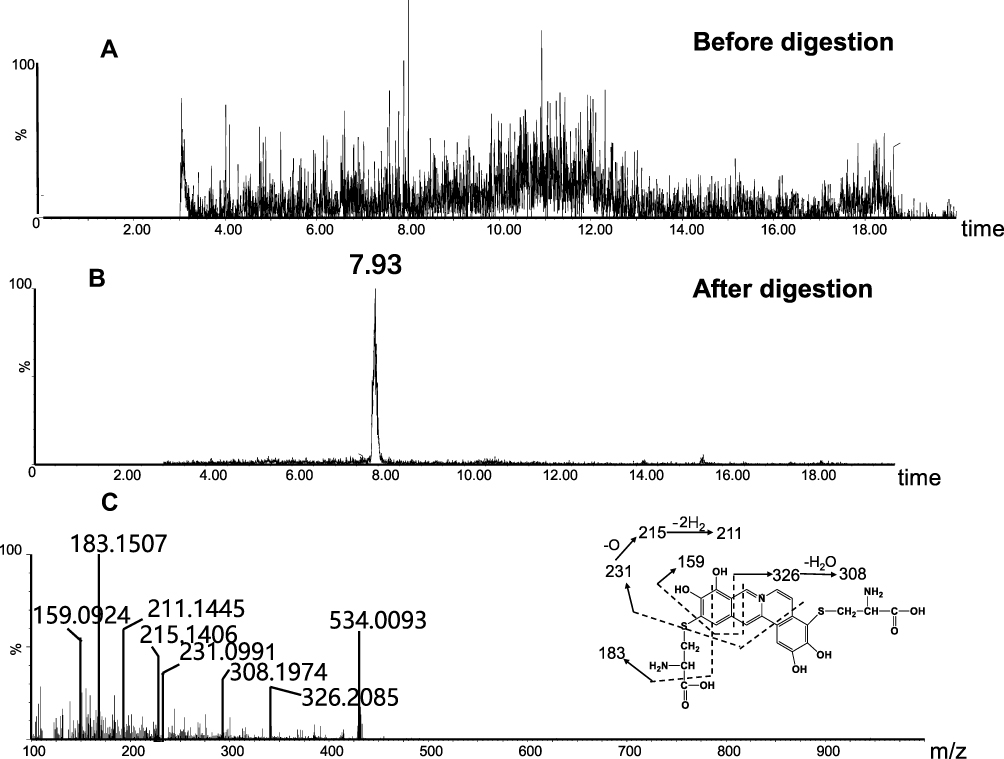 |
Figure 9 Identification of metabolites A3: BRB-treated mouse liver homogenates were proteolytically digested, followed by LC-MS/MS analysis. Extracted ion chromatogram of A3 obtained from LC-Q-TOF/MS before (A) or after exhaustive proteolytic digestion (B); MS/MS spectrum of A3 (C). |
In addition to A1 and A3, another two cys-based conjugates (A5 and A6) were detected at retention times of 5.64 minutes and 7.89 minutes (Figures 10B and 11B), respectively. The protonated molecular ion observed for A5 (m/z 425.0809) was consistent with the molecular weight of the elemental composition of C21H17N2O6S+ (calculated protonated molecular mass: 425.0807, Table 2). The MS/MS spectrum of A5 (Figure 10C) in positive mode showed the indicative characteristic fragment ions at m/z 307.0873 (loss of C18H13O4N), 322.0799 (loss of C18H14O2N2S or C18H14O4N2), 354.0072 (loss of C18H14O4N2S), 364.0455 (loss of C20H16O3N2S), 392.0556 (loss of C21H16O4N2S), 407.0776 (loss of C21H15O5N2S), indicating that A5 was a cys-based conjugate derived from metabolic activation of section B of dehydro-BRB. A6 exhibited its protonated molecular ion at m/z 532.0842 in positive ion mode, corresponding to the formula of C23H22N3O8S2+ (calculated protonated molecular mass: 532.0804, Table 2), which is 2 Da lower than A3. Based on the similar rationale as for the analysis of MS/MS spectra of the above cys-derived conjugates, A6 (Figure 11C) was proposed to be a bis-cys–derived conjugate resulting from metabolic activation of both section A and B of dehydro-BRB (scheme 2). No such conjugates described above were observed in the samples before proteolytic digestion (Figures 8A, 9A, 10A, 11A), indicating that the analytes detected solely resulted from the proteolytic digestion of the adducted protein.
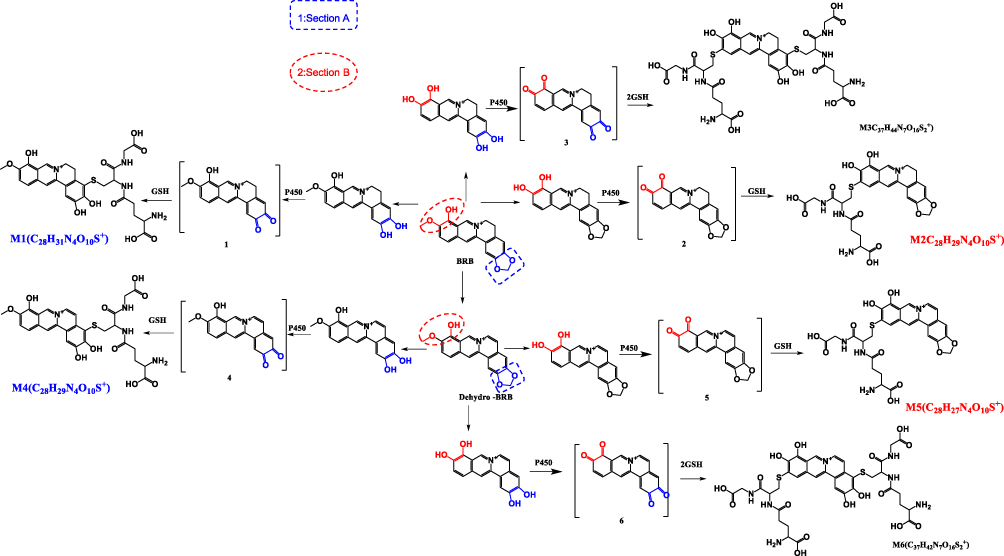 |
Scheme 1 Proposed metabolic activation pathways of BRB (M1-M6). |
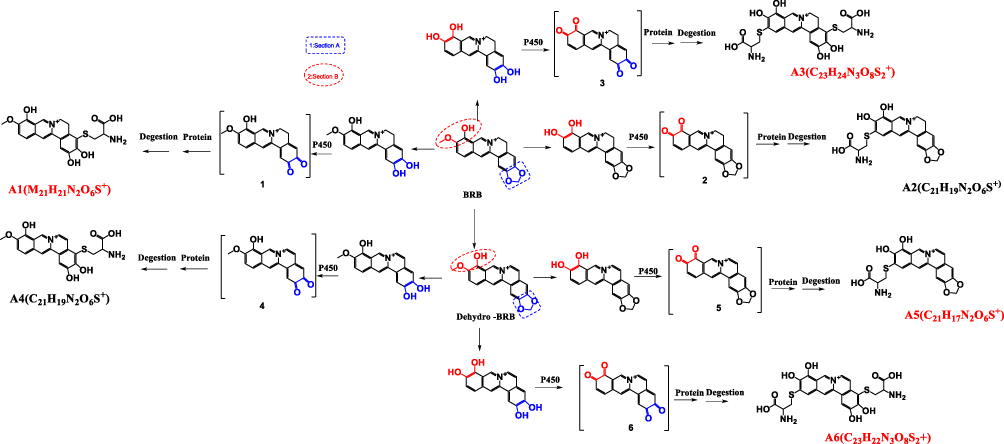 |
Scheme 2 Proposed pathways of protein and amino acid adduct formation as a result of BRB metabolism (A1-A6). |
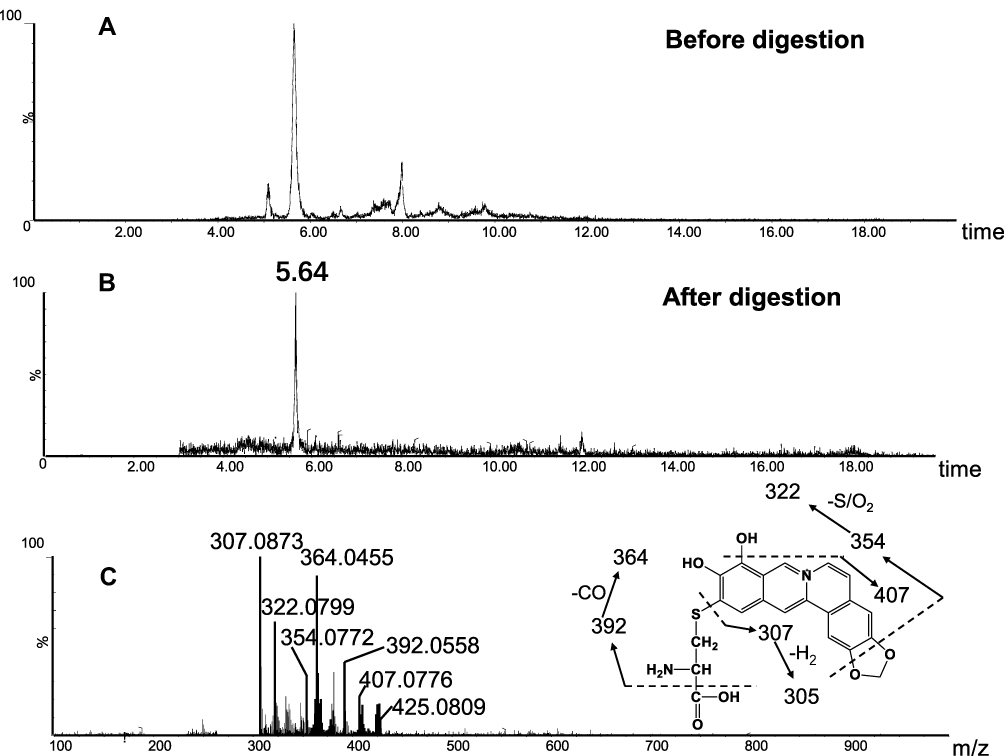 |
Figure 10 Identification of metabolites A5: BRB-treated mouse liver homogenates were proteolytically digested, followed by LC-MS/MS analysis. Extracted ion chromatogram of A5 obtained from LC-Q-TOF/MS before (A) or after exhaustive proteolytic digestion (B); MS/MS spectrum of A5 (C). |
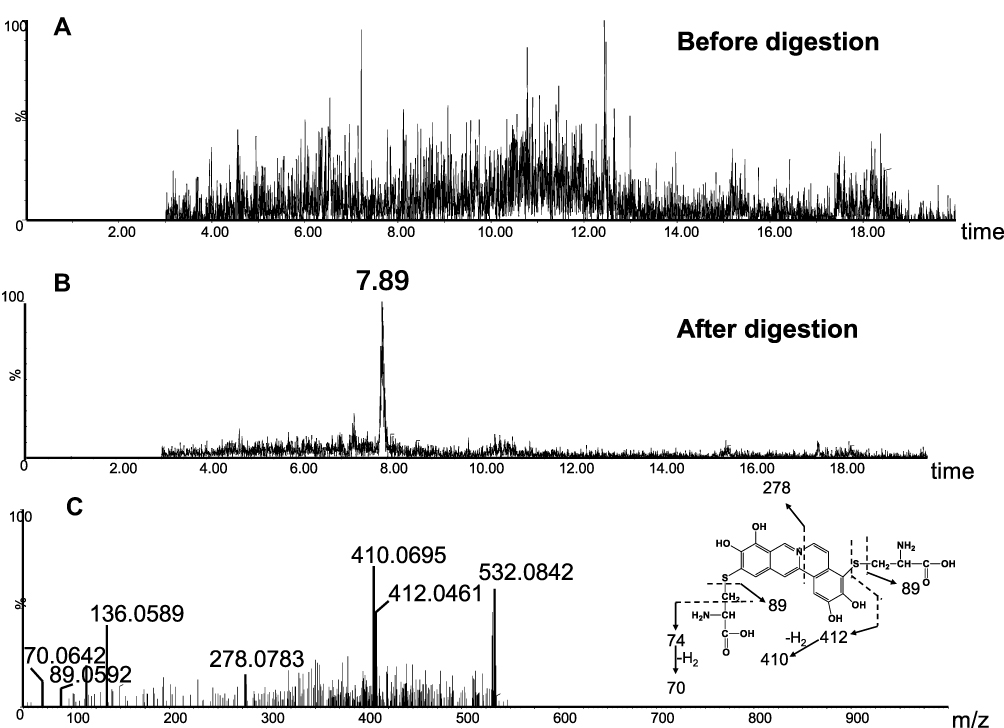 |
Figure 11 Identification of metabolites A6: BRB-treated mouse liver homogenates were proteolytically digested, followed by LC-MS/MS analysis. Extracted ion chromatogram of A6 obtained from LC-Q-TOF/MS before (A) or after exhaustive proteolytic digestion (B); MS/MS spectrum of A6 (C). |
Discussion
BRB, as a major metabolite of berberine, has been reported to induce kidney toxicities in experimental animals. Interestingly, the present study demonstrated that BRB also induced obvious hepatotoxicities in a time- and dose-dependent manner in mice (Figure 2A and B). The serum biochemical indexes, including both ALT and AST, reached maximum 2 h post the administration of a single dose of BRB, which is consistent with a report that BRB can be rapidly absorbed in mice and the time to reach the maximum concentration (Tmax) was about 1 h.29 The administration doses of BRB were 25, 50, 100 mg/kg, which were estimated with reference to the administration dose of BRB in pharmacokinetics or metabolism studies,30,31 as well as the administration dose and LD50 values of berberine, which are reported in the literature.32 With the increase of dose, the ALT levels were increasingly elevated (Figure 2C), while the AST levels were gradually decreased (Figure 2D). ALT is mainly distributed in hepatocyte plasma, which reflects the degree of damage of the liver cell membrane. As for AST, it is mainly distributed in hepatocyte plasma and hepatocyte mitochondria, that indicates hepatocyte injury. ALT can be used as the biochemical index to predict transient damage caused by drugs. The AST level always indicates liver cell damage in various liver diseases, including hepatitis B cirrhosis, fatty liver, alcoholic liver and other hepatobiliary diseases. ALT is more sensitive and accurate than AST in the evaluation of liver damage induced by drugs.33 Furthermore, regulation of AST isozymes during liver disease is complex, including differential, pretranslational and translational or posttranslational mechanisms.34
Sub-chronic hepatotoxicity study of BRB in rats showed that the AST and ALT levels were highest at the first one week, while these biochemical parameters were gradually reduced at the following five weeks (Figure 3A and B). A large number of literature can be referred to provide the possible reason for this experimental results. This phenomenon is common in drug-induced hepatotoxicity.35,36 Drug-induced liver disease (DILD) has been considered as the leading cause of acute liver failure worldwide.37 The liver is not only a major organ for metabolism of many drugs, but also an organ with strong innate immune system, which comprises enrichment of innate immune cells (such as macrophages, natural killer cells, natural killer T cells, NK cells, and NKT cells) and plays an important role in self-repair.38,39 Once toxic metabolites of xenobiotics were produced and induced toxicity, this may elicit sensitization to the adaptive immune system, inducing an immune response directed at hepatocytes as a result of hapten formation.40 Therefore, the repair and regeneration functions of liver may further modulate the development and severity of toxicity with the prolongation of BRB-administration.
Metabolic activation is generally considered as the initial step in the process of drug-induced toxicities. Sequential modification of key proteins or DNA by the electrophilic reactive intermediates is suggested to be an important mechanism of the toxic actions.41,42 A well-known example should be that metabolic activation of aristolochic acid I (AAI) either in the liver or directly in the renal cortex to form a reactive metabolite, a nitrenium/carbenium ion which can further covalently combine with DNA, is confirmed to be the underlying mechanism of AAI-induced nephrotoxicity. Except for forming DNA adducts by reactive metabolites, protein modification is also an important mechanism of toxic action.
In this study, we not only examined the hepatotoxicity of BRB, but also made lots of efforts to reveal the possible mechanism. As the initial step, liver microsomal incubations were employed to explore the metabolic activation of BRB. We speculated electrophilic quinone intermediates produced by oxidation reaction should be the main reactive metabolites of BRB (scheme 1). As expected, four GSH adducts resulting from metabolic activation of section A or B of BRB structure were detected in the microsomal incubation system supplemented with GSH as a trapping agent. However, both M3 and M6 derived from two molecules of GSH were not detected in our study. This indicated that it might be difficult for the reactive metabolite to be attacked by two molecules of GSH at the same time, which was probably related to steric hindrance. Generally, these reactive species are capable of alkylating key cellular proteins and then protein modification by quinone intermediates derived from BRB was investigated. As a result, four cys-based adducts derived from reaction of electrophilic metabolites of BBR and proteins were found in livers. This indicated that the reactive metabolites of BRB covalently modified the cysteine residues of proteins in liver, which may be the possible mechanism of BRB-induced toxicity. Two cys-based adducts, A2 and A4 were not detected in our in vivo study. On one hand, the production of protein adducts in vivo may depend on the physiological conditions and the steric hindrance. On the other hand, the formation and detection of cys-based adducts may be related to the protein digestion process and analysis conditions. Nevertheless, through the study mentioned above, we can conclude that both section A and section B of BRB can undergo metabolic activation, resulting in the formation of the corresponding protein adducts. Moreover, it was determined that the formation of protein adduction derived from BRB exhibited dose dependency in vivo (Figure S3), which is consistent with the dose-dependent trend of toxicity. Besides, the same quinone intermediates derived from BRB were also detected in rat liver microsomal incubations (data not shown).
Berberine, a main bioactive component found in Coptis chinensis Franch. (family Ranunculaceae), Phellodendron chinense Schneid. (family Rutaceae), Berberis vulgaris L. (family Berberidaceae), or Chelidonium majus L. (family Papaveraceae) has been widely accepted to be safe in clinical and significantly effective in the treatment of some clinical disorders, such as type-2 diabetes mellitus, neurodegenerative diseases, cancers and other diseases.16,43–45 BRB has an extremely similar structure with berberine, and is a demethylation metabolite of berberine at section B. We have also compared the metabolic activation of BRB and berberine by using GSH or cysteine as trapping agent in liver microsomal incubation. Interestingly, the production of GSH adducts or cysteine adducts derived from section B of BRB is significantly more than that of berberine (Figure S1 and S2). This provided further evidence that metabolic activation and subsequent protein modification could be one of the main reasons for BRB-induced toxicity. Moreover, this study implicated that quinone intermediates derived from section B of BRB should play a more important role in the toxic action.
Conclusion
In conclusion, we systematically investigated the hepatotoxicity of BRB, and it was found that exposure of BRB in an early stage can lead to hepatotoxicity. Additionally, the underlying mechanism was also clarified in the perspective of metabolic activation. This study provided clear evidence for the formation of quinone metabolites from BRB. The electrophilic metabolites can further covalently react with cysteine residues of protein to form protein adducts, which should be one of the main reasons for BRB-induced hepatotoxicity.
Acknowledgments
This work was supported by the Technology Major Project of China “Key New Drug Creation and Manufacturing Program” (2017ZX09301012-001), the “Major State Basic Research Development Program of China” (2014CB560706), the “National Nature Science Foundation of China” (Grant No. 81803615), and the “Program of 131 Talents of Tianjin” (YL19020231).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Dunnick JK, Singh B, Nyska A, et al. Investigating the potential for toxicity from long-term use of the herbal products, goldenseal and milk thistle. Toxicol Pathol. 2011;39(2):398–409. doi:10.1177/0192623310394211
2. Blowey DL. Nephrotoxicity of over-the-counter analgesics, natural medicines, and illicit drugs. Adolesc Med Clin. 2005;16(1):31–43. doi:10.1016/j.admecli.2004.10.001
3. Perazella MA. Pharmacology behind common drug nephrotoxicities. Clin J Am Soc Nephrol. 2018;13:1897–1908.
4. Randy L, Mark A. Aristolochic acid nephropathy: epidemiology, clinical presentation and treatment. Drug Saf. 2015;38(1):55–64. doi:10.1007/s40264-014-0244-x
5. Debelle DF, Vanherweghem JL, Nortier JL. Aristolochic acid nephropathy: A worldwide problem. Kidney Int. 2008;74(2):158–169. doi:10.1038/ki.2008.129
6. Jadot I, Declèves AE, Nortier J, et al. An integrated view of aristolochic acid nephropathy: update of the literature. Int J Mol Sci. 2017;18(2):297. doi:10.3390/ijms18020297
7. Bastek H, Zubel Z, Stemme K, et al. Comparison of aristolochic acid I derived DNA adduct levels in human renal toxicity models. Toxicology. 2019;420:29–38. doi:10.1016/j.tox.2019.03.013
8. Priestap HA, Santos CD, Quirk JM. Identification of a reduction product of aristolochic acid: implications for the metabolic activation of carcinogenic aristolochic acid. J Nat Prod. 2010;73(12):1979–1986. doi:10.1021/np100296y
9. Li WW, Lin DG, Gao HY, et al. Metabolic activation of furan moiety makes Diosbulbin B hepatotoxic. Arch Toxicol. 2016;90(4):863–872. doi:10.1007/s00204-015-1495-8
10. Wang JM, Ji LL, Liu H, et al. Study of the hepatotoxicity induced by Dioscoreabulbifera L. rhizome in mice. Biosci Trends. 2010;4:79–85.
11. Lin DG, Li CY, Peng Y, et al. Cytochrome P450–mediated metabolic activation of Diosbulbin B. Drug Metab Dispos. 2014;42(10):1727–1736. doi:10.1124/dmd.114.059261
12. Lin DJ, Li WW, Peng Y, et al. Role of metabolic activation in 8-Epidiosbulbin E acetate-induced liver injury: mechanism of action of the hepatotoxic furanoid. Chem Res Toxicol. 2016;29(3):359–366. doi:10.1021/acs.chemrestox.5b00501
13. Wang K, Lin DG, Guo XC, et al. Chemical identity of interaction of protein with reactive metabolite of Diosbulbin B in vitro and in vivo. Toxins. 2017;14(8):249.
14. Zhou SF, Xue CC, Wang GG, Wang G. Metabolic activation of herbal and dietary constituents and its clinical and toxicological implications: an update. Curr Drug Metab. 2007;8(6):526–553. doi:10.2174/138920007781368863
15. Tripathi AN, Chauhan L, Thankacha PP, et al. Quantum chemical and nuclear magnetic resonance spectral studies on molecular properties and electronic structure of berberine and berberrubine. Magn. Reson. Chem. 2007;45(8):647–655. doi:10.1002/mrc.2019
16. Wang K, Feng XC, Chai LW, et al. The metabolism of berberine and its contribution to the pharmacological effects. Drug Metab Rev. 2017;49(2):139–157. doi:10.1080/03602532.2017.1306544
17. Chen ML, Xian YF, Lp SU, et al. Chemical and biological differentiation of Cortex Phellodendri Chinensis and Cortex Phellodendri Amurensis. Planta Med. 2010;76(14):1530–1535. doi:10.1055/s-0030-1249774
18. Gilca M, Gaman L, Panait E, et al. Chelidonium majus-an integrative review: traditional knowledge versus mod-ern findings. Forsch Komplementmed. 2010;17(5):241–248. doi:10.1159/000321397
19. Zhou Y, Cao SJ, Wang Y, et al. Berberine metabolites could induce low density lipoprotein receptor up-regulation to exert lipid-lowering effects in human hepatoma cells. Fitoterapia. 2014;92:230–237. doi:10.1016/j.fitote.2013.11.010
20. Hu JP, Takahashi NT, Yamada T. Coptidis rhizoma inhibits growth and pro-teases of oralbacteria. Oral Dis. 2000;6(5):297–302. doi:10.1111/j.1601-0825.2000.tb00142.x
21. Wright CW, Marshall SJ, Russell PF, et al. In vitro antiplasmodial, antiamoebic, and cytotoxic activities of some monomeric isoquinoline alkaloids. J Nat Prod. 2000;63(12):1638–1640. doi:10.1021/np000144r
22. Baird AW, Taylor CT, Brayden DJ. Non-antibiotic anti-diarrhoeal drugs: factors affecting oral bioavailability of berberine and loperamide in intestinal Tissue.Adv. Drug Deliv Rev. 1997;23(1–3):111–120. doi:10.1016/S0169-409X(96)00429-2
23. Chen GR, Xu YB, Jing J, et al. The anti-sepsis activity of the components of Huanglian Jiedu Decoction with high lipid A-binding affinity[J]. Int Immunopharmacol. 2017;46:87–96. doi:10.1016/j.intimp.2017.02.025
24. Ma HL, Liu Y, Mai X, et al. Identification of the constituents and metabolites in rat plasma after oral administration of HuanglianShangqing pills by ultra high-performance liquid chromatography/quadrupole time-of-flight mass spectrometry[J]. J Pharm Biomed Anal. 2016;125:194–204. doi:10.1016/j.jpba.2016.03.038
25. Huang WY, Dong H. Coptidis Rhizoma-Contained Traditional Formulae for Insomnia: A Potential to Prevent Diabetes?[J]. Chin J Integr Med. 2018;24(10):785–788. doi:10.1007/s11655-018-3012-4
26. Yang N, Sun RB, Zhao YQ, et al. High fat diet aggravates the nephrotoxicity of berberrubine byinfluencingon its pharmacokinetic profile. Environ Toxicol Pharmacol. 2016;46:319–327. doi:10.1016/j.etap.2016.08.003
27. Jin X, Yan TH, Yan Y, et al. Design, synthesis, and anticancer activity of novel berberine derivatives prepared via CuaaC “click” chemistry as potential anticancer agents. Drug Des Devel Ther. 2014;8:1047–1059. doi:10.2147/DDDT.S63228
28. Wang K, Li WW, Chen JM, et al. Detection of cysteine- and lysine-based protein adductions by reactive metabolites of 2,5-dimethylfuran. Anal Chim Acta. 2015;896:93–101. doi:10.1016/j.aca.2015.09.017
29. Wang XQ, Wang SH, Ma JS, et al. Pharmacokinetics in rats and tissue distribution in mouse of berberrubine by UPLC-MS/MS. J Pharm Biomed Anal. 2015;115:368–374. doi:10.1016/j.jpba.2015.07.031
30. Wang K, Chai LW, Feng XC, et al. Metabolites identification of berberine in rats using ultra-high performance liquid chromatography/quadrupole time-of-flight mass spectrometry[J]. J Pharm Biomed Anal. 2017;139:73–86. doi:10.1016/j.jpba.2017.02.038
31. Tan XS, Ma JY, Feng R, et al. Tissue Distribution of Berberine and Its Metabolites after Oral Administration in Rats[J]. PLoS One. 2013;8.
32. Singh N, Sharma B. Toxicological Effects of Berberine and Sanguinarine[J]. Front Mol Biosci. 2018;19:5–21.
33. Kępka A, Zwierz P, Chojnowska S, et al. Relation of plasma carnitine and aminotransferases to alcohol dose and time of dependence[J]. Alcohol. 2019;81:62–69. doi:10.1016/j.alcohol.2019.04.004
34. Pol S, Nalpas B, Vassault A, et al. Hepatic activity and mRNA expression of aspartate aminotransferase isoenzymes in alcoholic and nonalcoholic liver disease[J]. Hepatology. 1991;14:620–625.
35. McGill MR, Jaeschke J. Animal models of drug-induced liver injury[J]. Biochim Biophys Acta Mol Basis Dis. 2019;1865(5):1031–1039. doi:10.1016/j.bbadis.2018.08.037
36. Jaeschke H, Williams CD, Ramachandran A, et al. Acetaminophen hepatotoxicity and repair: the role of sterile inflammation and innate immunity[J]. Liver Int. 2012;32(1):8–20. doi:10.1111/j.1478-3231.2011.02501.x
37. Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov. 2005;4(6):489–499. doi:10.1038/nrd1750
38. Yasemin HB, Ceylan AK, Ufuk KC. Liver immunology and herbal treatment. World J Hepatol. 2017;9(17):757–770. doi:10.4254/wjh.v9.i17.757
39. Gao B, Jeong WI, Tian ZG. Liver: an organ with predominant innate immunity. Hepatology. 2008;47(2):729–736. doi:10.1002/hep.22034
40. Gunawan B, Kaplowitz N. Clinical perspectives on xenobiotic-induced hepatotoxicity. Drug Metab Rev. 2004;36(2):301–312. doi:10.1081/DMR-120034148
41. Wang K, Wang H, Peng Y, et al. Identification of Epoxide-Derived Metabolite(s) of Benzbromarone[J]. Drug Metab Dispos. 2016;44(4):607–615. doi:10.1124/dmd.115.066803
42. Wang K, Lin DJ, Guo XC, et al. Chemical Identity of Interaction of Protein with Reactive Metabolite of Diosbulbin B In Vitro and In Vivo[J]. Toxins. 2017;9(8):249–264. doi:10.3390/toxins9080249
43. Lan JR, Zhao YY, Dong FX, et al. Meta-analysis of the effect and safety of berberine in the treatment of type 2 diabetes mellitus, hyperlipemia and hypertension. J Ethnopharmacol. 2015;161:69–81.
44. Ahmed T, Gilani AH, Abdollahi M, et al. Berberine and neurodegeneration: a review of literature. Pharmacol Rep. 2015;67(5):970–979. doi:10.1016/j.pharep.2015.03.002
45. Kaboli PJ, Rahmat A, Ismail P, et al. Targets and mechanisms of berberine, a natural drug with potential to treat cancer with special focus on breast cancer. Eur J Pharmacol. 2014;740:584–595. doi:10.1016/j.ejphar.2014.06.025
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.