")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 8
Metabolic abnormalities in pituitary adenoma patients: a novel therapeutic target and prognostic factor
Authors Zheng X, Li S, Zhang W, Yang H
Received 9 April 2015
Accepted for publication 11 June 2015
Published 5 August 2015 Volume 2015:8 Pages 357—361
DOI https://doi.org/10.2147/DMSO.S86319
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ming-Hui Zou
Xin Zheng,1 Song Li,1 Wei-hua Zhang,2 Hui Yang1
1Department of Neurosurgery, Xinqiao Hospital, Third Military Medical University, Shapingba District, Chongqing, People's Republic of China; 2Department of Biology and Biochemistry, University of Houston, Houston, TX, USA
Abstract: Metabolic abnormalities are common in cancers, and targeting metabolism is emerging as a novel therapeutic approach to cancer management. Pituitary adenoma (PA) is a type of benign tumor. Impairment of tumor cells' metabolism in PA seems not to be as apparent as that of other malignant tumor cells; however, aberrant hormone secretion is conspicuous in most PAs. Hormones have direct impacts on systemic metabolism, which in turn, may affect the progression of PA. Nowadays, conventional therapeutic strategies for PA do not include modalities of adjusting whole-body metabolism, which is most likely due to the current consideration of the aberrant whole-body metabolism of PA patients as a passive associated symptom and not involved in PA progression. Because systemic metabolic abnormalities are presented by 22.3%–52.5% PA patients and are closely correlated with disease progression and prognosis, we propose that assessment of metabolic status should be emphasized during the treatment of PA and that control of metabolic abnormalities should be added into the current therapies for PA.
Keywords: metabolic abnormalities, pituitary adenoma, therapeutic target, prognostic factor
Introduction and background
Tumor cross talks with its environment, macroenvironment, and microenvironment. The macroenvironment of tumors is a concept relative to that of tumor microenvironment. Al-Zhoughbi et al1 defined “tumor macroenvironment” as the pathological interaction between the tumor cells, as well as the tumor microenvironment, with other organs and systems of the body, which emphasizes the influence of tumors on the whole body. A number of studies have been done to understand the role of tumor microenvironment in tumor progression. Changes in microenvironment such as inflammatory cytokines, angiogenic factors, integrins, matrix metalloproteinases, and hypoxis are known to play important roles in tumor growth.2 Cytokines released by tumor cells can, on the one hand, remodel the microenvironment, and, on the other hand, also influence the homeostasis of the whole body. One of the most consequential changes of the whole body in response to impacts originating from tumors is the metabolic imbalance in tumor patients.
Alterations in metabolisms of protein, lipid, and carbohydrate are often associated with tumor progression. Fearon et al3 found that in lung or colorectal cancer patients, the whole-body protein synthesis was decreased. Increase of muscle protein degradation was observed in noncachectic sarcoma patients, and leucine metabolic abnormalities are specific to tumor growth in humans with high-grade sarcomas.4 Tumor can also directly affect the profile of plasma-free amino acids. Studies on different types of cancers such as breast cancer and non-small cell lung cancer have shown that each type of these cancers has its specific plasma-free amino acid profiles.5,6 Lipid is another important dysregulated metabolite in cancer patients. Dysregulated lipid metabolism is considered a hallmark of cancer.7 Cancer cells can use exogenous fatty acids for membrane synthesis and oncogenic signaling, which can ultimately influence the plasma lipid profile.8 Compared with body mass index (BMI)-matched healthy controls, hematological neoplasia patients exhibit lower concentrations of plasma cholesterol, high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C), and higher concentrations of triglyceride (TG).9 These observations confirm the influence of tumor on the whole-body metabolism.
The whole-body metabolic imbalance may affect the progression of cancer. Progression of cancer is also found to be associated with the so-called metabolic syndrome, which is defined as a syndrome composed of obesity, dyslipidemia, hyperglycemia associated with insulin resistance, and development of diabetes mellitus.10 Obesity has been reported as a cancer risk in colon cancer and rectal cancer,11 pancreatic cancer,12 ovarian cancer,13 breast cancer,14 and liver cancer.15 Recently, Bhaskaran et al16 conducted a population-based cohort study on 5.24 million adults in the United Kingdom, and found that BMI was associated with 17 most common cancers, although the effects varied substantially by tumor site. Hyperglycemia is also linked to cancers independent of BMI. Levine et al’s17 study in 1990 revealed the relationship of postload plasma glucose and 12-year cancer mortality, and the conclusion was that hyperglycemia was related to cancer mortality in men, but not in women. A significantly increased risk of pancreatic cancer and malignant melanoma was found in both male and female hyperglycemia patients.18 Dyslipidemia is another metabolic factor associated with risks of several cancers. Dyslipidemia presents low HDL-C, high LDL-C, and high serum TG levels. Low HDL-C level was associated with non-Hodgkin lymphoma,19 lung cancer,20 prostate cancer,21 and breast cancer.22 High serum TG was associated with postmenopausal breast and prostate cancer in a few studies.23,24 The pathophysiological mechanisms by which metabolic abnormalities participate in tumor progression is complicated. Braun et al25 summarized the potential causal factors. The key factors include insulin and insulin-like growth factor (IGF) system; inflammatory factors such as IL-6, TNF-α, and C-reactive protein; transcription factors such as NF-κB, HIF-1α, PPARs; and other factors such as COX-2, leptin, and adiponectin.25 In general, these observations indicate that tumor-associated whole-body metabolic alterations are not a passive element, but an active participant in tumor progression. Thus, correcting the whole-body imbalance of metabolism may be an effective approach for tumor management.
Pituitary adenoma (PA) is known to be often associated with whole-body imbalance of metabolism due to its aberrant secretion of hormones. In addition to the mass effects, the oversecretion of pituitary hormones is another key clinical manifestation. Even though 39% of PA are hormone silent, namely the nonfunctioning pituitary adenomas (NFPAs),26 yet because of the mass effects of NFPAs, the portal vessels and pituitary stalk can be compressed, which can lead to a decrease in dopamine delivery that can cause hyperprolactinemia in nearly 35% of NFPA patients.27
Pituitary hormones can affect whole-body metabolism directly or indirectly. The most common hormone-secreting pituitary tumors are prolactinomas (40%),28 and 63% of prolactinoma patients have weight gain.29 Prolactin can affect whole-body metabolism by regulating key enzymes and transporters that are associated with lipid and glucose metabolism. Prolactin can act as an autocrine or paracrine factor in human adipose tissue. Prolactin can either bind to its receptor, which induces the activation of the downstream pathways such as dimerization and phosphorylation of Jak2, recruitment of STAT 5a/b, finally alters the transcription of lipid-metabolism-related genes and the secretion of IL-6, adiponectin, and leptin.30 In addition, prolactin can also reduce the excretions of water, sodium, and potassium from the kidney.31 Growth hormone (GH) is another hormone that can significantly affect the whole-body metabolism. The predominant effect of GH is to stimulate lipid oxidation and lipolysis, switching energetic substrate from glucose and protein to lipid.32 In acromegaly patients, hyperinsulinemia and impaired glucose tolerance are quite common. In an acromegaly registry clinical study conducted in France, the prevalence of diabetes in these patients was 22.3%,33 and another study reported a 52.5% prevalence of diabetes in acromegaly patients.34 Additionally, impaired glucose metabolism was also found in 66% of acromegaly patients.35 GH acts through two pathways: one is to bind to GH receptors and then activate JAK2, MAPK, and STATs;36 the other is to induce the production of IGFs that can stimulate proteoglycan, glycosaminoglycan and protein synthesis.37 Adrenal corticosteroid is closely related to whole-body metabolic imbalance. Adrenocorticotropic hormone (ACTH) regulates the secretion of adrenal gland hormones directly. Glucocorticoids can increase blood glucose concentration by affecting glycogen, protein, and lipid metabolisms. Cortisol can stimulate glycogen deposition in the liver, inhibit glucose uptake and use in peripheral tissues, and activate lipolysis in adipose tissue, leading to the release of free fatty acids into circulation. High cortisol levels are thought to be related to the pathogenesis of metabolic syndrome.38 A high prevalence of metabolic syndrome exists in Cushing’s syndrome patients.39 95% of patients have obesity, 75% have hypertension, and 60% have glucose intolerance.40 Prolonged and excessive glucocorticoid exposure and activation of the hypothalamic–pituitary–adrenal axis are involved in the pathogenesis of metabolic syndrome.41 Polymorphisms of human glucocorticoid receptor gene are found to be associated with hypertension and obesity.42 Metabolic syndrome is even considered to be a mild form of Cushing’s syndrome.43 Interestingly, the prevalence of metabolic syndrome in NFPAs is up to 44.7%.39 In a cross-sectional study, TSH in the upper normal range was found to be related to more obese, higher TGs, and increased likelihood of the metabolic syndrome.44 And the thyroid hormone can bind to high-affinity thyroid hormone receptors, which can activate pathways such as NcoR/SMRT, MEK/STATs, ANT, UCPs, and Raf1/MEK/MAPK, thus affecting the synthesis, mobilization, and degradation of protein, carbohydrate, and lipid metabolism.45,46 Leptin is thought to play a role in the regulation of food intake and metabolism. A recent study shows that leptin-treated females and males had significantly elevated serum levels of luteinizing hormone and follicle-stimulating hormone, respectively (Figure 1).47
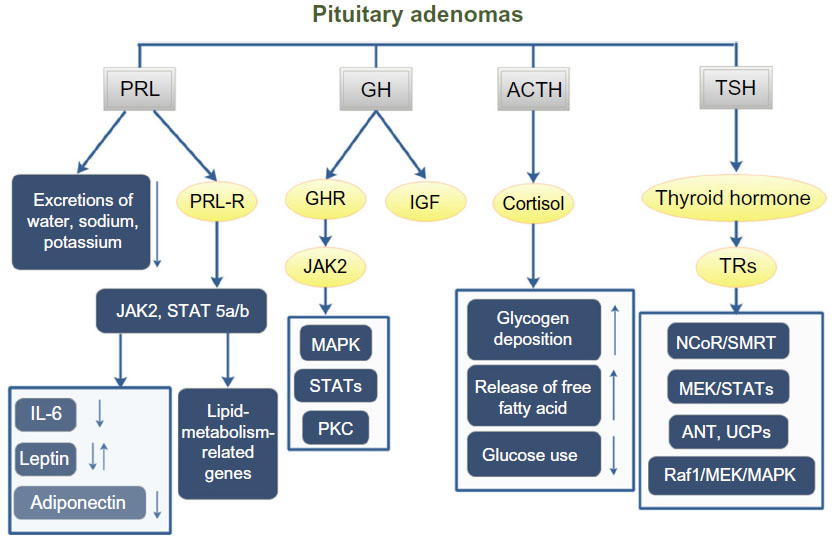 | Figure 1 PA affects the whole-body metabolism through the pituitary hormones. |
Currently, the first-line therapy for PA is surgery, which can be conducted transsphenoidally, transcranially, or in a combination of both. But prolactinomas are an exception.48 Surgery can achieve local control completely or partly by releasing the compression of vital structures. Owing to infiltration to the adjacent structures, tumor tissues can be residual, which may elevate hormone levels and lead to a need for repetitive surgeries or pharmacological treatment. Dopamine agonists and somatostatin analogs are the most widely used medications to treat PA. But some patients with invasive PA are resistant at the beginning or develop resistance to these drugs.28 When surgery and pharmacotherapy have failed, radiotherapy is the third-line therapy.48 With the current procedures of managing PA, the recurrence rate is still not ideal, which is often related to tumor size and cavernous sinus extension.49 In NFPA, the recurrence rate from nondetectable residual tumor is approximately 12%, and from the detectable tumor residuals is approximately 46%.50 With successful surgeries, hormonal control cannot be achieved in every case. In Wang et al’s51 study, the hormonal control rate was 69% of ACTH adenomas, 66% of GH adenomas, 85% of PRL adenomas, and 86% of TSH adenomas.
Imbalance of body metabolism can affect the prognosis of PA. A high rate of postoperative or postmedical therapy imbalance of body metabolism exists. Often, after 6 months of treatment with dopamine agonists, the BMI of prolactinoma patients does not change at all.52 Acromegaly patients with abnormal glucose metabolism need more combined medical therapies than surgery alone.53
Hypothesis
Based on the close association between PA and whole-body metabolic alterations, we propose that it is necessary to assess the status of whole-body metabolism and monitor the metabolic abnormalities before and after surgical or medical therapies, and control of imbalance of body metabolism should be added to the current procedures of treating PA.
Prospects
The metabolic abnormalities of PA patients should not be ignored or just considered as an associated symptom. The effect of pituitary hormones on body metabolism is apparent. But the relationship between the imbalance of whole-body metabolism and prognosis of PA has not gained enough attention. In fact, the prevalence rate of metabolic abnormalities in PA is high. Current therapies cannot relieve the metabolic abnormalities completely, and assessing the changes of metabolic status before and after therapies is not a standard practice. The change of metabolic indexes is a chronic process. Only after 12 months of treatment by dopamine agonists do the prolactinoma patients tend to lose weight, and can cabergoline induce weight loss.52 In the treatment of acromegaly, different drugs affect the body metabolism to different degrees. Somatostatin analog Octreotide failed to change glucose tolerance significantly even though the insulin levels tended to be decreased by the treatment. Pegvisomant, a GH analog and functioning as a specific GH receptor antagonist, can effectively reduce plasma glucose concentrations.54
Metabolic syndrome is a complicated disease. Treatment of metabolic syndrome-associated PA is not simple. Grundy has reviewed the therapies of metabolic syndrome.55 Effective lifestyle therapies, including weight reduction, increasing physical activity, and antiatherogenic diet composition, are the first recommended way to control or slow down the progression of metabolic syndrome. Postponing the initiation of drug therapy as long as possible is preferred.
PA is benign, but has severe clinical complications. The mass effects of tumors can be severe or even fatal such as PA apoplexy, but they can always be remitted immediately by surgeries. Compared with its other life-threatening complications, hormonal disorders seem to be moderate. But even after surgeries, many patients will still suffer endocrine disorders. Postsurgery medical treatments are needed. As metabolic abnormalities are involved in all stages of PA development, the assessment and interference of whole-body metabolism before and after surgery should be considered in order to bring about better outcomes of PA patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Al-Zhoughbi W, Huang J, Paramasivan GS, et al. Tumor macroenvironment and metabolism. Semin Oncol. 2014;41:281–295. | |
Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010;29: 285–293. | |
Fearon KC, Hansell DT, Preston T, et al. Influence of whole body protein turnover rate on resting energy expenditure in patients with cancer. Cancer Res. 1988;48:2590–2595. | |
Inculet RI, Stein TP, Peacock JL, et al. Altered leucine metabolism in noncachectic sarcoma patients. Cancer Res. 1987;47:4746–4749. | |
Cascino A, Muscaritoli M, Cangiano C, et al. Plasma amino acid imbalance in patients with lung and breast cancer. Anticancer Res. 1995;15: 507–510. | |
Maeda J, Higashiyama M, Imaizumi A, et al. Possibility of multivariate function composed of plasma amino acid profiles as a novel screening index for non-small cell lung cancer: a case control study. BMC Cancer. 2010;10:690. | |
Nomura DK, Cravatt BF. Lipid metabolism in cancer. Biochim Biophys Acta. 2013;1831:1497–1498. | |
Louie SM, Roberts LS, Mulvihill MM, Luo K, Nomura DK. Cancer cells incorporate and remodel exogenous palmitate into structural and oncogenic signaling lipids. Biochim Biophys Acta. 2013;1831: 1566–1572. | |
Kuliszkiewicz-Janus M, MaŁecki R, Mohamed AS. Lipid changes occuring in the course of hematological cancers. Cell Mol Biol Lett. 2008;13:465–474. | |
Jaggers JR, Sui X, Hooker SP, et al. Metabolic syndrome and risk of cancer mortality in men. Eur J Cancer. 2009;45:1831–1838. | |
Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86:556–565. | |
Aune D, Greenwood DC, Chan DS, et al. Body mass index, abdominal fatness and pancreatic cancer risk: a systematic review and non-linear dose-response meta-analysis of prospective studies. Ann Oncol. 2012;23:843–852. | |
Olsen CM, Nagle CM, Whiteman DC, et al. Body size and risk of epithelial ovarian and related cancers: a population-based case-control study. Int J Cancer. 2008;123:450–456. | |
Jensen A, Sharif H, Olsen JH, Kjaer SK. Risk of breast cancer and gynecologic cancers in a large population of nearly 50,000 infertile Danish women. Am J Epidemiol. 2008;168:49–57. | |
Larsson SC, Wolk A. Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br J Cancer. 2007;97: 1005–1008. | |
Bhaskaran K, Douglas I, Forbes H, et al. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5.24 million UK adults. Lancet. 2014;384(9945):755–765. | |
Levine W, Dyer AR, Shekelle RB, Schoenberger JA, Stamler J. Post-load plasma glucose and cancer mortality in middle-aged men and women. 12-year follow-up findings of the Chicago Heart Association Detection Project in Industry. Am J Epidemiol. 1990;1:54–62. | |
Stattin P, Björ O, Ferrari P, et al. Prospective study of hyperglycemia and cancer risk. Diabetes Care. 2007;561–567. | |
Morimoto Y, Conroy SM, Ollberding NJ, et al. Erythrocyte membrane fatty acid composition, serum lipids, and non-Hodgkin’s lymphoma risk in a nested case-control study: the multiethnic cohort. Cancer Causes Control. 2012;3:1693–1703. | |
Kucharska-Newton AM, Rosamond WD, Schroeder JC, et al. HDL-cholesterol and the incidence of lung cancer in the Atherosclerosis Risk in Communities (ARIC) study. Lung Cancer. 2008;61:292–300. | |
Kotani K, Sekine Y, Ishikawa S, et al. High-density lipoprotein and prostate cancer: an overview. J Epidemiol. 2013;23:313–319. | |
Kucharska-Newton AM, Rosamond WD, Mink PJ, et al. HDL-cholesterol and incidence of breast cancer in the ARIC cohort study. Ann Epidemiol. 2008;18:671–677. | |
Gaard M, Tretli S, Urdal P. Risk of breast cancer in relation to blood lipids: a prospective study of 31,209 Norwegian women. Cancer Causes Control. 1994;5:501–509. | |
Wuermli L, Joerger M, Henz S, et al. Hypertriglyceridemia as a possible risk factor for prostate cancer. Prostate Cancer Prostatic Dis. 2005;8:316–320. | |
Braun S, Bitton-Worms K, LeRoith D. The link between the metabolic syndrome and cancer. Int J Biol Sci. 2011;7:1003–1015. | |
Buurman H, Saeger W. Subclinical adenomas in postmortem pituitaries: classification and correlations to clinical data. Eur J Endocrinol. 2006;154:753–758. | |
Greenman Y, Stern N. Non-functioning pituitary adenomas. Best Pract Res Clin Endocrinol Metab. 2009;23:625–638. | |
Gillam MP, Molitch ME, Lombardi G, Colao A. Advances in the treatment of prolactinomas. Endocr Rev. 2006;27:485–534. | |
Colao A, Sarno AD, Cappabianca P, et al. Gender differences in the prevalence, clinical features and response to cabergoline in hyperprolactinemia. Eur J Endocrinol. 2003;148:325–331. | |
Ben-Jonathan N, Hugo ER, Brandebourg TD, LaPensee CR. Focus on prolactin as a metabolic hormone. Trends Endocrinol Metab. 2006;17:110–116. | |
Horrobin DF. Prolactin as a regulator of fluid and electrolyte metabolism in mammals. Fed Proc. 1980;39:2567–2570. | |
Rabinowitz D, Zierler KL. A metabolic regulating device based on the actions of human growth hormone and of insulin, singly and together, on the human forearm. Nature. 1963;199:913–915. | |
Fieffe S, Morange I, Petrossians P, et al. Diabetes in acromegaly, prevalence, risk factors, and evolution: data from the French Acromegaly Registry. Eur J Endocrinol. 2011;164:877–884. | |
Dreval AV, Trigolosova IV, Misnikova IV, et al. Prevalence of diabetes mellitus in patients with acromegaly. Endocr Connect. 2014;3:93–98. | |
Bondanelli M, Bonadonna S, Ambrosio MR, et al. Cardiac and metabolic effects of chronic growth hormone and insulin-like growth factor I excess in young adults with pituitary gigantism. Metabolism. 2005;54:1174–1180. | |
Waters MJ, Brooks AJ, Chhabra Y. A new mechanism for growth hormone receptor activation of JAK2, and implications for related cytokine receptors. Jak-Stat. 2014;3:e29569. | |
Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. | |
Park SB, Blumenthal JA, Lee SY, Georgiades A. Association of cortisol and the metabolic syndrome in Korean men and women. J Korean Med Sci. 2011;26:914–918. | |
Webb SM, Mo D, Lamberts SW, et al. Metabolic, cardiovascular, and cerebrovascular outcomes in growth hormone-deficient subjects with previous cushing’s disease or non-functioning pituitary adenoma. J Clin Endocrinol Metab. 2010;95:630–638. | |
Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367:1605–1617. | |
Anagnostis P, Athyros VG, Tziomalos K, Karagiannis A, Mikhailidis DP. Clinical review: the pathogenetic role of cortisol in the metabolic syndrome: a hypothesis. J Clin Endocrinol Metab. 2009;94: 2692–2701. | |
Michailidou Z, Coll AP, Kenyon CJ, et al. Peripheral mechanisms contributing to the glucocorticoid hypersensitivity in proopiomelanocortin null mice treated with corticosterone. J Endocrinol. 2007;194:161–170. | |
Krikorian A, Khan M. Is metabolic syndrome a mild form of Cushing’s syndrome? Rev Endocr Metab Disord. 2010;11:141–145. | |
Ruhla S, Weickert MO, Arafat AM, et al. A high normal TSH is associated with the metabolic syndrome. Clin Endocrinol (Oxf). 2010;72:696–701. | |
Bassett JHD, Harvey CB, Williams GR. Mechanisms of thyroid hormone receptor-specific nuclear and extra nuclear actions. Mol Cell Endocrinol. 2003;213:1–11. | |
Pucci E, Chiovato L, Pinchera A. Thyroid and lipid metabolism. Int J Obes Relat Metab Disord. 2000;24(Suppl 2):S109–S112. | |
Barash IA, Cheung CC, Weigle DS, et al. Leptin is a metabolic signal to the reproductive system. Endocrinology. 1996;137:3144–3147. | |
Colao A, Grasso LFS, Pivonello R, Lombardi G. Therapy of aggressive pituitary tumors. Expert Opin Pharmacother. 2011;12: 1561–1570. | |
Shirvani M, Motiei-Langroudi R. Transsphenoidal surgery for growth hormone-secreting pituitary adenomas in 130 patients. World Neurosurg. 2014;81:125–130. | |
Chen Y, Wang CD, Su ZP, et al. Natural history of postoperative nonfunctioning pituitary adenomas: a systematic review and meta-analysis. Neuroendocrinology. 2012;96:333–342. | |
Wang F, Zhou T, Wei S, et al. Endoscopic endonasal transsphenoidal surgery of 1,166 pituitary adenomas. Surg Endosc. 2014;29(6):1270–1280. | |
Dos Santos Silva CM, Barbosa FR, Lima GA, et al. BMI and metabolic profile in patients with prolactinoma before and after treatment with dopamine agonists. Obes (Silver Spring). 2011;19:800–805. | |
Cheng S, Al-Agha R, Araujo PB, et al. Metabolic glucose status and pituitary pathology portend therapeutic outcomes in acromegaly. PLoS One. 2013;8:e73543. | |
Møller N, Jørgensen JOL. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev. 2009;30:152–177. | |
Grundy SM. Drug therapy of the metabolic syndrome: minimizing the emerging crisis in polypharmacy. Nat Rev Drug Discov. 2006;5: 295–309. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.