")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Mesenchymal stem cell-based NK4 gene therapy in nude mice bearing gastric cancer xenografts
Authors Zhu Y, Cheng M, Yang Z, Zeng C, Chen J, Xie Y, Luo S, Zhang K, Zhou S, Lu N
Received 20 July 2014
Accepted for publication 22 August 2014
Published 9 December 2014 Volume 2014:8 Pages 2449—2462
DOI https://doi.org/10.2147/DDDT.S71466
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Yin Zhu,1,* Ming Cheng,2,* Zhen Yang,3 Chun-Yan Zeng,3 Jiang Chen,3 Yong Xie,3 Shi-Wen Luo,3 Kun-He Zhang,3 Shu-Feng Zhou,4 Nong-Hua Lu1,3
1Department of Gastroenterology, 2Department of Orthopedics, 3Institute of Digestive Disease, The First Affiliated Hospital of Nanchang University, Jiangxi, People’s Republic of China; 4Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA
*These authors contributed equally to this work
Abstract: Mesenchymal stem cells (MSCs) have been recognized as promising delivery vehicles for gene therapy of tumors. Gastric cancer is the third leading cause of worldwide cancer mortality, and novel treatment modalities are urgently needed. NK4 is an antagonist of hepatocyte growth factor receptors (Met) which are often aberrantly activated in gastric cancer and thus represent a useful candidate for targeted therapies. This study investigated MSC-delivered NK4 gene therapy in nude mice bearing gastric cancer xenografts. MSCs were transduced with lentiviral vectors carrying NK4 complementary DNA or enhanced green fluorescent protein (GFP). Such transduction did not change the phenotype of MSCs. Gastric cancer xenografts were established in BALB/C nude mice, and the mice were treated with phosphate-buffered saline (PBS), MSCs-GFP, Lenti-NK4, or MSCs-NK4. The tropism of MSCs toward gastric cancer cells was determined by an in vitro migration assay using MKN45 cells, GES-1 cells and human fibroblasts and their presence in tumor xenografts. Tumor growth, tumor cell apoptosis and intratumoral microvessel density of tumor tissue were measured in nude mice bearing gastric cancer xenografts treated with PBS, MSCs-GFP, Lenti-NK4, or MSCs-NK4 via tail vein injection. The results showed that MSCs migrated preferably to gastric cancer cells in vitro. Systemic MSCs-NK4 injection significantly suppressed the growth of gastric cancer xenografts. MSCs-NK4 migrated and accumulated in tumor tissues after systemic injection. The microvessel density of tumor xenografts was decreased, and tumor cellular apoptosis was significantly induced in the mice treated with MSCs-NK4 compared to control mice. These findings demonstrate that MSC-based NK4 gene therapy can obviously inhibit the growth of gastric cancer xenografts, and MSCs are a better vehicle for NK4 gene therapy than lentiviral vectors. Further studies are warranted to explore the efficacy and safety of the MSC-based NK4 gene therapy in animals and cancer patients.
Keywords: gastric cancer, gene therapy, tumor xenograft, hepatocyte growth factor, lentivirus, angiogenesis, apoptosis
Introduction
Stomach or gastric cancer is the fifth most common cancer and the third leading cause of death from cancer globally with approximately 952,000 new cases and 723,000 deaths making up 7% of all cancer cases and 9% of deaths.1,2 Almost two-thirds of gastric cancer cases occur in developing countries and 42% in the People’s Republic of China accounting for 3.99% of all deaths.3 The Republic of Korea had the highest rate of stomach cancer, followed by Mongolia, Japan, Guatemala, People’s Republic of China, Tajikistan, Kazakhstan, Kyrgyzstan, Albania, and Belarus. There are about 22,220 new cases of stomach cancer and 10,990 deaths every year in the United States. In the United Kingdom, 7,089 people were diagnosed with stomach cancer and 4,830 deaths due to gastric cancer were recorded in 2011.3 Primary treatment modalities for stomach cancer include surgery, chemotherapy, radiation therapy, and biological therapy. Despite advances in past decades in treatment of early gastric cancer, early-stage disease accounts for only 10% to 20% of all cases diagnosed and most gastric cancer patients are still diagnosed with advanced stage disease and their 5 years relative survival rate is as low as <10%.2–5 Therefore, it is necessary to identify novel therapeutic strategies for later stage gastric cancer. Gene therapies for solid tumors have been extensively investigated in animal models, but in clinical trials, very few gene therapies have demonstrated antitumor effects on advanced solid tumours.6 This poor outcome relates, at least in part, to our inability to deliver therapeutic agents specifically and efficiently to the tumor tissues.7 Selective gene transfer to the target organ/cells, eg, the stomach cancer, is a major challenge for gene therapy.8
Towards this end, our research is focused on mesenchymal stem cells (MSCs) as a gene delivery vehicle. These plastic, adherent cells isolated from bone marrow are capable of self-renewal and have the potential to differentiate into mesenchymal and non-mesenchymal tissues.9,10 Thus, they may be advantageous for tumor gene therapy because MSCs exhibit tropism to the sites of tissue damage as well as cancer lesions.11,12 Migration of MSCs to tumor lesions is thought to be due to similarities in the inflammatory milieu produced by healing wounds and tumors, evoking the notion that “tumors are wounds that never heal”.13 To date, MSCs have been investigated in the treatment of a number of diseases, such as myocardial infarction, Alzheimer’s disease, Parkinson’s disease, Crohn’s disease, diabetes, stroke, pulmonary arterial hypertension, and cancer.14–17 Furthermore, MSCs can be used for autologous transplantation to avoid host immune response.14,18,19 The homing capacity of MSCs has been demonstrated with almost all tested human cancer cell lines.20–22
Hepatocyte growth factor (HGF) was first identified and cloned as a mitogenic polypeptide for hepatocytes.23 Subsequent studies have revealed that HGF is a multifunctional growth factor that stimulates mitogenesis and morphogenesis in a variety of epithelial and endothelial cells.24 The receptor for HGF is the c-Met proto-oncogene product.25 HGF also mediates tumor–stromal interactions that confer invasive, angiogenic, and metastatic potentials to cancer through HGF/c-Met receptor binding.26–29 NK4 is composed of the N-terminal hairpin and subsequent four kringle domains of HGF.30–32 NK4 not only acts as a competitive antagonist for HGF but also independently acts as a potent angiogenesis inhibitor.30,33,34 Considering these bifunctional properties (angiogenesis inhibitor and HGF antagonist), NK4 gene therapy could potentially be an ideal treatment for human cancer.32,35 For example, virus vector-mediated NK4 gene therapy suppressed tumor growth and metastasis in nude mice.36–38 However, the systemic administration of viral vectors often causes severe adverse effects such as severe liver toxicity and immune responses.39 In this regard, we investigated MSCs as a gene delivery vehicle carrying NK4 gene therapy to gastric cancer xenografts in nude mice in comparison to a lentiviral vector carrying NK4 and other controls in this study.
Materials and methods
Culture and identification of human MSCs
Human MSCs were isolated by adherence purification as described previously by Mohr et al40 from the bone marrow of young patients with femur fracture during open reduction and internal fixation in our hospital. These patients were otherwise healthy before hospitalization. The separated MSCs were subcultured at a density of 6,000–8,000 cell/cm2 in a low-glucose Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (HyClone, Thermo Fisher Scientific), 2 mmol/L L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific) and used for experiments during passages 3 to 5. The cell markers CD34, CD44, CD45, and CD105 were used to identify the MSCs upon fluorescence-activated cell sorter (FACS, Becton, Dickinson and Company, NJ, USA) with specific antibodies (eBioscience, San Diego, CA, USA). This study was approved by the Ethics Committee of the First Affiliated Hospital, Nanchang University, Nanchang, Jiangxi, People’s Republic of China, and each patient signed an informed consent form.
Cell lines and culture
A human gastric carcinoma cell line MKN45 was kindly provided by Dr Dai-Ming Fan of Xi-Jing Hospital of Digestive Diseases, Xi’an, Shanxi, People’s Republic of China, and cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Thermo Fisher Scientific). Human fibroblasts were kindly provided by Dr Li-Xin Liao of the Department of Burn Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People’s Republic of China, and cultured in low-glucose DMEM. A normal human gastric epithelial cell line, GES-1, was cultured in high glucose DMEM. All media were supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum. Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2/95% air.
NK4 lentiviral vector construction, production, and cell transduction
The NK4 gene was acquired by polymerase chain reaction from human HGF complementary DNA (cDNA) (German Resource Center RZPD, Berlin, Germany). The primers used to amplify A fragment (521 bp) of NK4 cDNA gene from human HGF cDNA included: forward, 5′-GAGGATCCCCGGGTACCGGTCGCCACCAT GTGGGTGACCAAACTCC-3′, and reverse, 5′-CGAAGGCAAAAAGCTGTGTTCGTGT GGTATCATGG-3′; and the primers to amplify B fragment (976 bp) of NK4 cDNA gene from HGF cDNA: forward, 5′-CACAGCTTTTTGCCTTCGAGCTATCGGGGTAAAGACC-3′, reverse, 5′-TCACCATGGTGGCGACCGGGACTATTGTAGGTGTGGTATC-3′. A and B fragments were used as the templates for amplification of full-length NK4 cDNA gene (1,479 bp), and the primers were: forward, 5′-GAGGATCCCCGGGTACCGGCGCCACCATGTGGGTGACCAAACTCC-3′, and reverse, 5′-TCACCATGGTGGCGACCGGGACTATTGTAGGTGTGGTATC-3′. A lentiviral plasmid pGC-FU carrying the enhanced green fluorescent protein (GFP) gene (GeneChem Co., Ltd., Shanghai, People’s Republic of China) was used as a backbone for subcloning the NK4 fragment. Purified polymerase chain reaction products containing the NK4 gene coding sequence were ligated with pGC-FU-GFP vector by In-Fusio convertase (BD Biosciences, San Jose, CA, USA). The recombinant pGC-FU-GFP-NK4-plasmids, the construction plasmids Helper1.0, and the envelope plasmids Helper2.0 (GeneChem Co., Ltd.,) were cotransfected into human embryonic kidney epithelial 293T cells mediated by Lipofectamine 2000 (Thermo Fisher Scientific). Lentiviral vectors carrying the NK4 fragment (Lenti-NK4) or GFP (Lenti-GFP) were produced and the lentiviral titer was detected as described previously.41
After production of these lentiviral vectors, MSCs were transduced with Lenti-NK4 (MSCs-NK4) or Lenti-GFP (MSCs-GFP) at a multiplicity of infection (MOI) of 50. NK4 constructed in the lentiviral plasmid was in a secreting form and NK4 was continuously expressed and released from MSCs. Expression of NK4 was detected by enzyme linked immunosorbent assay (ELISA) and Western blotting assay as previously described.39 GFP expression was analyzed by flow cytometry using the FACSCalibur system (Becton Dickinson Co., Franklin Lakes, NJ, USA) or a fluorescence microscope (LSM700, Carl Zeiss Meditec AG, Jena, Germany).
Protein extraction and Western blotting analysis
MSCs-NK4, MSCs-GFP, or MSCs were lysed in a buffer containing 0.5% Lubrol-PX, 50 mM KCl, 2 mM CaCl2, 20% glycerol, 50 mM Tris-HCl at pH 7.4, 0.1% protease, and 1% phosphatase inhibitors (Sigma-Aldrich Co., St Louis, MO, USA), and all cellular lysates were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The proteins were transferred onto a nitrocellulose membrane (Bio-Rad Laboratories Inc., Hercules, CA, USA). After incubation with a rabbit polyclonal anti-HGF antibody at a dilution of 1:1,000 (Santa Cruz Biotechnology Inc., Dallas, TX, USA), the blots were washed with phosphate-buffered saline (PBS) and then incubated with a goat anti-rabbit Immunoglobulin G conjugated with horseradish peroxidase (Santa Cruz Biotechnology Inc.). Bands were visualized by enhanced chemiluminescence (Thermo Fisher Scientific). Recombinant human HGF (Peprotech, Rocky Hill, NJ, USA) was used as a positive control.
Cell migration assay
The tropism of MSCs to gastric cancer cells was determined using a Transwell migration assay with 8 μm pore size membranes (Corning Incorporated, Corning, NY, USA). MNK45 or GES-1 cells were cultured for 24 hours in serum-free medium and then plated into the bottom wells. MSCs, MSCs-NK4, or human fibroblasts (2×104) in 300 μL serum-free medium were seeded into the upper chamber and cultured for 24 hours at 37°C. The MSCs, MSCs-NK4, or human fibroblasts that had attached to the top side of the membrane were removed by a cotton swab, and the cells that had migrated onto the bottom side were fixed with 4% paraformaldehyde, stained using crystal violet, and counted (five fields per well for triplicate wells) at 10× magnification with an Olympus BH-2 microscope (Olympus Corporation, Tokyo, Japan).
Nude mouse xenograft cancer model
BALB/C nude mice (6–8 weeks old) from the National Institutes for Food and Drug Control Laboratory Animal Center (Beijing, People’s Republic of China) were bred and kept under standard pathogen-free conditions. All animal studies were performed in accordance with institutional guidelines under the approved protocols. Xenografts were initiated by subcutaneous injection of 2×107 MKN45 cells in 200 μL PBS into the right flank near the axillary fossa. The mouse was euthanized when the longest diameter of the tumor reached 10 mm, and the tumor was aseptically dissected and mechanically minced. Then 2 mm3 pieces of tumor tissue were transplanted into the right flank near the axillary fossa of mice. Tumors were measured every 3 days with calipers. When tumors were 1.5 cm in the longest diameter, mice were randomly assigned to one of four experimental groups.
Treatment of gastric cancer nude mouse xenografts
To evaluate the therapeutic effects of MSCs-NK4 in vivo, tumor-bearing nude mice were injected with MSCs-GFP or MSCs-NK4 (6×105 cells in 0.2 mL PBS), Lenti-NK4 (3×107 viral particles), or PBS (0.2 mL) in the lateral tail vein for systemic administration at 1, 7, 14, and 21 days (eight animals per group). Three different diameters of each tumor were measured twice a week. The tumor volume was calculated using the formula: π/6 (0.5326) × length (mm) × width (mm) × height (mm).42 Animals were sacrificed on the 28th day after injections, and none of the mice had to be sacrificed during experiment due to tumor ulceration, bleeding, or moribund state with excessive weight loss exceeding 25% of initial weight before the experiment. Tumor xenografts were excised and weighed to assess tumor burden. Results were evaluated as mean tumor volume or weight ± standard error (SEM).
Histopathological examination of tumor xenografts
Tumor xenografts were fixed in 4% buffered formalin and embedded in paraffin for sectioning and hematoxylin and eosin (H & E) staining. Histopathology was evaluated blindly under a light microscope by a pathologist. Tumor necrosis was scored according to the method described previously.43 In brief, necrotic tissues observed in 1%–20% of the regions of interest were scored as 1, and 21%–40%, 41%–60%, 61%–80%, and 81%–100% of necrotic tissues were scored as 2, 3, 4, and 5, respectively. In contrast, if necrosis occupied <1% of the regions of interest, the score was 0. Next, the mean regions of the interest were calculated and designated as the score of the tumor xenograft. Partial tumor xenografts were snap-frozen for fluorescence microscopy observation of MSCs in fresh cryosections (6–8 μm).
Immunohistochemistry
Paraffin-embedded tumor sections (4 μm) were used for immunostaining of proliferating cell nuclear antigen (PCNA) with a monoclonal primary mouse anti-human PCNA antibody at a dilution of 1:100 (from Santa Cruz Biotechnology Inc.). The proliferative index was defined as the percentage of positively stained cells among 100 nuclei from five randomly selected fields at 200× magnification. The microvessel density was assessed using a monoclonal mouse anti-human CD31 antibody (1:100; Santa Cruz Biotechnology Inc.). Any distinct area of positive staining for CD31 was counted as a single vessel. The number of vessels was counted in five fields at 200× magnification, and the results were expressed as the mean number of vessels ± SEM. The negative control sections for PCNA and CD31 were obtained by omitting the primary antibody incubations.
TUNEL apoptosis assay
Cellular apoptosis was detected by terminal deoxynucleotidyl tranferase-mediated dUTP nick end-labeling (TUNEL) using an in situ apoptosis detection kit (Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s instructions. For the positive control, the section was treated with DNase I, whereas the negative control was prepared with the omission of terminal deoxynucleotidyl transferase incubation. The total number of apoptotic cells in five randomly selected fields (400× magnification) per tumor xenograft was counted under a microscope. The apoptotic index was expressed as the percentage of positively stained cells among 100 nuclei per field.
Statistical analysis
Data are presented as the mean ± SEM. One-way analysis of variance (ANOVA) was used to compare the means from multiple experimental groups, followed by Dunnett’s post hoc test. Statistical differences were considered to be significant when P<0.05 in a two-tailed test.
Results
Isolation and culture of human MSCs
Attached cells were observed on day three after the initial plating of human MSCs. Our data showed that the cultures underwent an initial lag phase growth lasting approximately 6 days, during which the colonies were seen to arise from single cells. Cells had a typical spindle shape, consistent with the morphology reported by others (Figure 1A and B).44,45 Cells were primarily expanded when they reached confluence after 10–14 days’ culture. A population of fibroblast-like MSCs were observed microscopically after cells were expanded in three passages (Figure 1C) and verified using flow cytometry. The isolated cells were negative for typical hematopoietic antigens CD45 and CD34 but were positive for CD44 and CD105 in each culture (Figure 1D). Therefore, the isolated and cultured cells exhibited typical properties of human MSCs as described previously.44–49
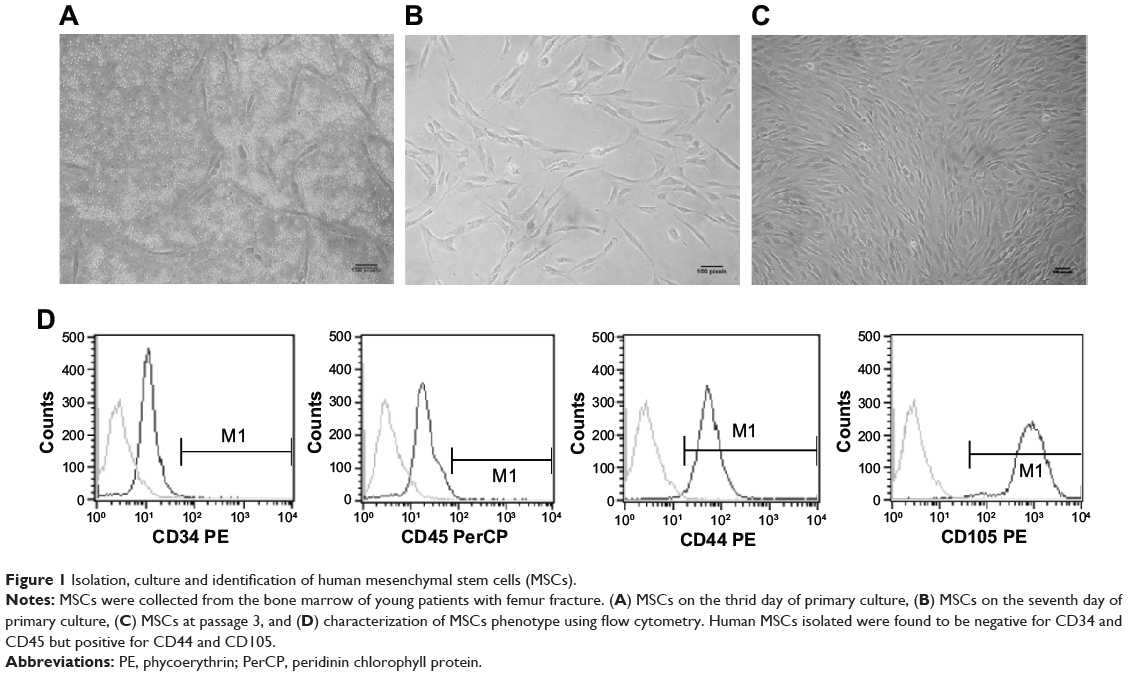
|
Figure 1 Isolation, culture and identification of human mesenchymal stem cells (MSCs). |
Transduction efficiency of MSCs with lentiviral vectors and expression of NK4 in Lenti-NK4-transduced MSCs
Next, MSCs were stably transduced with Lenti-NK4 or Lenti-GFP, optimized using different MOIs (10–100). The data showed that GFP expression in the MSCs was increased with increasing MOI. MSCs tolerated an MOI of 50, and this MOI was used for further experiments, producing a more than 85% successful transduction rate at day three with 4.26% cell death only (Figure 2A and B). Moreover, the NK4 level in cell culture supernatants was also significantly increased with increasing MOI up to 100 (P<0.001, by one-way ANOVA; Figure 2C). At an MOI of 50, the NK4 level in the cell culture supernatants of MSCs-NK4 was increased and reached the highest level at day five (2473.38±155.82 pg/mL, Figure 2D). GFP expression in MSCs-NK4 was also increased over 3 days of incubation (Figure 2E). The Western blot data showed that the NK4 protein was also present in Lenti-NK4-infected MSC lysates, but not in that of Lenti-GFP or non-infected MSCs (Figure 2F). The phenotype of the MSCs-NK4 and MSCs-GFP was the same as the non-transduced MSCs, suggesting that the lentiviral-mediated transduction did not alter the phenotypes of MSCs (Figure 3).
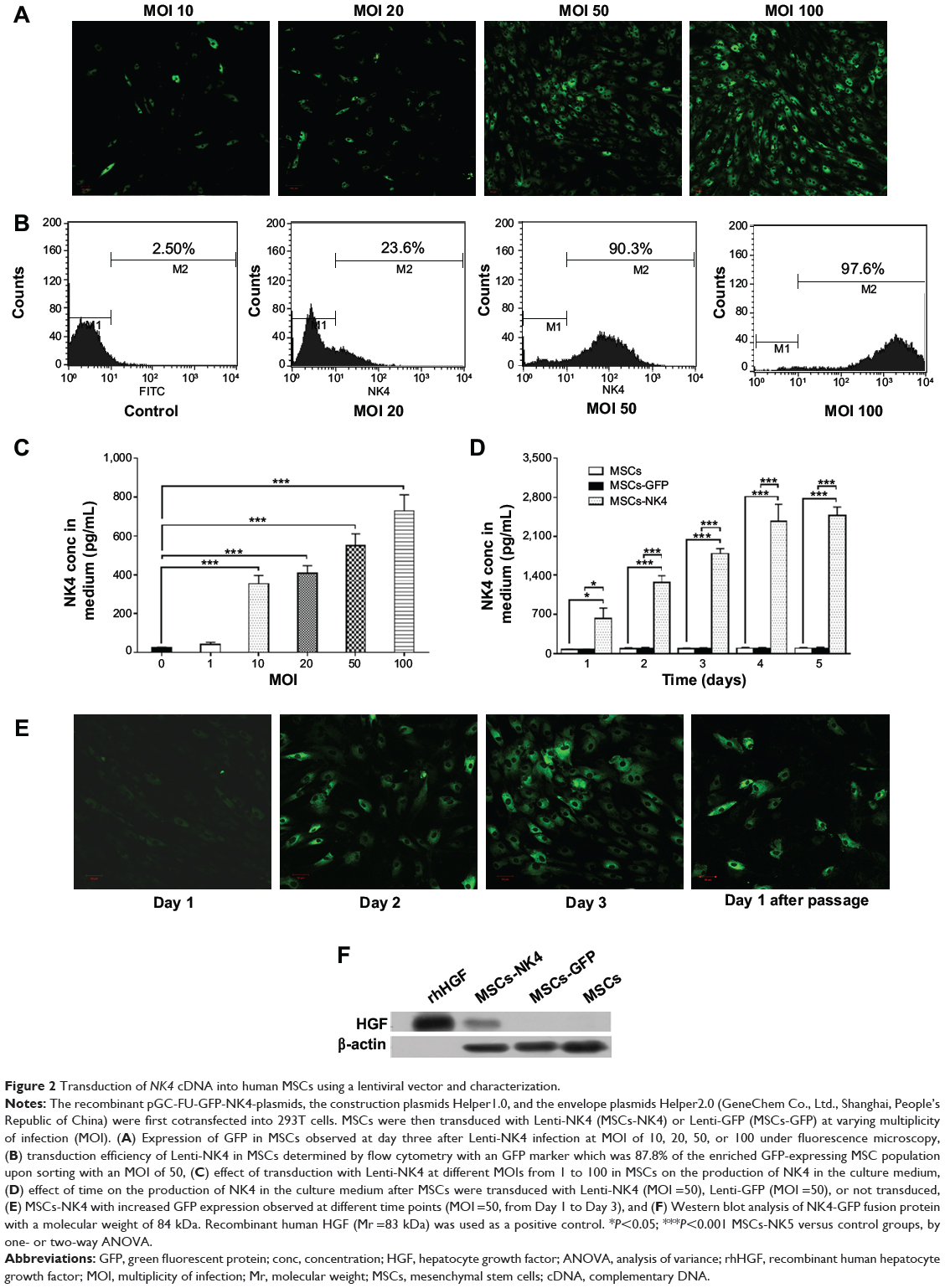
|
Figure 2 Transduction of NK4 cDNA into human MSCs using a lentiviral vector and characterization. |
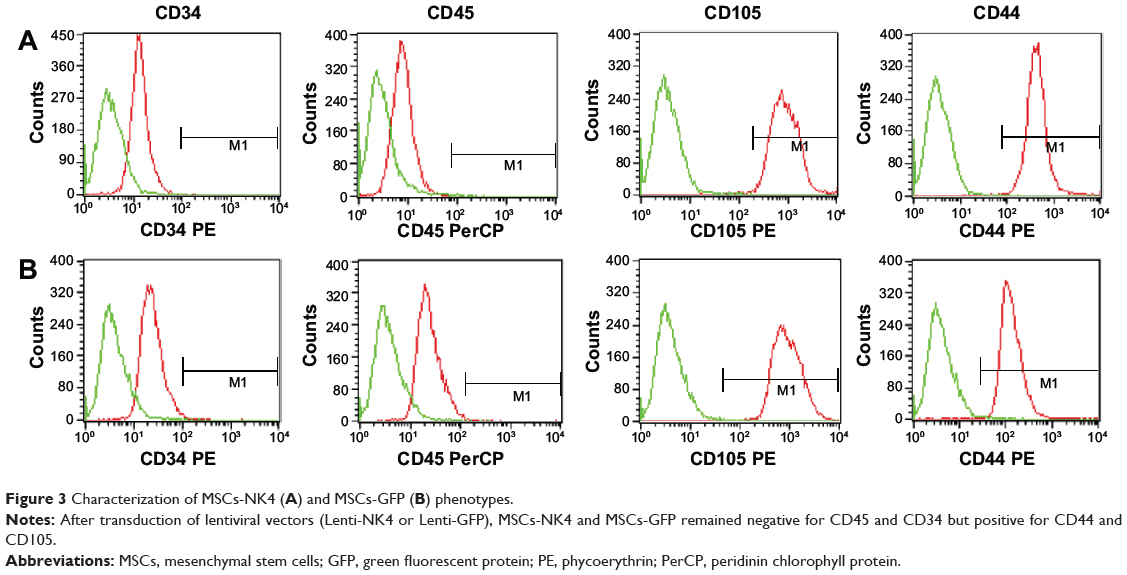
|
Figure 3 Characterization of MSCs-NK4 (A) and MSCs-GFP (B) phenotypes. |
Tropism of MSCs toward gastric cancer cells in vitro
It has been reported that factors released from cancer cells may be potential chemoattractants for MSC tropism.11 To test the ability of the isolated MSCs to migrate toward gastric cancer cells, an in vitro cell migration assay was performed. Transwell cell culture with MKN45 cells to attract MSCs was used, whereas an immortalized human gastric epithelial cell line GES-1 and the medium alone were used as controls. The data showed that migration of MSCs-NK4 was significantly stimulated by the medium conditioned with MKN45 cells compared to GES-1-conditioned medium or medium alone (P<0.001, by one-way ANOVA; Figure 4A and B). We also confirmed that non-infected MSCs significantly migrated toward MKN45 cells compared to GES-1 cells or medium alone (P<0.01; Figure 4A and B). These data indicate that human gastric cancer cells are capable of stimulating the migration of MSCs and the migratory ability of MSCs was not affected by lentiviral transduction. Medium conditioned by MKN45 cells significantly stimulated the migration of MSCs and MSCs-NK4 but not human fibroblasts. There was no significant increase in the migration of human fibroblasts to MKN45 cells compared to GES-1 cells or medium alone (P>0.05, by one-way ANOVA; Figure 4A and B). These data suggest that MSCs could migrate preferably toward gastric cancer cells, but human fibroblasts do not show this characteristic.
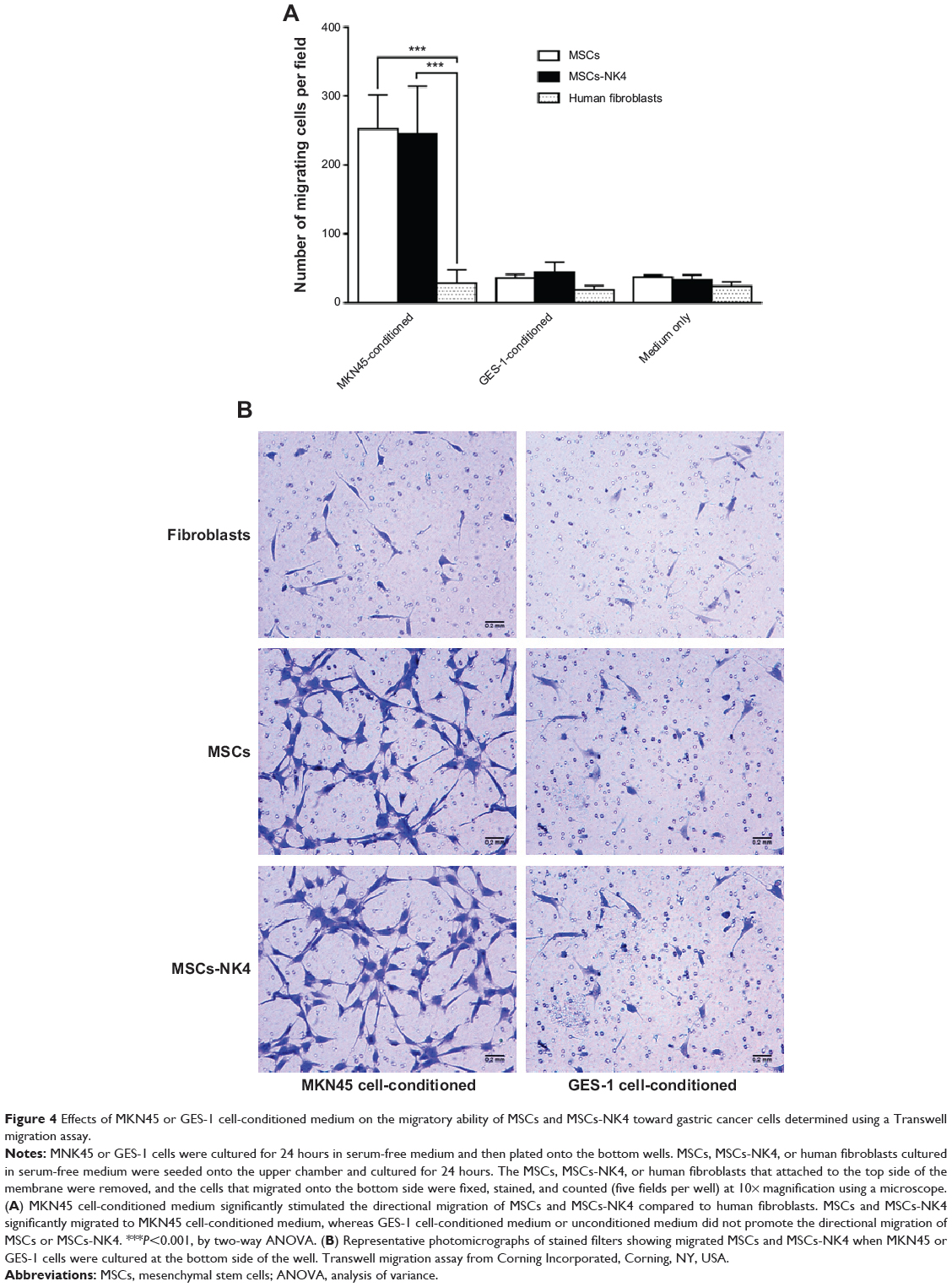
|
Figure 4 Effects of MKN45 or GES-1 cell-conditioned medium on the migratory ability of MSCs and MSCs-NK4 toward gastric cancer cells determined using a Transwell migration assay. |
Effects of systemic administration of MSCs-NK4 on tumor growth in nude mice bearing gastric cancer xenografts
To assess the effects of MSCs-NK4 on the treatment of gastric cancer xenografts, we first established gastric cancer cell MKN45 tumor xenografts in nude mice and then systemically administrated MSCs-NK4 into nude mice. Among all treatment groups, MSCs-NK4 produced the smallest tumor volume in nude mice (Figure 5A and B). The average tumor volume in MSCs-NK4 treated mice was significantly decreased compared to that in nude mice treated with PBS or MSCs-GFP at day 21, and this antitumor effect continued through day 28 (P<0.05 or 0.001, by two-way ANOVA; Figure 5A–C). However, there were no detectable differences in tumor volume between Lenti-NK4 treated mice, PBS injected mice, and MSCs-GFP treated mice at days 7, 14, and 28 (P>0.05 by two-way ANOVA; Figure 5A–C). The tumor mass in MSCs-NK4 treated mice was smaller than that in Lenti-NK4 treated mice (P<0.05 by two-way ANOVA; Figure 5A–C). The tumor growth inhibition rate in MSCs-NK4– and Lenti-NK4 treated mice was 52.2% and 29.4%, respectively at day 28. The tumor weight in MSCs-NK4 treated mice was the lowest among all groups (1.94±0.67 g versus 2.16±0.49 g in Lenti-NK4 treated mice, 2.87±0.86 g in MSCs-GFP treated mice, or 2.84±0.65 g PBS treated mice), although did not achieve statistical significance (Figure 5D). These data demonstrate that NK4-secreting MSCs significantly inhibited the growth of gastric cancer xenografts in nude mice.
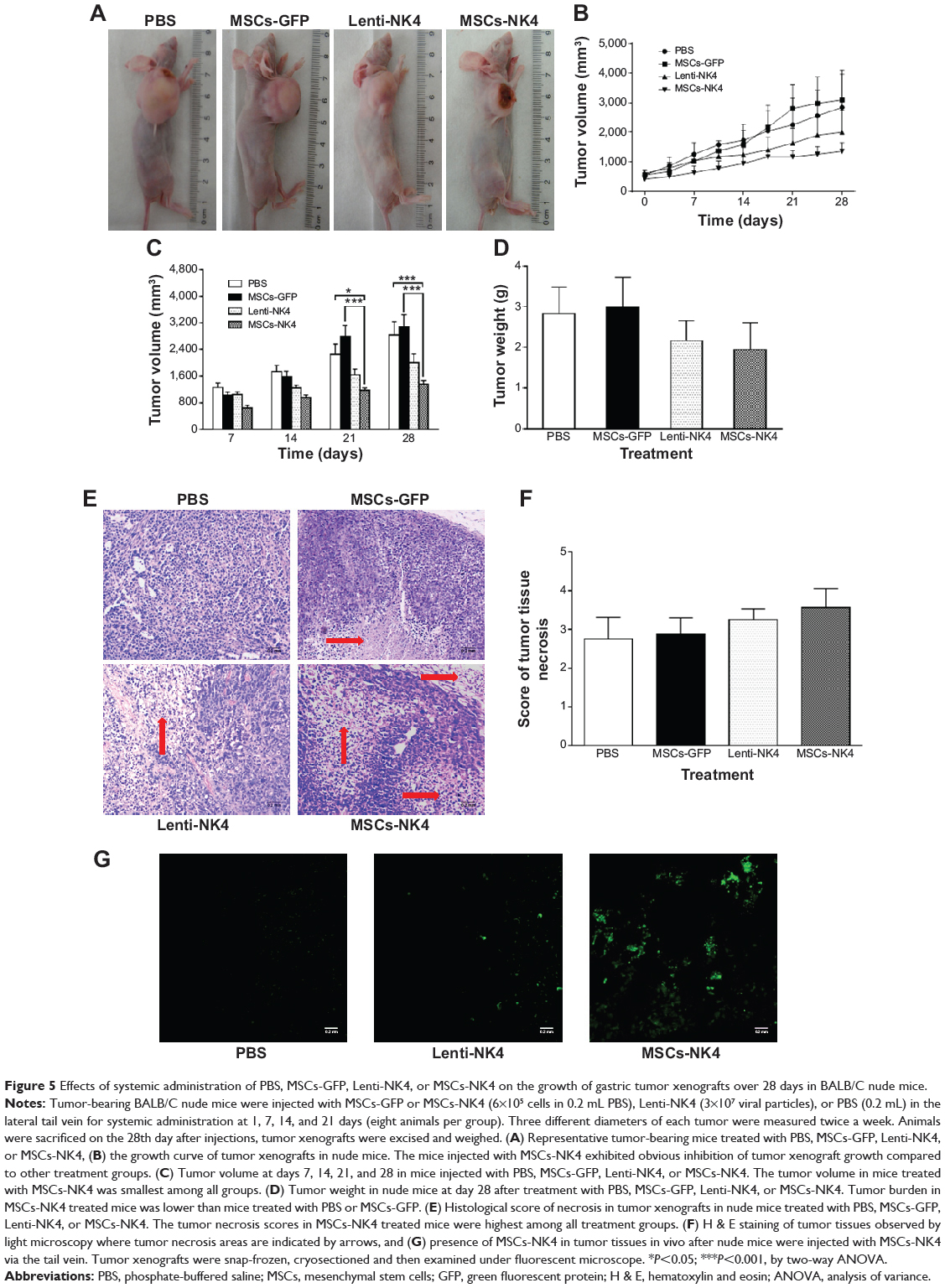
|
Figure 5 Effects of systemic administration of PBS, MSCs-GFP, Lenti-NK4, or MSCs-NK4 on the growth of gastric tumor xenografts over 28 days in BALB/C nude mice. |
Moreover, tumor sections displayed necrosis at different degrees in each group of mice (Figure 5E). The scores of tumor tissue necrosis were highest in the MSCs-NK4 treated mice among all treatment groups of mice, although did not achieve statistical significance (Figure 5F). These data suggest that MSC-based NK4 gene therapy was able to induce tumor necrosis.
To determine the presence of MSCs-NK4 in tumor xenografts of nude mice, frozen sections of tumor xenografts were analyzed by fluorescence microscopy. Green fluorescence was obviously observed within tumor tissues treated with MSCs-NK4, whereas only faint fluorescent signal was visible in cryosections from Lenti-NK4 treated mice (Figure 5G). These results indicate that MSCs-NK4 migrated and accumulated in tumor tissues after systemic administration to the circulation in vivo. Therefore, MSCs are a better vehicle for NK4 gene therapy than lentiviral vectors.
Inhibition of intratumoral angiogenesis and induction of apoptosis in MSCs-NK4 treated tumor xenografts
The microvessel density of MSCs-NK4 treated tumor xenografts was 5.6±1.47 per microscopic field and less than that of the MSCs-GFP or PBS treated mice (16.4±6.16 and 17.4±9.38/high power field, respectively), but did not achieve statistical significance probably due to remarkable standard errors (Figure 6A and B). The microvessel density was also decreased in Lenti-NK4 treated tumor xenografts (12.3±2.65/high power field) compared to MSCs-GFP or PBS treated mice. Furthermore, the apoptotic index, as determined by the percentage of TUNEL-positive cells, was significantly increased in MSCs-NK4 treated tumor xenografts (7.31%±1.9%) compared to mice treated with PBS (P<0.05 by one-way ANOVA, Figure 6C and D). The apoptotic index in Lenti-NK4 treated tumor xenografts was also higher (4.2%±1.3%) compared to PBS or MSCs GFP treated mice.
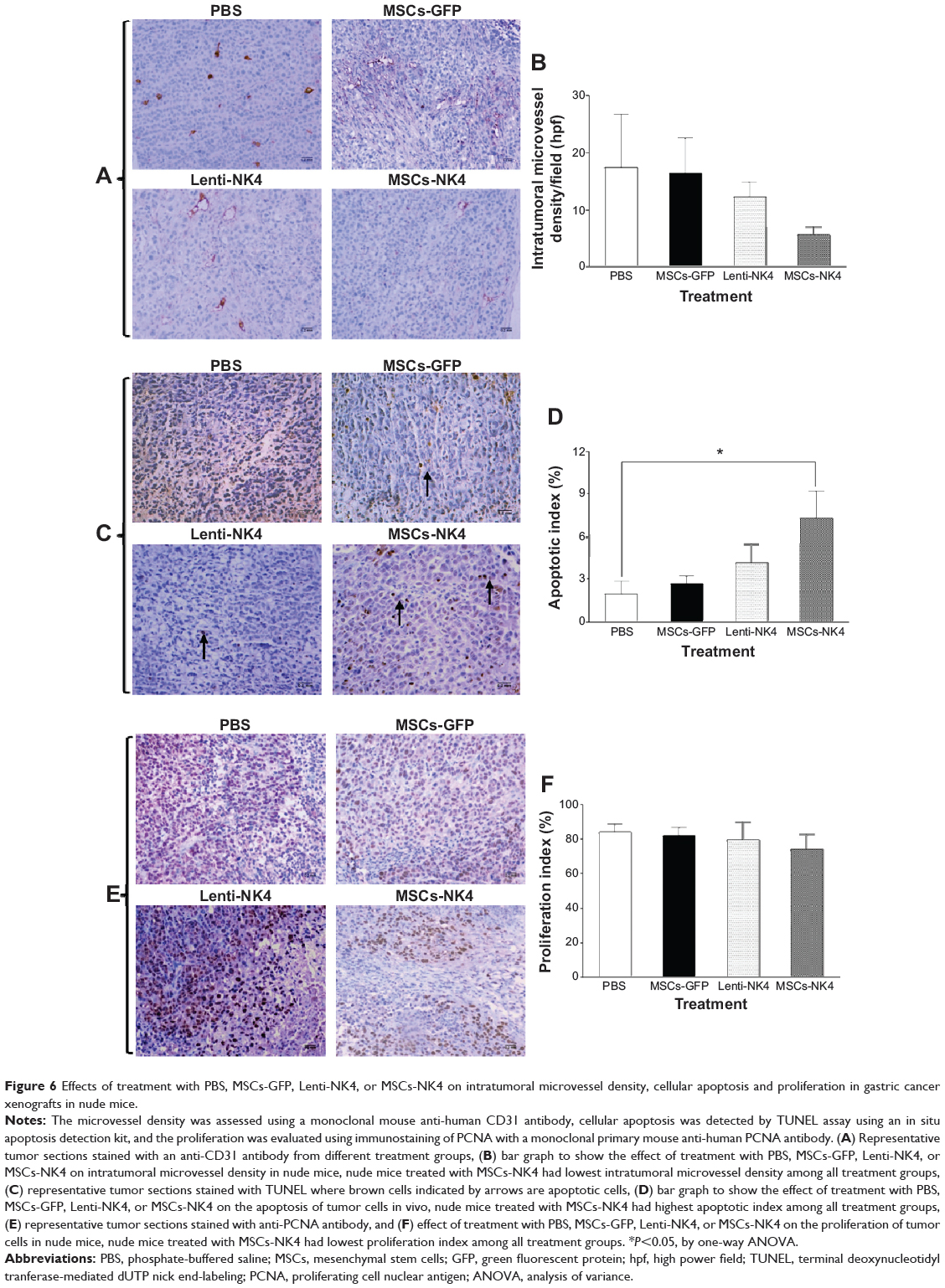
|
Figure 6 Effects of treatment with PBS, MSCs-GFP, Lenti-NK4, or MSCs-NK4 on intratumoral microvessel density, cellular apoptosis and proliferation in gastric cancer xenografts in nude mice. |
Tumor cell proliferation was evaluated as the percentage of PCNA-positive cells, and no significant differences in the number of PCNA-positive cells in tumors were observed between MSCs-NK4 or Lenti-NK4 treated mice and control groups (P>0.05 by one-way ANOVA; Figure 6E and F). These results indicate that MSCs-NK4 effectively inhibited the growth of gastric cancer xenografts by suppressing tumor-associated angiogenesis and inducing tumor cell apoptosis.
Discussion
Tyrosine kinase receptors are interesting candidates for targeted cancer therapy, as they are often aberrantly activated in human cancers. Among these receptors with tyrosine kinase activity, Met is a “master gene” controlling the growth and invasion of cancer cells and protection from apoptosis and angiogenesis in the tumor microenvironment.28,32,50 Met can also be genetically selected for the long-term maintenance of the primary transformed phenotype, and some tumors such as gastric cancer appear to be dependent on (or “addicted” to) sustained Met activity for their growth, survival, and invasion. Because of its dual role as an adjuvant, pro-metastatic gene for some tumor types and as a necessary oncogene for others, Met is a versatile candidate for targeted cancer therapy.51 To date, a number of drugs has been developed, including chemical kinase inhibitors and monoclonal antibodies targeting either the ligand HGF or the receptor Met.28 In this study, we investigated whether systemic administration of MSCs transduced with NK4 (an antagonist of Met) using lentiviral vectors could inhibit the growth of gastric cancer xenografts in nude mice. First, we isolated and cultured human MSCs, and these MSCs expressed CD44 and CD105 but without CD45 and CD34 expression. An in vitro cell migration assay showed that the isolated MSCs could migrate preferably to gastric cancer cells, but human fibroblasts did not exhibit this characteristic. Next, we stably transduced the MSCs with lentiviral vectors carrying NK4 or GFP. NK4 was expressed in the conditioned medium and MSC lysates. We then established gastric cancer xenografts in nude mice and treated these mice with MSCs or lentiviral vectors carrying NK4 cDNA for up to 28 days. The data showed that systemic MSCs-NK4 injection significantly inhibited the growth of gastric cancer xenografts. The microvessel density of tumor xenografts was decreased, but tumor cell apoptosis was significantly increased in nude mice treated with MSCs-NK4 compared to PBS treated mice. The current data suggest that MSC-delivered NK4 gene therapy was better than lentiviral vectors carrying NK4 cDNA for the suppression of gastric cancer xenografts and that NK4 gene therapy inhibited angiogenesis of tumor xenografts and induced apoptosis of tumor cells. Future mechanistic and clinical studies using MSC-based gene therapy are warranted to test the usefulness and safety of MSCs as a gene delivery vehicle for human gastric cancer therapy.
Gene and cellular therapy has become a promising strategy in the treatment of human cancer in recent years.7,22,52,53 However, although significant advances were made for gene therapy of human cancers, major hurdles continue to limit its potential clinical use, eg, tumor tropism of vectors and stimulation of host immune response. In particular, viral vectors are associated with immune recognition, mutagenic integration of viruses into the human genome, and inflammatory toxicity and rapid clearance by liposomes.54 Thus, a better gene delivery vehicle could help advance this field. In this regard, MSCs are particularly attractive because they have tumor-targeting properties and are easily isolated, expanded in culture, and genetically manipulated using currently available molecular techniques.15–17,20,21,55–59 Moreover, MSCs expressing transgenes can be maintained for a long period of time (up to 3 months) in vivo because of their hypo-immunogenic properties and production of immunosuppressive molecules.60–62 Thus, MSCs are suggested to have a potential clinical use for cancer gene therapy. At present, various vectors are available to transduce genes into MSCs, including retroviral, adenoviral, and lentiviral vectors.16,22,59 Adenoviral vectors are the most commonly used vectors with high titers. However, the transduction efficiency of MSCs by conventional adenoviral vectors has been shown to be only up to 20% because of the low expression of Coxsackie-adenoviral receptors in MSCs.57,63 In contrast, retroviral vectors can integrate into the host genome and permit stable or long-term transgene expression. However, a major drawback of retroviral vectors is that they exclusively transduce dividing cells, and the majority of tumor cells do not divide within a given treatment window.64 In addition, lentiviral vectors have the ability to transduce both dividing and non-dividing cells as well as to integrate into the genome of target cells, which guarantees long-term expression of the therapeutic genes.65,66 Lentiviral vectors have been successfully applied to generate potent dendritic cell based anticancer vaccines and to deliver cancer-specific receptors to T-cells.66 In our current study, we transduced MSCs using lentiviral vectors carrying NK4, and these MSCs were able to infiltrate tumor lesions and self-renew on site. Thus, these cells exhibited better antitumor activity than that of directly delivered plasmids or viral vectors.
Furthermore, in our current study, we isolated human MSCs for in vitro expansion. These MSCs expressed several typical cell surface markers including CD44 and CD45. Moreover, the phenotype of MSCs was not altered after transduction of NK4, indicating that introduction of the NK4 gene into MSCs by the lentiviral vector did not change MSC morphology and function, consistent with the results of our previous study.41 MSCs can also achieve a high gene transduction efficiency (>85%) 3 days after exposure to lentiviral vectors. Expression of NK4 was detected in both MSCs-NK4 and the conditioned medium, suggesting that MSCs-NK4 can stably express and continually secrete NK4. Most importantly, MSCs can respond to inflammatory cytokine and chemokine signaling at the site of injury by migrating, proliferating, and differentiating to aid in wound repair.11,67 Through the release of a constant stream of inflammatory signals, tumors can be characterized as “wounds that never heal”.13 Tumors release inflammatory cytokines and chemokines with the ability to recruit supportive host cells and the potential to activate and recruit MSCs.68,69 Thus, MSCs engineered to express tumor killing proteins could be used to destroy tumors and prevent their invasion and metastases. Indeed, our previous study showed that MSCs can migrate to gastric cancer cells in vitro and in vivo.70 In the current study, we found that MSCs migrated to gastric cancer cells but not to normal cells in vitro and were present in tumor xenografts in nude mice, indicating that MSCs are a useful and safe vehicle for gene therapy.
In addition, our current study compared MSC-based NK4 gene therapy with lentiviral vectors carrying NK4. We found that MSC-based NK4 gene therapy had better effects on the inhibition of tumor xenograft growth than did direct injection of recombinant Lenti-NK4. Indeed, a previous study39 showed that in the Colon-26 (C-26) lung metastasis model, MSC-based NK4 gene therapy inhibited progression of multiple lung metastases and prolonged survival. In contrast, high-dose recombinant Ad-NK4 (2.0×109 infectious units [ifu]/mouse) reduced the numbers of metastatic tumors but did not improve survival. This was probably due to the severe liver damage caused by the adenoviruses. However, low doses of recombinant Ad-NK4 (5.0×107 ifu/mouse) had no antitumor effect.39 Adenovirus-mediated induction of high levels (109 ifu/mouse) of circulating NK4 significantly inhibited in vivo tumor growth and distant metastasis.36 Therefore, it is possible that the dose of recombinant Lenti-NK4 (3×107 transduction units/mouse, equivalent to lentiviral vector-infected MSCs) delivered in our current study was less than the effective concentration needed. Lentiviral vectors have no tropism to tumor lesions and may not reach the required high levels at the tumor site, thus leading to the poor antitumor effect observed in this study.
NK4-induced anti-angiogenic effects are reproduced in various types of cancer.37,71–76 Our current data show that the microvessel density in tumor xenografts treated with MSCs-NK4 was lowest among all treatment groups of mice. Moreover, the cancer cell apoptosis was obviously increased in MSCs-NK4 treated mice. However, our current study is only proof-of-principle, and more research is needed before these findings can be translated into the clinical use of MSCs as a suitable and safe delivery vehicle for gene therapy. It would be interesting to check whether MSC-based NK4 gene therapy could block Met-mediated signaling and suppress the activities of downstream signaling molecules. In future studies, we will further test the efficacy and safety of MSCs as the gene delivery vehicle for cancer treatment in animals and then isolate MSCs from patients for in vitro expansion and gene transduction before injection back into the patients.
Acknowledgments
We would like to thank Fang Chu and Liang Li of Jiangxi Institute of Materia Medica (Nanchang, Jiangxi, People’s Republic of China) for their help with the animal experiments. This study was supported by grants from the Natural Science Foundation of China (Grant No 81060198) and National Science and Technology Major Projects for “Major New Drug Innovation and Development” of People’s Republic of China (Grant No 2011ZX09302-007-03) and the Technology Support Program of Jiangxi Provincial Department of Science and Technology, Nanchang, Jiangxi, People’s Republic of China (Grant No 2010BSA12800).
Disclosure
The authors declare no conflicts of interest in this work.
References
Ferlay J, Soerjomataram I, Ervik M, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No 11 [homepage on the Internet]. Lyon, France. International Agency for Research on Cancer; 2013. Available from: http://globocan.iarc.fr. Accessed September 11, 2014. | ||
Hartgrink HH, Jansen EP, van Grieken NC, van de Velde CJ. Gastric cancer. Lancet. 2009;374(9688):477–490. | ||
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Takahashi T, Saikawa Y, Kitagawa Y. Gastric cancer: current status of diagnosis and treatment. Cancers (Basel). 2013;5(1):48–63. | ||
DeSantis CE, Lin CC, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64(4):252–271. | ||
Ortiz R, Melguizo C, Prados J, et al. New gene therapy strategies for cancer treatment: a review of recent patents. Recent Pat Anticancer Drug Discov. 2012;7(3):297–312. | ||
Wang D, Gao G. State-of-the-art human gene therapy: Part I Gene delivery technologies. Discov Med. 2014;18(97):67–77. | ||
Wadhwa R, Song S, Lee JS, Yao Y, Wei Q, Ajani JA. Gastric cancer - molecular and clinical dimensions. Nat Rev Clin Oncol. 2013;10(11):643–655. | ||
Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276(5309):71–74. | ||
Bianco P, Riminucci M, Gronthos S, Robey PG. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19(3):180–192. | ||
Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15(10):730–738. | ||
Bao B, Ahmad A, Li Y, et al. Targeting CSCs within the tumor microenvironment for cancer therapy: a potential role of mesenchymal stem cells. Expert Opin Ther Targets. 2012;16(10):1041–1054. | ||
Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–1659. | ||
Alagesan S, Griffin MD. Autologous and allogeneic mesenchymal stem cells in organ transplantation: what do we know about their safety and efficacy? Curr Opin Organ Transplant. 2014;19(1):65–72. | ||
Bronckaers A, Hilkens P, Martens W, et al. Mesenchymal stem/stromal cells as a pharmacological and therapeutic approach to accelerate angiogenesis. Pharmacol Ther. 2014;143(2):181–196. | ||
Farini A, Sitzia C, Erratico S, Meregalli M, Torrente Y. Clinical applications of mesenchymal stem cells in chronic diseases. Stem Cells Int. 2014;2014:306573. | ||
Sharma RR, Pollock K, Hubel A, McKenna D. Mesenchymal stem or stromal cells: a review of clinical applications and manufacturing practices. Transfusion. 2014;54(5):1418–1437. | ||
Stagg J, Galipeau J. Mechanisms of immune modulation by mesenchymal stromal cells and clinical translation. Curr Mol Med. 2013;13(5):856–867. | ||
Atoui R, Chiu RC. Immune responses after mesenchymal stem cell implantation. Methods Mol Biol. 2013;1036:107–120. | ||
Lotfinegad P, Shamsasenjan K, Movassaghpour A, Majidi J, Baradaran B. Immunomodulatory nature and site specific affinity of mesenchymal stem cells: a hope in cell therapy. Adv Pharm Bull. 2014;4(1):5–13. | ||
Serakinci N, Fahrioglu U, Christensen R. Mesenchymal stem cells, cancer challenges and new directions. Eur J Cancer. 2014;50(8):1522–1530. | ||
Uchibori R, Tsukahara T, Ohmine K, Ozawa K. Cancer gene therapy using mesenchymal stem cells. Int J Hematol. 2014;99(4):377–382. | ||
Nakamura T, Nishizawa T, Hagiya M, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342(6248):440–443. | ||
Montesano R, Matsumoto K, Nakamura T, Orci L. Identification of a fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell. 1991;67(5):901–908. | ||
Bottaro DP, Rubin JS, Faletto DL, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251(4995):802–804. | ||
Takami T, Kaposi-Novak P, Uchida K, et al. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine induced hepatocarcinogenesis. Cancer Res. 2007;67(20):9844–9851. | ||
Marx-Stoelting P, Borowiak M, Knorpp T, Birchmeier C, Buchmann A, Schwarz M. Hepatocarcinogenesis in mice with a conditional knockout of the hepatocyte growth factor receptor c-Met. Int J Cancer. 2009;124(8):1767–1772. | ||
Vigna E, Comoglio PM. Targeting the oncogenic Met receptor by antibodies and gene therapy. Oncogene. Epub 2014 Jun 2. | ||
Lee YH, Morrison BL, Bottaro DP. Synergistic signaling of tumor cell invasiveness by hepatocyte growth factor and hypoxia. J Biol Chem. 2014;289(30):20448–20461. | ||
Matsumoto K, Nakamura T, Sakai K, Nakamura T. Hepatocyte growth factor and Met in tumor biology and therapeutic approach with NK4. Proteomics. 2008;8(16):3360–3370. | ||
Matsumoto K, Nakamura T. NK4 (HGF-antagonist/angiogenesis inhibitor) in cancer biology and therapeutics. Cancer Sci. 2003;94(4):321–327. | ||
Mizuno S, Nakamura T. HGF-MET cascade, a key target for inhibiting cancer metastasis: the impact of NK4 discovery on cancer biology and therapeutics. Int J Mol Sci. 2013;14(1):888–919. | ||
Matsumoto K, Nakamura T. Mechanisms and significance of bifunctional NK4 in cancer treatment. Biochem Biophys Res Commun. 2005;333(2):316–327. | ||
Suzuki Y, Sakai K, Ueki J, et al. Inhibition of Met/HGF receptor and angiogenesis by NK4 leads to suppression of tumor growth and migration in malignant pleural mesothelioma. Int J Cancer. 2010;127(8):1948–1957. | ||
Parikh RA, Wang P, Beumer JH, Chu E, Appleman LJ. The potential roles of hepatocyte growth factor (HGF)-MET pathway inhibitors in cancer treatment. Onco Targets Ther. 2014;7:969–983. | ||
Kishi Y, Kuba K, Nakamura T, et al. Systemic NK4 gene therapy inhibits tumor growth and metastasis of melanoma and lung carcinoma in syngeneic mouse tumor models. Cancer Sci. 2009;100(7):1351–1358. | ||
Ogura Y, Mizumoto K, Nagai E, et al. Peritumoral injection of adenovirus vector expressing NK4 combined with gemcitabine treatment suppresses growth and metastasis of human pancreatic cancer cells implanted orthotopically in nude mice and prolongs survival. Cancer Gene Ther. 2006;13(5):520–529. | ||
Kubota T, Taiyoh H, Matsumura A, et al. NK4, an HGF antagonist, prevents hematogenous pulmonary metastasis by inhibiting adhesion of CT26 cells to endothelial cells. Clin Exp Metastasis. 2009;26(5):447–456. | ||
Kanehira M, Xin H, Hoshino K, et al. Targeted delivery of NK4 to multiple lung tumors by bone marrow-derived mesenchymal stem cells. Cancer Gene Ther. 2007;14(11):894–903. | ||
Mohr A, Lyons M, Deedigan L, et al. Mesenchymal stem cells expressing TRAIL lead to tumour growth inhibition in an experimental lung cancer model. J Cell Mol Med. 2008;12(6B):2628–2643. | ||
Zhu Y, Cheng M, Lu N, Luo S, Xie Y, Liu D. [Construction of NK4 gene lentiviral vector and its expression in bone mesenchymal stem cells]. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. 2011;28(5):976–981. Chinese. | ||
Fu J, Malm IJ, Kadayakkara DK, Levitsky H, Pardoll D, Kim YJ. Preclinical evidence that PD1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res. 2014;74(15):4042–4052. | ||
Tsuji S, Kawano S, Sawaoka H, et al. Evidences for involvement of cyclooxygenase-2 in proliferation of two gastrointestinal cancer cell lines. Prostaglandins Leukot Essent Fatty Acids. 1996;55(3):179–183. | ||
Azizi SA, Stokes D, Augelli BJ, DiGirolamo C, Prockop DJ. Engraftment and migration of human bone marrow stromal cells implanted in the brains of albino rats – similarities to astrocyte grafts. Proc Natl Acad Sci U S A. 1998;95(7):3908–3913. | ||
Conget PA, Minguell JJ. Phenotypical and functional properties of human bone marrow mesenchymal progenitor cells. J Cell Physiol. 1999;181(1):67–73. | ||
Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65(8):3307–3318. | ||
Beckermann BM, Kallifatidis G, Groth A, et al. VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br J Cancer. 2008;99(4):622–631. | ||
Shinojima N, Hossain A, Takezaki T, et al. TGF-β mediates homing of bone marrow-derived human mesenchymal stem cells to glioma stem cells. Cancer Res. 2013;73(7):2333–2344. | ||
Alt E, Yan Y, Gehmert S, et al. Fibroblasts share mesenchymal phenotypes with stem cells, but lack their differentiation and colony-forming potential. Biol Cell. 2011;103(4):197–208. | ||
Lordick F. Targeting the HGF/MET pathway in gastric cancer. Lancet Oncol. 2014;15(9):914–916. | ||
Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7(6):504–516. | ||
Rice J, Ottensmeier CH, Stevenson FK. DNA vaccines: precision tools for activating effective immunity against cancer. Nat Rev Cancer. 2008;8(2):108–120. | ||
Nabel GJ. Genetic, cellular and immune approaches to disease therapy: past and future. Nat Med. 2004;10(2):135–141. | ||
Wu TL, Zhou D. Viral delivery for gene therapy against cell movement in cancer. Adv Drug Deliv Rev. 2011;63(8):671–677. | ||
Aboody KS, Najbauer J, Danks MK. Stem and progenitor cell-mediated tumor selective gene therapy. Gene Ther. 2008;15(10):739–752. | ||
Colter DC, Class R, DiGirolamo CM, Prockop DJ. Rapid expansion of recycling stem cells in cultures of plastic-adherent cells from human bone marrow. Proc Natl Acad Sci U S A. 2000;97(7):3213–3218. | ||
Conget PA, Minguell JJ. Adenoviral-mediated gene transfer into ex vivo expanded human bone marrow mesenchymal progenitor cells. Exp Hematol. 2000;28(4):382–390. | ||
Hong IS, Lee HY, Kang KS. Mesenchymal stem cells and cancer: Friends or enemies? Mutat Res Fundam Mol Mech Mutagen. Epub 2014 Feb 7. | ||
Amara I, Touati W, Beaune P, de Waziers I. Mesenchymal stem cells as cellular vehicles for prodrug gene therapy against tumors. Biochimie. Epub 2014 Jun 27. | ||
Lee K, Majumdar MK, Buyaner D, Hendricks JK, Pittenger MF, Mosca JD. Human mesenchymal stem cells maintain transgene expression during expansion and differentiation. Mol Ther. 2001;3(6):857–866. | ||
Eliopoulos N, Al-Khaldi A, Crosato M, Lachapelle K, Galipeau J. A neovascularized organoid derived from retrovirally engineered bone marrow stroma leads to prolonged in vivo systemic delivery of erythropoietin in nonmyeloablated, immunocompetent mice. Gene Ther. 2003;10(6):478–489. | ||
Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood. 2005;105(7):2821–2827. | ||
Mizuguchi H, Sasaki T, Kawabata K, Sakurai F, Hayakawa T. Fiber-modified adenovirus vectors mediate efficient gene transfer into undifferentiated and adipogenic-differentiated human mesenchymal stem cells. Biochem Biophys Res Commun. 2005;332(4):1101–1106. | ||
Cavazza A, Moiani A, Mavilio F. Mechanisms of retroviral integration and mutagenesis. Hum Gene Ther. 2013;24(2):119–131. | ||
Van Damme A, Thorrez L, Ma L, et al. Efficient lentiviral transduction and improved engraftment of human bone marrow mesenchymal cells. Stem Cells. 2006;24(4):896–907. | ||
Emeagi PU, Goyvaerts C, Maenhout S, Pen J, Thielemans K, Breckpot K. Lentiviral vectors: a versatile tool to fight cancer. Curr Mol Med. 2013;13(4):602–625. | ||
Inoue H, Murakami T, Ajiki T, Hara M, Hoshino Y, Kobayashi E. Bioimaging assessment and effect of skin wound healing using bone-marrow-derived mesenchymal stromal cells with the artificial dermis in diabetic rats. J Biomed Opt. 2008;13(6):064036. | ||
Barcellos-de-Souza P, Gori V, Bambi F, Chiarugi P. Tumor microenvironment: bone marrow-mesenchymal stem cells as key players. Biochim Biophys Acta. 2013;1836(2):321–335. | ||
Goubran HA, Kotb RR, Stakiw J, Emara ME, Burnouf T. Regulation of tumor growth and metastasis: the role of tumor microenvironment. Cancer Growth Metastasis. 2014;7:9–18. | ||
Zhu Y, Cheng M, Xie Y, et al. Chemotaxis of bone marrow derived-mesenchymal stem cells to gastric cancer cells in vivo and in vitro. Chinese Journal of Cancer Biotherapy. 2011;6:647–652. | ||
Jie JZ, Wang JW, Qu JG, Hung T. Suppression of human colon tumor growth by adenoviral vector-mediated NK4 expression in an athymic mouse model. World J Gastroenterol. 2007;13(13):1938–1946. | ||
Son G, Hirano T, Seki E, et al. Blockage of HGF/c-Met system by gene therapy (adenovirus-mediated NK4 gene) suppresses hepatocellular carcinoma in mice. J Hepatol. 2006;45(5):688–695. | ||
Murakami M, Nagai E, Mizumoto K, et al. Suppression of metastasis of human pancreatic cancer to the liver by transportal injection of recombinant adenoviral NK4 in nude mice. Int J Cancer. 2005;117(1):160–165. | ||
Wen J, Matsumoto K, Taniura N, Tomioka D, Nakamura T. Inhibition of colon cancer growth and metastasis by NK4 gene repetitive delivery in mice. Biochem Biophys Res Commun. 2007;358(1):117–123. | ||
Buhles A, Collins SA, van Pijkeren JP, et al. Anti-metastatic effects of viral and non-viral mediated Nk4 delivery to tumours. Genet Vaccines Ther. 2009;7:5. | ||
Kubota T, Taiyoh H, Matsumura A, et al. Gene transfer of NK4, an angiogenesis inhibitor, induces CT26 tumor regression via tumor-specific T lymphocyte activation. Int J Cancer. 2009;125(12):2879–2886. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.