")
Back to Journals » Drug Design, Development and Therapy » Volume 10
MEK inhibitors and their potential in the treatment of advanced melanoma: the advantages of combination therapy
Authors Tran K, Cheng M, Mitra A, Ogawa H, Shi V, Olney L, Kloxin A, Maverakis E
Received 1 August 2015
Accepted for publication 8 October 2015
Published 21 December 2015 Volume 2016:10 Pages 43—52
DOI https://doi.org/10.2147/DDDT.S93545
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Khiem A Tran,1,* Michelle Y Cheng,1,* Anupam Mitra,1 Hiromi Ogawa,1 Vivian Y Shi,1 Laura P Olney,1 April M Kloxin,2 Emanual Maverakis1
1Department of Dermatology, University of California, Davis, Sacramento, CA, USA; 2Department of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE, USA
*These authors contributed equally to this work
Abstract: The treatment of melanoma has improved markedly over the last several years with the advent of more targeted therapies. Unfortunately, complex compensation mechanisms, such as those of the mitogen-activated protein kinase (MAPK) pathway, have limited the clinical benefit of these treatments. Recently, a better understanding of melanoma resistance mechanisms has given way to intelligently designed multidrug regimes. Herein, we review the extensive pathways of BRAF inhibitor (vemurafenib and dabrafenib) resistance. We also review the advantages of dual therapy, including the addition of an MEK inhibitor (cobimetinib or trametinib), which has proven to increase progression-free survival when compared to BRAF inhibitor monotherapy. Finally, this review touches on future treatment strategies that are being developed for advanced melanoma, including the possibility of triple therapy with immune checkpoint inhibitors and the work on optimizing sequential therapy.
Keywords: cobimetinib, trametinib, vemurafenib, dabrafenib, BRAF inhibitor, MAPK pathway
Introduction
Traditional chemotherapies have been well studied for metastatic melanoma with no evidence supporting survival benefit. Melanoma oncogenesis involves both DNA damage from ultraviolet light and genetic predispositions.1,2 In the last several years, successful targeted therapies have revolutionized the treatment of advanced melanomas by targeting the mitogen-activated protein kinase (MAPK) pathway. The MAPK pathway is a critical regulator of cell proliferation and survival. BRAF and its downstream target, MEK, are kinases in the MAPK pathway, and therefore play an important role in cell proliferation.3,4 The discovery of somatic BRAF V600 mutations in melanomas initiated the development of targeted therapies. BRAF inhibitors were the first pharmacological agents used clinically, but tumor resistance has limited their benefit. To overcome these resistance mechanisms, MEK inhibitors have been used in combination and their results are promising. This review explores the resistance pathways of BRAF inhibitors and the role of MEK inhibitors in combating BRAF inhibitor resistance in advanced BRAF-mutant melanomas.
The start of targeted therapy
After the BRAF V600 mutation was identified in melanoma,4 BRAF inhibitors were developed for advanced melanomas harboring this mutation. In 2011, the US Food and Drug Administration (FDA) approved the BRAF inhibitor vemurafenib for treatment of unresectable or metastatic melanoma with BRAF(V600E) mutations.5 In patients with advanced melanoma, the median progression-free survival (PFS) with single-agent vemurafenib ranges from 5 to 7 months, and the median overall survival is approximately 16 months, which is 7 months more than with chemotherapy.6–9
In 2013, dabrafenib became the second FDA-approved BRAF inhibitor with similar indications.10 Although a significant difference in overall survival was not observed, patients treated with single-agent dabrafenib demonstrated an improved median PFS compared to those treated with dacarbazine.11
Development of BRAF inhibitor resistance through reactivation of the MAPK pathway
Unfortunately, the clinical benefit of a BRAF inhibitor is limited by intrinsic and acquired resistance. Reactivation of the MAPK pathway is a major contributor to treatment failure in BRAF-mutant melanoma (Figure 1). In fact, a study of resistance mechanisms showed that reactivation of MAPK signaling drives BRAF inhibitor resistance in roughly 80% of melanoma tumors.12,13
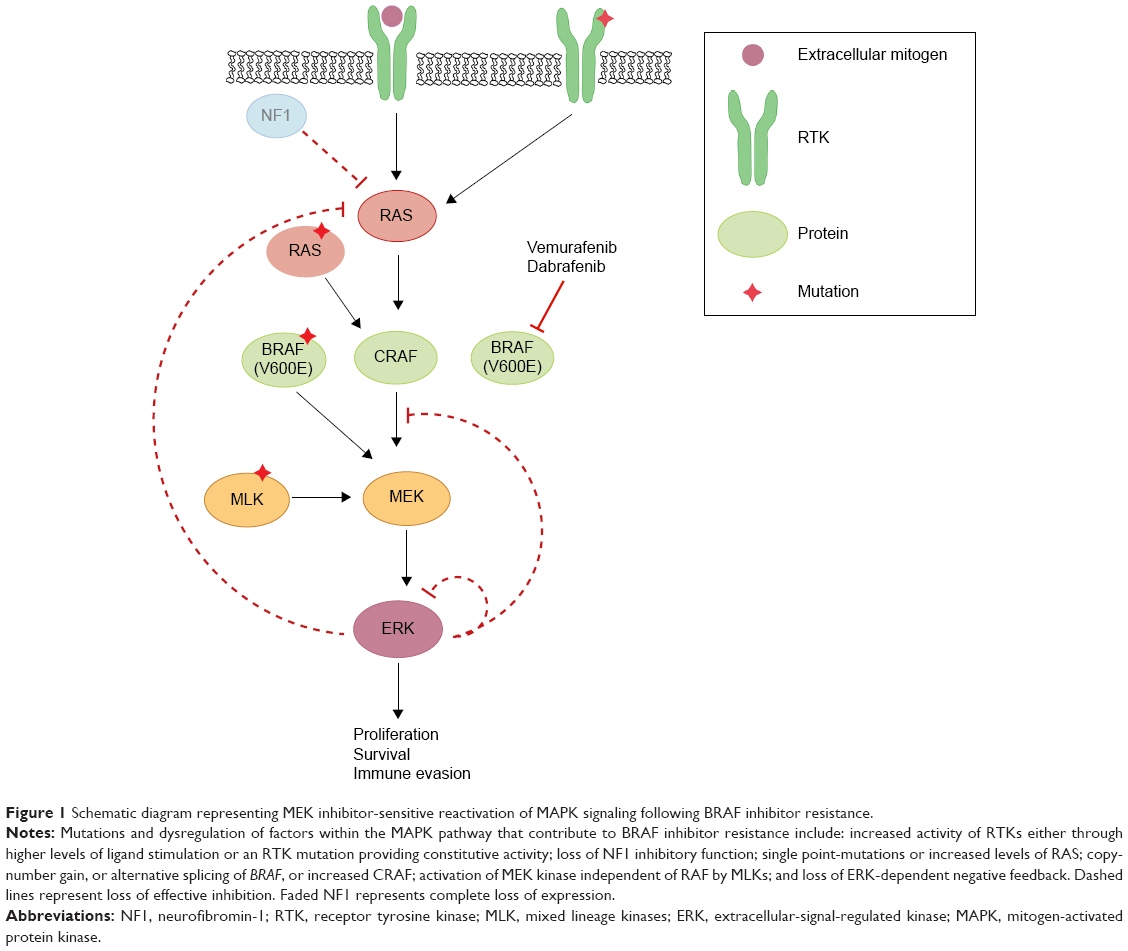 | Figure 1 Schematic diagram representing MEK inhibitor-sensitive reactivation of MAPK signaling following BRAF inhibitor resistance. |
Reactivation of MAPK through receptor tyrosine kinase activation and ERK rebound
Receptor tyrosine kinases (RTKs), upstream of RAS in the MAPK pathway, consist of growth factor receptors for ligands such as epidermal growth factor (EGF), fibroblast growth factor (FGF), insulin-like growth factor (IGF), and platelet-derived growth factor receptor (PDGFR). Stimulation of RTKs activates RAS, which triggers downstream signaling cascades. Mutations in the genetic coding or regulation of expression of these enzymes have been shown to induce and promote resistance to BRAF inhibition, including invasion and metastasis.14–17 One study demonstrated that increased levels of basic FGF induced higher FGF receptor 3 (FGFR3) activity, thus reactivating MAPK in vemurafenib-resistant cells in culture.17 The authors also discovered a constitutively active mutant of FGFR3 promoted BRAF inhibitor resistance through the MAPK pathway. Adding bFGF led to RAS activation, upregulated extracellular-signal-regulated kinase (ERK) activation, and was responsive to both pan-RAF and MEK inhibitors.
Negative feedback of MAPK is directly mediated by ERK-dependent phosphorylation of enzymes in the pathway including RAF, RTK, and SOS.18,19 Additionally, ERK activation induces expression of negative regulators of MAPK.20,21 In BRAF(V600E) cancers, the MAPK pathway is hyper-stimulated, which suppresses ERK-dependent feedback inhibition. When a BRAF(V600E)-mutant melanoma is treated with a RAF inhibitor, there is induction of RAS-GTP accompanied by a rebound in phospho-ERK (pERK).22 This rise in RAS-GTP levels promotes CRAF dimerization and the subsequent phosphorylation of MEK, and thereby decreases the effectiveness of RAF inhibitors. Although CRAF dimers are insensitive to vemurafenib, ERK rebound through CRAF remains sensitive to MEK inhibition.
Further findings demonstrate that various growth factor ligands (EGF, hepatocyte growth factor [HGF], neuregulin [NRG], FGF) can antagonize vemurafenib sensitivity via ligand-induced sensitization,22,23 which promotes ERK rebound and desensitization to BRAF inhibition.24 These findings suggest that enhancement of RTK signaling is due to relief of ERK-dependent feedback inhibition. Combined inhibition of the MAPK pathway could theoretically increase the degree of response duration compared to RAF inhibition alone.
Reactivation of MAPK through RAS
HRAS, KRAS, and NRAS are RAS isoforms within the MAPK pathway. While BRAF is mutated in 50% of melanomas, NRAS and KRAS are mutated in about 20% and 2%, respectively.25 Although BRAF inhibitors can induce metastasis in RAS mutants by reactivation of MEK/ERK signaling, this mechanism can be blocked by MEK inhibition.25 Mutations in KRAS and NRAS reactivate the MAPK pathway, activating tumor growth as well as accelerating preexisting but previously benign secondary tumors.26,27 Melanomas were found to acquire resistance to BRAF(V600E) inhibition by acquisition of activating NRAS mutations as well as NRAS upregulation through increased copy number.16 Additionally, although NRAS mutations concurrent with BRAF V600 mutations are rare, they can promote BRAF inhibitor resistance through reactivation of MAPK via preference for CRAF.28 Others have shown that some mutations in BRAF or KRAS alone do not induce melanoma, but dual mutations can promote CRAF-dependent resistance.29 Knockdown of NRAS in mutant melanoma cells resulted in decreased pERK with a predominantly apoptotic response, suggesting MAPK dependence in mutated NRAS melanomas.30 Indeed, sensitization to MEK inhibition is a hallmark trait of NRAS-mutated BRAF inhibitor-resistant cells.16
Reactivation of MAPK through silencing of NF1
RAS proteins are activated when guanine exchange factors bind GTP and inactivated by GTPase-activating proteins. Active RAS facilitates dimerization and activation of RAF kinases, which stimulates MEK and ERK. RAS proteins are inactivated by GTPase-activating proteins such as neurofibromin-1 (NF1), a GTP-binding factor that facilitates GTP hydrolysis.31 Consequently, NF1 is a negative regulator of the MAPK pathway. Genome-scale RNA interference and screening of melanoma tumors identified inactivation of NF1 as a mechanism of resistance to BRAF inhibition,32 due to hyperactivation through CRAF dimerization.32–34 Furthermore, mutant NF1 alleles identified in melanomas with relatively short response times were thought to confer either a selective intrinsic or acquired advantage. NF1 silencing in BRAF-mutant melanoma cell lines in the presence of a BRAF inhibitor led to increased active GTP-bound RAS, sustained phosphorylation of ERK, and proliferation.34 Mutants with complete loss of NF1 exhibited resistance to BRAF inhibitors. However, NF1 null cells treated with a MEK inhibitor displayed decreased proliferation, highlighting MEK dependence.34
Reactivation of MAPK through RAF
Mutations inducing upregulation of BRAF(V600E) were also found to confer resistance to BRAF inhibitor therapy. A copy-number gain of mutated BRAF(V600E) provided a form of resistance yet was still dependent on MEK/ERK signaling.35 Splice variants of BRAF(V600E) were found to be a driving force in vemurafenib resistance in mutant melanoma cell culture as well as in patient tumors.36 The causative alternative splicing event was shown to be an in-frame deletion confined to the mutant BRAF allele. Structurally, this promoted constitutive dimerization of mutant BRAF which bestowed BRAF inhibitor resistance, even in the absence of RAS activation. This mutant also retained sensitivity to MEK inhibition, demonstrating that additional downstream pathway inhibitors are necessary to delay or inhibit resistance. Furthermore, potential resistance to BRAF inhibition could be induced by switching tumor dependency from BRAF to increasing expression of CRAF.37
Paradoxical oncogenesis is a concern in BRAF inhibitor-treated melanoma due to transactivation of RAF isoforms. Vemurafenib and dabrafenib bind to the highly active monomeric mutant BRAF(V600) to inhibit its function. However inhibitor-bound mutant BRAF can still dimerize with uninhibited CRAF to activate MAPK, consequently leading to proliferation and tumor progression,38,39 highlighting the importance of additional downstream inhibitors.
Reactivation of MAPK through MEK and MEK-activating kinase
Mixed lineage kinases (MLK1–4) are MEK kinases that are able to independently reactivate the MEK/ERK pathway, even in the presence of RAF inhibitors. Studies of acquired resistance in melanoma tumors demonstrated that BRAF inhibitor resistance correlates with MLK upregulation,40,41 as well as a gain of function mutation in MLK1,41 leading to patient tumor progression. Furthermore, MLK expression in the presence of vemurafenib prevented apoptosis, contributing an additional mechanism of survival advantage by MLK in BRAF inhibitor resistance.41
Immune evasion and survival through alteration of tumor microenvironment
Recent data suggest that BRAF(V600E) contributes to immune escape.42,43 Expression of melanoma antigens Mart1, Tyrp1, Tyrp2, and GP100 were significantly increased after treatment of BRAF-mutant melanoma cell lines with BRAF inhibitor, facilitating T-cell cytotoxicity.42 BRAF inhibition was associated with a decrease in immunosuppressive cytokines,42,43 as well as a significant increase in CD8+ T-cell infiltrate,42,44 and T-cell recognition of melanoma.45 Thus, compared to no treatment, BRAF inhibitor therapy provides a more favorable tumor microenvironment, supporting the idea of potential synergy of BRAF-targeted therapy and immunotherapy.46
PDL1 is an inducible cell surface protein, one of two ligands for programmed-death receptor 1 (PD1), a molecule that negatively regulates T-cell immune responses. Increased PDL1 expression in cancer cells is a significant escape mechanism from host immunity.47 Although BRAF inhibition induced immune evasion through increased PDL1, BRAF inhibitor-treated tumors exhibited increased markers for T-cell exhaustion signifying a dysfunctional immune response.42 At time of resistance, melanoma tumor antigen expression and CD8+ T-cell infiltrate are decreased. As MAPK reactivation leads to higher PDL1 expression in BRAF inhibitor-resistant cells, MEK inhibition expectedly and significantly reduced PDL1 in BRAF inhibitor-resistant melanoma cells48,49 and induced apoptosis.48 In another cancer model, MEK inhibition reversed PDL1-mediated inhibition of cytotoxic T-cell function against acute myeloid leukemia cells.50
Use of MEK inhibitors in BRAF-mutant melanoma
Given that the majority of BRAF inhibitor resistance occurs through reactivation of MAPK, several potent, non-ATP competitive MEK inhibitors were developed (eg, trametinib and cobimetinib)51 that show no off-target effects on other kinases and are currently being used in clinical trials. MEK inhibitors are able to target MAPK-dependent tumors and exhibit distinct efficacies against BRAF- and KRAS-mutant melanomas.52,53
Cobimetinib is a potent selective oral MEK inhibitor with antitumor activity and it demonstrates 100-fold stronger potency against mutated BRAF-mutant versus KRAS-mutant cell lines.52 Cobimetinib binds strongly to pMEK, making it highly potent in inhibiting BRAF(V600E) melanoma, since mutant BRAF constantly produces high levels of pMEK. On the other hand, trametinib displays higher binding affinity to unphosphorylated MEK.54 However, both inhibitors are able to inhibit downstream ERK signaling, particularly effective in BRAF-mutant tumor cells. Studies reveal that BRAF inhibitor-resistant tumor cells are highly sensitive to MEK inhibition and demonstrate that targeted pharmacological MEK inhibition may be a highly effective therapeutic alternative in BRAF inhibitor-resistant melanoma.53 In light of these findings, focus has turned to dual inhibition of BRAF and its downstream target, MEK.
Clinical trials
Single-agent MEK inhibitors
Trametinib, a competitive allosteric MEK inhibitor, demonstrated promise as monotherapy in a Phase III trial. The median PFS of patients who received trametinib was significantly longer than that of patients who received chemotherapy (4.8 vs 1.5 months; hazard ratio [HR] 0.45; P<0.001). At 6 months, the rate of overall survival was significantly higher as well in the trametinib group (81% vs 67%; HR 0.54; P=0.01).55 In a small sample cohort, selumetinib, another MEK inhibitor, elicited tumor suppression in BRAF-mutated melanomas, but only in low phosphor-Akt melanomas. This observation further supports that resistance can also be caused by overactivation of the PI3K/Akt pathway.56 Although cobimetinib has not been evaluated as a single agent in clinical trials, the maximum tolerated dose is 60 mg daily on the 21-day on/7-day off dosing schedule. At this dose, cobimetinib is generally well tolerated and the most frequent adverse effects include diarrhea, rash, nausea, fatigue, dry skin, and peripheral edema.57
Combination therapy
The results of combination therapy of MEK inhibitors and BRAF inhibitors are promising. New clinical trials confirm improved PFS with concurrent inhibition of BRAF and MEK. coBRIM (NCT01689519), a Phase III, randomized study, compared patients with BRAF-mutated melanomas treated with vemurafenib 960 mg twice daily versus vemurafenib plus cobimetinib 60 mg daily for 21 days followed by 7 days off. The group on combination therapy displayed prolonged median PFS (9.9 months vs 6.2 months). At the time of interim analysis, the median overall survival had yet to be reached for both groups (Table 1).58 The coBRIM study is scheduled to conclude in 2017 and will provide highly anticipated data on the survival benefits of combination vemurafenib and cobimetinib therapy. With the promising results of the coBRIM study, the FDA in November 2015 approved cobimetinib for its utility in combination with vemurafenib in patients with advanced BRAF V600 mutation-positive melanomas.
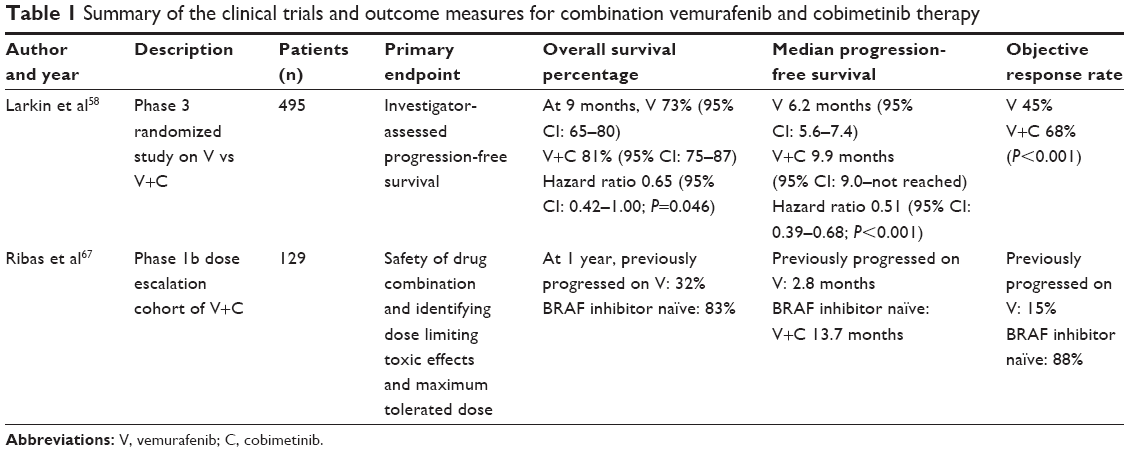 | Table 1 Summary of the clinical trials and outcome measures for combination vemurafenib and cobimetinib therapy |
Other clinical trials focused on different BRAF and MEK inhibitor combinations suggested similar findings.59–61 In fact, the latest Phase III clinical trial of dabrafenib (BRAF inhibitor) and trametinib versus monotherapy with vemurafenib showed increased median PFS (11.4 months vs 7.3 months; HR 0.56; 95% confidence interval [CI]: 0.46–0.69; P<0.001) and improved overall survival at 12 months with combination therapy (72% vs 65%; HR 0.69; 95% CI: 0.53–0.89; P=0.005).60 Additionally, the double-blinded COMBI-d trial compared dabrafenib and trametinib vs dabrafenib and placebo. Interim results demonstrated an increased median PFS as well (9.3 months vs 8.8 months; HR 0.75; 95% CI: 0.57–0.99; P=0.035).62
Cutaneous adverse events
An additional benefit of combination therapy is the decreased frequency of cutaneous adverse events compared to single-agent MEK or BRAF inhibitor therapy.63 Although BRAF inhibitors decrease MAPK pathway activity in BRAF-mutant cells, paradoxical activation can take place in BRAF wild-type cells with upstream mutations. This leads to the development of keratoacanthomas and squamous cell carcinomas early in BRAF inhibitor treatment.27,64,65 Co-targeting with MEK inhibitors appears to decrease this undesired proneoplastic adverse effect that BRAF inhibitors have on keratinocytes.26 For example, only 3.5% of patients on combination therapy developed cutaneous squamous cell carcinoma and keratoacanthoma, whereas 19.7% of those on vemurafenib developed these adverse events.58 Therefore, transitioning from monotherapy to combination therapy would decrease concerns for undesired cutaneous adverse events.
Future studies
With recent positive outcomes, dual BRAF and MEK inhibition therapy will likely be the future of advanced melanoma therapy. Further clinical trials are under way and we anticipate that combination therapy will become the standard of care over monotherapy.
Specific patient populations
Investigation into the potential of dual BRAF and MEK inhibitor therapy in specific patient populations is currently under way. coBRIM-B, a multicenter Phase II study, plans to evaluate the utility of combination vemurafenib and cobimetinib in metastatic melanoma patients with brain involvement (NCT02230306). Additionally COMBI-AD, a double-blinded Phase II study, is currently evaluating the efficacy of combination dabrafenib and trametinib as adjuvant treatment in patients with completely resected advanced melanoma (NCT01682083). These studies may broaden the utility of combination therapy in melanoma patients.
Sequential therapy
The future of melanoma therapy may not hinge solely on combination therapy, but also on finding an optimal sequential regime to combat melanoma as it evolves.66 In a Phase Ib study of 66 patients with advanced melanoma who progressed on vemurafenib, transitioning to combination therapy resulted in a limited response with a median PFS of 2.8 months. The clinical significance of this modest response is unclear since the study was not designed to directly compare effects of sequential therapy with a combination regime. However, the response is undeniably worse than the response seen with combination vemurafenib and cobimetinib in BRAF inhibitor-naïve patients, who had a median PFS of 13.7 months.67
Another study looked at the effect of trametinib in patients who were previously treated with BRAF inhibitor vs those who were treated with chemotherapy and/or immunotherapy. Trametinib monotherapy had minimal clinical activity when administered as sequential therapy in patients with BRAF-mutant melanoma who had failed BRAF inhibitor therapy.68 Therefore, the therapeutic role of adding a MEK inhibitor after BRAF inhibitor resistance has already occurred is unclear, and further investigation on sequential therapy is required to address this option fully.
Paradox-breakers
Recently, new compounds have been developed that inhibit both BRAF and CRAF as well as SRC family kinases (SFKs) termed “paradox-breakers”. These inhibitors were shown to be active in treatment-naïve BRAF-mutant tumors, BRAFi-resistant tumors, and cells isolated from a patient with BRAF/MEK inhibitor-resistant melanoma.69,70 The MAPK pathway was upregulated in each of these cases, demonstrating that these inhibitors are useful in the context of high pERK levels. Moreover, NRAS-mutant tumors reliant on CRAF are highly sensitive to these inhibitors. Future clinical studies will be required to ascertain treatment strategies for these compounds.
Resistance mechanisms not responsive to BRAF or MEK inhibitors
Despite these clinical benefits, the emergence of resistance to BRAF inhibitors and eventually to MEK inhibitors restricts the therapeutic efficacy of these kinase inhibitors (Figure 2). In an attempt to overcome this resistance, more potential molecules targeting other aberrant signaling pathways are in clinical development.71,72
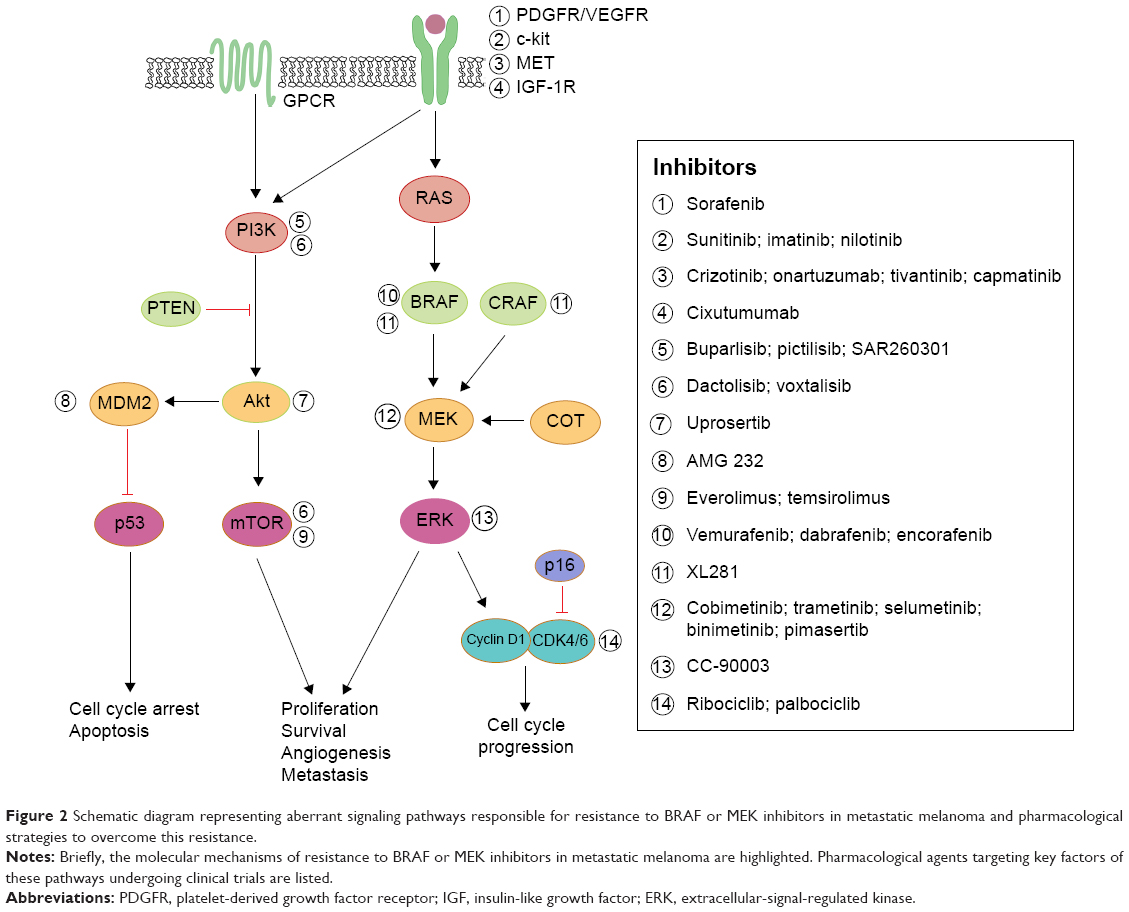 | Figure 2 Schematic diagram representing aberrant signaling pathways responsible for resistance to BRAF or MEK inhibitors in metastatic melanoma and pharmacological strategies to overcome this resistance. |
A myriad of studies have determined pathways other than MAPK that can confer resistance. Those upregulated mechanisms are briefly discussed here. An increase in RTK ligands, either through autocrine production from tumor cells, paracrine secretion from stroma, or systemic production, promotes resistance to kinase inhibitors.24,73 BRAF inhibitor resistance mediated by EGFR was shown in both cell culture studies and patient melanoma.15,74 Similarly, IGF-1R75 and MET receptor23 were found to reactivate both the MAPK and PI3K/AKT pathway, suggesting that inhibition of MEK is only one step of overcoming resistance to BRAF inhibition. Indeed, patient-derived samples revealed tumor-associated upregulation of PDGFR316,74 in vemurafenib-resistant cells, which was shown to be SOX10 and TGFBR2 dependent.74 MEK inhibition is ineffective in these resistant strains, demonstrating that MEK/MAPK is not the key downstream pathway here. Both cyclin D1 as well as PTEN were implicated in BRAF inhibitor resistance.76,77
Single codon mutations in MEK were found in tumors from relapsed patients while on treatment with a BRAF28,78 or MEK inhibitor.79 A recent study of vemurafenib-resistance tumors revealed a MEK mutation at codon 56 which produced the highest levels of pERK (compared to other MEK mutations).28 Overall, these MEK mutations confer BRAF inhibitor resistance and exhibited robust resistance to MEK inhibition,40,79 highlighting the necessity for further downstream inhibition at the level of ERK.80 As others have suggested,16 these findings support specific treatment strategies for patients who relapse on vemurafenib and require precise combinations of targeting agents to optimize inhibition of proliferation.
With these sources of dual-therapy resistance, targeting BRAF and/or MEK may also require targeting other upregulated enzymes to achieve durable therapeutic responses.81 Clinical trials are currently under way to assess safety and efficacy of MEK inhibition (cobimetinib, pimasertib, trametinib) combined with single-agent PI3K/Akt/mTOR pathway inhibitors (BKM120, GDC-0941, uprosertib, BEZ235, SAR245409) (NCT01363232, NCT01138085, NCT01337765, NCT01390818, and NCT00996892, respectively). Further, trials simultaneously inhibiting both BRAF and MEK, as well as a third inhibitor targeting individual enzymes (Akt, MDM2, Bcl-2, or MET) are also taking place for advanced melanoma patients (NCT01902173, NCT02110355, NCT01989585, or NCT01974258, respectively). Finally, a Phase I trial is under way in relapsed or refractory BRAF(V600)-mutated melanoma with a selective ERK inhibitor, CC-90003 (NCT02313012).
In a different combined-therapy approach using a preclinical model, intermittent combination therapy with MEK inhibitor (cobimetinib) and PI3K inhibitor (GDC-0941) was sufficient to demonstrate a robust antitumor effect against BRAF/KRAS-mutated melanoma, compared to continuous exposure of these agents.82 Taking one step further, “adaptive sequential therapy”83 is a new therapeutic strategy of using targeted inhibitors based upon the patients’ resistance mechanism. An ongoing Phase II clinical trial for advanced metastatic melanoma provides BRAF inhibitor therapy until progression; upon progression, patients will be treated with an interventional drug targeting enzymes within the upregulated resistance pathway (NCT01820364).
Targeted treatment in combination with immunotherapy
In spite of the impressive results of targeted inhibition of MAPK, further advancements are needed to improve treatment outcomes for all patients in metastatic melanoma. As immunotherapy has displayed positive results in melanoma,46,84,85 it was suggested to synergistically augment the immune system by combining immune checkpoint blockades with MAPK inhibition.86,87 A representative combination therapy is the use of BRAF inhibitor combined with antibodies that prevent inhibitory signaling, such as anti-CTLA-4 (ipilimumab) or anti-PD1 (pembrolizumab). The first clinical trial combining MAPK inhibition with immunotherapy involved ipilimumab and vemurafenib (NCT01400451); however, this combination therapy was terminated due to substantial liver toxicities.88 Investigations with other combinations of MAPK inhibitors, anti-PDL1 immunotherapies, cytokines (IL-2, IFN-alpha 2b), or adoptive cell transfer therapy are still ongoing in clinical trials.89
Although immunotherapies are highlighted by recent success in advanced melanoma,89–92 toxic effects in combination with kinase-targeted therapy remain unpredictable. Thus, the dosing, timing, and adverse effects of combination treatments involving immunotherapy need to be carefully studied. Through further studies, prolonged antitumor response using combination MAPK inhibition with immunomodulators may be achievable for patients with advanced melanoma. With their high antitumor activity and synergistic properties, these therapies have a high potential for improving clinical outcomes.
Conclusion
With the discovery of BRAF-mutant melanomas, the development of new targeted therapeutics has increased tremendously in the last several years. These drugs hold great potential in improving the prognosis of advanced melanoma, but are limited by intrinsic and acquired resistance. BRAF inhibitors have demonstrated tumor response and increased median PFS in advanced BRAF-mutant melanomas. However, their utility is limited by numerous, complex, and often overlapping resistance pathways. Usage of MEK inhibitors in conjunction with BRAF inhibitors offers a method of combating this resistance. However, acquired resistance may also hinder dual therapy. Whether the best strategy to prolong survival is through optimization of triple therapy or sequential therapy will need to be further explored.
Disclosure
The authors report no conflicts of interest in this work.
References
Jhappan C, Noonan FP, Merlino G. Ultraviolet radiation and cutaneous malignant melanoma. Oncogene. 2003;22(20):3099–3112. | ||
Maverakis E, Miyamura Y, Bowen MP, Correa G, Ono Y, Goodarzi H. Light, including ultraviolet. J Autoimmun. 2010;34(3):J247–J257. | ||
Peyssonnaux C, Eychène A. The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell. 2001;93(1–2):53–62. | ||
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | ||
Kim G, McKee AE, Ning YM, et al. FDA Approval summary: vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin Cancer Res. 2014;20(19):4994–5000. | ||
Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved Survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. | ||
Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–714. | ||
McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–332. | ||
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–2526. | ||
FDA Approval for Dabrafenib [webpage on the Internet]. Bethesda: National Cancer Institute; [updated January 16, 2014]. Available from: http://www.cancer.gov/about-cancer/treatment/drugs/fda-dabrafenib. Accessed October 4, 2015. | ||
Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. | ||
Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20(7):1965–1977. | ||
Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32(19):2373–2379. | ||
Abel EV, Basile KJ, Kugel CH 3rd, et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J Clin Invest. 2013;123(5):2155–2168. | ||
Girotti MR, Pedersen M, Sanchez-Laorden B, et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013;3(2):158–167. | ||
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. | ||
Yadav V, Zhang X, Liu J, et al. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J Biol Chem. 2012;287(33):28087–28098. | ||
Dougherty MK, Müller J, Ritt DA, et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17(2):215–224. | ||
Douville E, Downward J. EGF induced SOS phosphorylation in PC12 cells involves P90 RSK-2. Oncogene. 1997;15(4):373–383. | ||
Eblaghie MC, Lunn JS, Dickinson RJ, et al. Negative feedback regulation of FGF signaling levels by Pyst1/MKP3 in chick embryos. Curr Biol. 2003;13(12):1009–1018. | ||
Hanafusa H, Torii S, Yasunaga T, Nishida E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol. 2002;4(11):850–858. | ||
Lito P, Pratilas CA, Joseph EW, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22(5):668–682. | ||
Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–504. | ||
Paraiso KH, Fedorenko IV, Cantini LP, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102(12):1724–1730. | ||
Sanchez-Laorden B, Viros A, Girotti MR, et al. BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal. 2014;7(318):ra30. | ||
Oberholzer PA, Kee D, Dziunycz P, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol. 2012;30(3):316–321. | ||
Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366(3):207–215. | ||
Trunzer K, Pavlick AC, Schuchter L, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31(14):1767–1774. | ||
Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140(2):209–221. | ||
Eskandarpour M, Kiaii S, Zhu C, Castro J, Sakko AJ, Hansson J. Suppression of oncogenic NRAS by RNA interference induces apoptosis of human melanoma cells. Int J Cancer. 2005;115(1):65–73. | ||
Li Y, Bollag G, Clark R, et al. Somatic mutations in the neurofibromatosis 1 gene in human tumors. Cell. 1992;69(2):275–281. | ||
Whittaker SR, Theurillat JP, Van Allen E, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3(3):350–362. | ||
Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–867. | ||
Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74(8):2340–2350. | ||
Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. | ||
Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387–390. | ||
Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68(12):4853–4861. | ||
Gibney GT, Messina JL, Fedorenko IV, Sondak VK, Smalley KS. Paradoxical oncogenesis – the long-term effects of BRAF inhibition in melanoma. Nat Rev Clin Oncol. 2013;10(7):390–399. | ||
Freeman AK, Ritt AA, Morrison DK. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell. 2013;49(4):751–758. | ||
Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. | ||
Marusiak AA, Edwards ZC, Hugo W, et al. Mixed lineage kinases activate MEK independently of RAF to mediate resistance to RAF inhibitors. Nat Commun. 2014;5:3901. | ||
Frederick DT, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225–1231. | ||
Khalili JS, Liu S, Rodríguez-Cruz TG, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18(19):5329–5340. | ||
Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18(5):1386–1394. | ||
Boni A, Cogdill AP, Dang P, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70(13):5213–5219. | ||
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | ||
Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. | ||
Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013;19(3):598–609. | ||
Atefi M, Avramis E, Lassen A, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014;20(13):3446–3457. | ||
Berthon C, Driss V, Liu J, et al. In acute myeloid leukemia, B7-H1 (PD-L1) protection of blasts from cytotoxic T cells is induced by TLR ligands and interferon-gamma and can be reversed using MEK inhibitors. Cancer Immunol Immunother. 2010;59(12):1839–1849. | ||
Rice KD, Aay N, Anand NK, et al. Novel carboxamide-based allosteric MEK inhibitors: discovery and optimization efforts toward XL518 (GDC-0973). ACS Med Chem Lett. 2012;3(5):416–421. | ||
Hatzivassiliou G, Haling JR, Chen H, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501(7466):232–236. | ||
Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439(7074):358–362. | ||
Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17(5):989–1000. | ||
Flaherty KT, Robert C, Hersey P, et al; ETRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. | ||
Catalanotti F, Solit DB, Pulitzer MP, et al. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin Cancer Res. 2013;19(8):2257–2264. | ||
Rosen L, LoRusso P, Ma WW, et al. A first-in-human phase 1 study to evaluate the MEK1/2 inhibitor GDC-0973 administered daily in patients with advanced solid tumors [abstract]. In: Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research; 2011 April 2–6; Orlando, FL. Philadelphia: AACR. Cancer Res. 2011;71(8 Suppl):Abstract 4716. | ||
Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–1876. | ||
Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877–1888. | ||
Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–39. | ||
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. | ||
Long GV, Stroyakovsky DL, Gogas H, et al. COMBI-d: a randomized, double-blinded, Phase III study comparing the combination of dabrafenib and trametinib to dabrafenib and trametinib placebo as first-line therapy in patients (pts) with unresectable or metastatic BRAFV600E/K mutation-positive cutaneous melanoma. J Clin Oncol. 2014;32:5s(suppl; abstr 9011). | ||
Sanlorenzo M, Choudhry A, Vujic I, et al. Comparative profile of cutaneous adverse events: BRAF/MEK inhibitor combination therapy versus BRAF monotherapy in melanoma. J Am Acad Dermatol. 2014;71(6):1102–1109.e1. | ||
Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431–435. | ||
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–430. | ||
Curti BD. Rapid evolution of combination therapy in melanoma. N Engl J Med. 2014;371(20):1929–1930. | ||
Ribas A, Gonzalez R, Pavlick A, et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAF(V600)-mutated melanoma: a phase 1b study. Lancet Oncol. 2014;15(9):954–965. | ||
Kim KB, Kefford R, Pavlick AC, et al. Phase II study of the MEK1/MEK2 inhibitor trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31(4):482–489. | ||
Girotti MR, Lopes F, Preece N, et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell. 2015;27(1):85–96. | ||
Le K, Blomain ES, Rodeck U, Aplin AE. Selective RAF inhibitor impairs ERK1/2 phosphorylation and growth in mutant NRAS, vemurafenib-resistant melanoma cells. Pigment Cell Melanoma Res. 2013;26(4):509–517. | ||
Hao M, Song F, Du X, et al. Advances in targeted therapy for unresectable melanoma: new drugs and combinations. Cancer Lett. 2015;359(1):1–8. | ||
Niezgoda A, Niezgoda P, Czajkowski R. Novel approaches to treatment of advanced melanoma: a review on targeted therapy and immunotherapy. Biomed Res Int. 2015;2015:851387. | ||
Cheng H, Terai M, Kageyama K, et al. Paracrine effect of NRG1 and HGF drives resistance to MEK inhibitors in metastatic uveal melanoma. Cancer Res. 2015;75(13):2737–2748. | ||
Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508(7494):118–122. | ||
Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. | ||
Nathanson KL, Martin AM, Wubbenhorst B, et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res. 2013;19(17):4868–4878. | ||
Smalley KS, Lioni M, Dalla Palma M, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther. 2008;7(9):2876–2883. | ||
Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29(22):3085–3096. | ||
Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106(48):20411–20416. | ||
Hatzivassiliou G, Liu B, O’Brien C, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012;11(5):1143–1154. | ||
Jaiswal BS, Janakiraman V, Kljavin NM, et al. Combined targeting of BRAF and CRAF or BRAF and PI3K effector pathways is required for efficacy in NRAS mutant tumors. PLoS One. 2009;4(5):e5717. | ||
Hoeflich KP, Merchant M, Orr C, et al. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res. 2012;72(1):210–219. | ||
Spagnolo F, Ghiorzo P, Orgiano L, et al. BRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategies. Onco Targets Ther. 2015;8:157–168. | ||
Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299–308. | ||
Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–546. | ||
Hu-Lieskovan S, Robert L, Homet Moreno B, Ribas A. Combining targeted therapy with immunotherapy in BRAF-mutant melanoma: promise and challenges. J Clin Oncol. 2014;32(21):2248–2254. | ||
Menzies AM, Long GV. Systemic treatment for BRAF-mutant melanoma: where do we go next? Lancet Oncol. 2014;15(9):e371–e381. | ||
Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365–1366. | ||
McDermott D, Lebbé C, Hodi FS, et al. Durable benefit and the potential for long-term survival with immunotherapy in advanced melanoma. Cancer Treat Rev. 2014;40(9):1056–1064. | ||
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. | ||
Maverakis E, Cornelius LA, Bowen GM, et al. Metastatic melanoma – a review of current and future treatment options. Acta Derm Venereol. 2015;95(5):516–524. | ||
Shi VY, Tran K, Patel F, et al. 100% complete response rate in patients with cutaneous metastatic melanoma treated with intralesional interleukin (IL)-2, imiquimod, and topical retinoid cominbation therapy: results of a case series. J Am Acad Dermatol. 2015;73(4):645–654. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.