")
Back to Journals » OncoTargets and Therapy » Volume 13
Meg3 Induces EMT and Invasion of Glioma Cells via Autophagy
Authors Yang Z, Bian E, Xu Y, Ji X, Tang F, Ma C, Wang H, Zhao B
Received 22 November 2019
Accepted for publication 8 January 2020
Published 31 January 2020 Volume 2020:13 Pages 989—1000
DOI https://doi.org/10.2147/OTT.S239648
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
This paper has been retracted.
Zhihao Yang,1,2,* Erbao Bian,1,2,* Yadi Xu,1,2 Xinghu Ji,1,2 Feng Tang,1,2 Chunchun Ma,1,2 Hongliang Wang,1,2 Bing Zhao1,2
1Department of Neurosurgery, The Second Affiliated Hospital of Anhui Medical University, Hefei 230601, People’s Republic of China; 2Cerebral Vascular Disease Research Center, Anhui Medical University, Hefei 230601, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Bing Zhao
Department of Neurosurgery, The Second Affiliated Hospital of Anhui Medical University, 678 Fu Rong Road, Hefei, Anhui Province 230601, People’s Republic of China
Tel +86 551 63869502
Fax +86 551 63869400
Email [email protected]
Background: Glioma is one of the most common malignant tumors. Glioblastoma (grade IV) is considered the most malignant form of human brain tumors. Maternal expression gene 3 (Meg3) encodes a non-coding RNA (ncRNA) that plays an important role in the development and progression of cancer. However, the role of Meg3 in glioma cells remains largely unclear.
Methods: Reverse transcription-quantitative (RT-q) PCR was conducted to evaluate the mRNA expression related to cell autophagy and EMT while protein expression was detected by Western blotting. Staining of acidic vacuoles and immunofluorescence staining were used to detect autophagy. The ability of cells to migrate and invade was detected by Transwell migration and invasion assays.
Results: In the present study, it was found that the overexpression of Meg3 induced EMT, migration and invasion of glioma cells, whereas Meg3 overexpression induced autophagy of glioma cells. More importantly, the inhibition of autophagy impaired the EMT of glioma cells. In addition, Meg3-induced EMT, migration and invasion could be partially reversed by autophagy inhibitors, chloroquine (CQ) and Lys05, in glioma cells.
Conclusion: All data suggest that Meg3 induces EMT and invasion of glioma cells via autophagy. Overall, the findings of the present study demonstrate the importance of Meg3 in the molecular etiology of glioma, which also indicate its potential applications in the treatment of glioma.
Keywords: long non-coding RNA, Meg3, EMT, invasion, autophagy, glioma
Introduction
Glioma is one of the most frequent malignant tumors with a high recurrence rate.1 According to the classification of WHO, gliomas could be categorized into four distinct histopathological grades, grades I, II, III and IV. Glioblastoma (grade IV) is considered the most malignant form of human brain tumors.2 Due to the feature of the invasive growth of glioma, it has no perceptible boundaries with the normal brain tissue3,4 and is difficult to be completely resected, whereas simple to revert owing to resistance to radiotherapy as well as chemotherapy.5–7 Despite substantial advances in the understanding of the molecular status of this tumor type, new effective treatment is still necessary. As such, it is important to identify new mechanisms associated with the development of glioma, as well as to establish possible therapeutic targets for its treatment.
Long non-coding RNA (lncRNA) is a transcript with more than 200 nucleotides and has little coding power for functional proteins. Increasing evidences have shown that lncRNA could regulate gene expression at different levels, including transcription, post-transcription and epigenetic regulation.8–10 The abnormal expression of lncRNA has been found in a variety of cancer types. For example, some studies have shown that lncRNA participated in the promotion of tumor growth, angiogenesis and metastasis through various mechanisms.11,12 However, other studies showed that lncRNA inhibited the development and progression of cancer.13 Recently, several studies have shown that Meg3 played different roles in different cancer types. For example, the overexpression of Meg3 inhibited epithelial-mesenchymal transition (EMT), migration and invasion of cervical cancer.14 Similarly, in human pancreatic cancer, Meg3 knockdown promoted cell migration and invasion, and induced EMT.15 However, Meg3 contributes to the EMT phenotype, migration and invasion of HCC (Hepatocellular carcinoma) cells.16 Nevertheless, the role of Meg3 in EMT and invasion has not been well explored in glioma cells.
Autophagy is a conservative cellular pathway that can remove dysfunctional or damaged organelles.17 Cells redigest their own organelles and proteins, therefore maintaining macromolecule synthesis during autophagy. Currently, the role of autophagy in cancer is still controversial, since they may inhibit tumors in the development of cancer, but also promote cell survival during the progression of cancer.18 Recently, some studies indicate the association between tumor autophagy and tumor EMT and invasion. The inhibition of autophagy can damage the EMT and invasion of cancer cells.19
According to a study, EMT is a pivotal regulator of metastasis, by promoting the invasion of tumor cells and the spread to distant organs.20 In human non-small cell lung cancer cells, Fasone inhibits migration and invasion by attenuating EMT.21 The depletion of lncRNA DNM3OS inhibits the migration and invasion of gastric cancer cells by suppressing snail-mediated EMT.22 In human U251 glioma cells, β-Asarone suppressed EMT and invasion through the inhibition of the splicing factor HnRNP A2/B1.23 Furthermore, it is noteworthy that there is growing evidence that autophagy inhibitors could enhance the efficacy of treatment of different cancer types.24,25
The present study demonstrated that Meg3 induced EMT, migration, invasion and autophagy of glioma cells. Additionally, autophagy inhibitors reversed Meg3-induced EMT, migration and invasion. These results showed that Meg3 may be a potential therapeutic target for glioma.
Materials and Methods
Primary Cell Isolation from Patient-Derived Tumor Tissue
Tumor tissues were dissected from patients, ≥18 years of age with primary GBM tumors, during surgery in the Neurosurgery Department of The Second Affiliated Hospital of Anhui Medical University (Hefei, China) and collected in sterile Hibernation media and transported to the laboratory on ice within 1 hr. Patient-derived tumor tissue was cut into small pieces with a scalpel and digested for 30 min at 37°C enzymatically in a mixture consisting of Papain (20 µ/mL, #LK003176, Worthington) and DNase (2000 µ/mL, #LK003170, Worthington). Ovomucoid inhibitor (10 mg/mL, #LK003182, Worthington) was used to stop the enzymatic activity at room temperature and an erythrocyte lysis was performed for further 20 min at room temperature. Seed the primary GBM cells into a GelTrex or Matrigel-coated plastic flask and cultivate in RPMI 1640 (R8758, Sigma) supplemented with 10% FCS (F7524, Sigma) and 1% penicillin/streptomycin (P0781, Sigma). Two primary cell lines (T76, H34) isolated from patient tumor tissue were included in this experimental study. This study was approved by the Research Ethics Committee of The Second Affiliated Hospital of Anhui Medical University. Informed consent was obtained from all the patients.
Cell Culture and Transfection
The human glioma U87, U251MG and A172 cell lines were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences, Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China), where they were characterized by DNA fingerprinting and isozyme detection. All cell lines were cultured in Dulbecco’s Modified Eagle’s medium (DMEM; Hyclone; GE Healthcare Life Sciences)/high sugar supplemented 10% fetal bovine serum (FBS, Gibco; Thermo Fisher Scientific, Inc.), 100 U/mL penicillin and 100 U/mL streptomycin (Hyclone; GE Healthcare Life Sciences). The cells were maintained at 37°C, and the humidity was maintained at 5% CO2. Based on this culture state, the medium of the cells was changed every 2 days.
Cell transfection was implemented in glioma cells (2×105 cells per 200 mm2 dish) cultivated in penicillin/streptavidin-free DMEM, containing 10% FBS for 12 h. The Meg3 plasmid (pcDNA3.1-Meg3) was constructed by subcloning the full-length Meg3 coding sequence into pcDNA3.1(+) and confirming by sequencing. All overexpression plasmids were selected by culturing the cells in the presence of puromycin (1 μg/mL) for 72 h following transfection. Cell transfection was performed utilizing Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) in accordance with manufacturer’s protocol in Opti-MEM (Gibco; Thermo Fisher Scientific, Inc.).
Reverse Transcription-Quantitative (RT-q) PCR
TRIZOL reagent (Invitrogen; Thermo Fisher Scientific, Inc.) was used to extract total RNA from glioma cells. Meanwhile, a nanodrop spectrophotometer (IMPLEN GmbH) was used to determine the concentration and quality of RNA by 260/280 nm absorbance. Next, after utilizing PrimeScript RT Master mix (fully real-time) (Takara Biotechnology Co., Ltd.) to reverse transcribe RNA into high-capacity DNA, quantitative PCR (Takara Biotechnology Co., Ltd.) was performed using Maxima SYBR Green/ROX qPCR Master mix (Takara Biotechnology Co., Ltd.). In brief, the total volume of each PCR reaction mixture is 20 μL, containing 10 μL 2X SYBR Green Master mix, 0.4 μL ROX Dye, 1 μL sense and antisense primers (5 μmol/μL), 1 μL cDNA (10 ng) and 7.6 μL of nuclease-free water. A total of 45 cycles were performed, denatured at 95 °C for 15 sec, annealed at 60°C for 30 sec, and extended at 72°C for 30 sec. For relative quantification, 2−ΔΔCt was calculated and utilized as an indicator of relative expression level. This was calculated by subtracting the CT values of control genes from the CT values of corresponding genes. RT-qPCR was conducted using the following primers according to the manufacturer’s protocol: MEG3 forward, 5′-ATCATCCGTCCACCTCCTTGTCTTC-3′; MEG3 reverse, 5′-GTATGAGCATAGCAAAGGTCAGGGC-3′; LC3B forward, 5′-GCAGTGGAGGGGATGACC-3′; LC3B reverse, 5′-TGACGACGAGGATTGCTCTG-3′; P62 forward, 5′-ATCGGAGGATCCGAGTGT-3′; P62 reverse, 5′-TGGCTGTGAGCTGCTCTT-3′; ATG3 forward, 5′-ATGGGAGTTGGCGAAGGCAAGT-3′; ATG3 reverse, 5′-AGCTCCACGTATCGAAGACAGC-3′; ATG5 forward, 5′-TCAGCCACTGCAGAGGTGTTT-3′; ATG5 reverse, 5′-GGCTGCAGATGGACAGTTGCA-3′; LAMP1 forward, 5′-TGACAAGGCTTCTCAACATC-3′; LAMP1 reverse, 5′-CATTCATCCCGAACTGG-3′; ZEB1 forward, 5′-ACTCTGATTCTACACCGC-3′; ZEB1 reverse, 5′-TGTCACATTGATAGGGCTT-3′; ZEB2 forward, 5′-TGAGGATGACGGTATTGC-3′; ZEB2 reverse, 5′-ATCTCGTTGTTGTGCCAG-3′; BECLIN-1 forward, 5′-AGCACCATGCAGGTGAGCTT-3′; BECLIN-1 reverse 5′-TGACACGGTCCAGGATCTTG-3′; GAPDH forward, 5′-AGCAAGAGCACAAGAGGAAG-3′; GAPDH reverse, 5′-GGTTGAGCACAGGGTACTTT-3′. GAPDH was performed as an internal control.
Western Blot Analysis
RIPA pyrolysis buffer and PMSF (Beyotime, China) were used to lyse glioma cells. After the preparation of all the extracts, the protein concentration was determined by BCA protein assay kit (Wuhan Boster Biological Technology, Ltd.). The whole cell extract (25 mg or 45 mg) was then separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The gel was run under 80V for 30 min and then at 120V for 70 min, followed by transfer to PVDF membrane (EMD Millipore). After blocking the binding of non-specific proteins, the nitrocellulose membrane was imprinted at room temperature and incubated for 1 h with the first resistance diluted in TBS/Tween20 (0.075%) containing 5% skim milk. Rabbit Polyclonal Anti-LC3B (cat. no. D163557; Cell Signaling Technology, Inc.) was diluted 1:1000. Rabbit polyclonal anti-P62 (BIOSS) was diluted 1:1000. Rabbit polyclonal anti-BECLIN-1 (Santa Cruz Biotechnology, Inc.) was diluted 1:1000. Rabbit polyclonal anti-LAMP1 (ProteinTech Group, Inc.) was diluted 1:1000. Rabbit polyclonal anti-ZEB1 (cat. no. ab124512; Abcam) was diluted 1:1000. Rabbit polyclonal anti-ZEB2 (cat. no. ab138222; Abcam) was diluted 1:1000. Mouse polyclonal anti-beta-actin (Santa Cruz Biotechnology, Inc.) was diluted 1:1000. After incubation with the first antibody, the imprinting was washed in TBS/Tween-20 four times, and then incubated for 2 h with goat anti-mouse or mouse anti-rabbit horseradish peroxidase-conjugated antibodies containing 1:10,000 dilution of TBS/Tween-20, including 5% milk. Following washes in TBS/Tween-20, the imprinting was washed with distilled water and the protein was detected by enhanced chemiluminescence system. Protein observation was done using ECL-Chemiluminescence kit (ECL-plus, Thermo Fisher Scientific, Inc.).
Immunofluorescence Staining
Cells were inoculated on the cover slide of a 6-well plate and cultured overnight. Subsequently, the cells were immobilized with 4% paraformaldehyde (cat. no. ST476; Beyotime Institute of Biotechnology) in PBS. The cells were permeated with 0.5% Triton X-100 (cat. no. ST795; Beyotime Biotechnology) in PBS and incubated with rabbit anti-LC3B antibody (cat. no. D163557; 1:200; Cells Signaling Technology, Inc.) in 5% bovine serum albumin (cat. no. AR0004; BOSTER) in PBS. FITC-conjugated anti-rabbit IgG (cat. no. 150077; Abcam) was used to detect primary antibody. DAPI was used to incubate cells to stain the nuclei in darkness. The slides were observed under a fluorescence microscope and the images were obtained by using a charge coupled device (CCD) digital color camera (cat. no. DP71; Olympus).
Staining of Acidic Vacuoles
Cells were cultured overnight on 6-well flat cover slides, washed with PBS, stained with acridine orange (1 ug/mL in PBS) for 15 min at 37°C. Then, cells were washed with PBS and observed with a fluorescence microscope (Olympus Corporation), utilizing the CEIN filter set.
Migration and Invasion Assays
Cells were plated (1×104 for migration assay; 8×104 cells for invasion assay) on top of polycarbonate transwell filter without matrigel for transwell assay or covered in matrigel for transwell matrix permeation assay in the top chamber of QCM 24-pore cell invasion assay (Cell Biolabs, Inc.). For the migration assay, cells were suspended in serum-free medium which was in the basement chamber. For the invasion assay, cells were suspended in serum-free medium and serum-supplemented medium was used as chemical attractant in the basement chamber. Cells were incubated for 8 h (transwell assay) or 48 h (invasion assay) at 37°C. Non-migrating or non-invasive cells were removed from the parietal chamber with a cotton swab. The migrated and invaded cells on the submucosal surface were immobilized in 100% methanol for 10 mins, air dried, and then stained with 0.005% (w/v) crystal violet, and quantified under a microscope. The number of migrated cells was quantified by counting those in five random fields of each membrane. Three independent experiments were performed.
Statistical Analysis
The experimental data are presented in the form of the mean ± SD. Student’s t-test and the one-way analysis of variance (ANOVA, Tukey) was performed by SPSS 17.0 statistical software. P<0.05 was used to indicate a statistically significant difference.
Results
Meg3 Induces EMT, Migration and Invasion in Glioma Cells
To explore the functions of Meg3 in glioma, the expression of Meg3 was detected in several glioma cell lines and patient-derived glioma cell lines. The results showed that the abundance of Meg3 in U87 and U251 glioma cell lines was low (Figure S1A); thus, U87 and U251 cell lines were selected to perform subsequent experiments. We speculated that it was difficult to observe the phenotype of cells by knockdown of Meg3. Therefore, we observed the phenotype of cells in overexpressed Meg3. Meg3 plasmid was transfected into U87 and U251 glioma cells. As shown in Figure S1B, Meg3 expression was markedly increased in glioma cells transfected with Meg3 plasmid compared with vector. Meg3 overexpression induced a spindle-shaped morphology, a more mesenchymal cell-like morphology in U87 and U251 glioma cells after culturing for 3 weeks compared with vector groups (Figure 1A). RT-qPCR assays demonstrated significantly increased mRNA levels of ZEB1 and ZEB2, known as EMT markers, in U87 and U251 glioma cells that were transfected with Meg3 plasmid compared with vector groups (Figure 1B). In addition, Western blot assay results showed that the overexpression of Meg3 resulted in remarkedly increased protein expression of ZEB1 and ZEB2 (Figure 1C and D). By transwell assays, it was found that the overexpression of Meg3 significantly increased the migration and invasion of U87 and U251 glioma cells (Figure 1E). These findings indicate that Meg3 is a promoter of glioma EMT, migration and invasion.
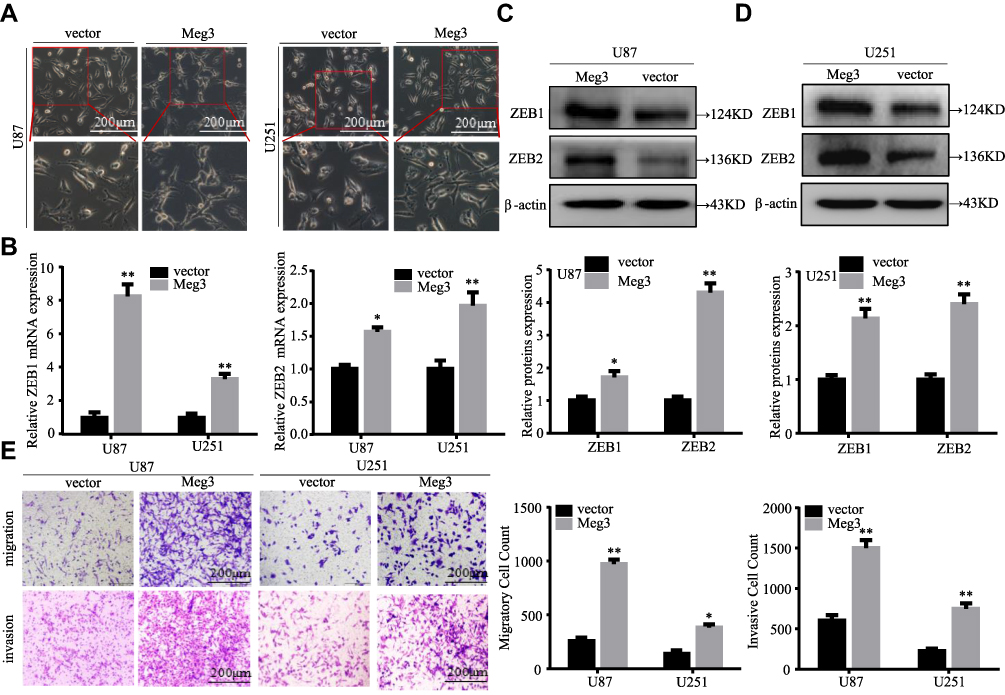 |
Figure 1 Meg3 induces EMT, migration, and invasion in glioma cells. (A) Morphological changes were observed in U87 and U251 glioma cells at 3 weeks following transfection with Meg3 plasmid. (B) The mRNA expression levels of EMT markers (ZEB1 and ZEB2) in glioma cells transfected with Meg3 plasmids and vector were examined by RT-qPCR. (C and D) The protein expression levels of EMT markers (ZEB1 and ZEB2) in glioma cells transfected with Meg3 plasmids and vector were tested by Western blotting. (E) The number of migrated and invasive cells were observed in glioma cells transfected with Meg3 plasmids and vector. *P<0.05, **P<0.01 vs vector. |
Meg3 Induces Autophagy in Glioma Cells
Staining the cells with acridine orange indicated that Meg3 overexpression resulted in increased amounts of acidic vacuoles, which is consistent with the formation of autophagosomes compared with cells transfected with the vector group (Figure 2A). Moreover, an elevated immunofluorescence signal of LC3B was observed in U87 and U251 glioma cells transfected with Meg3 plasmid compared with vector (Figure 2B). As shown in Figure 3A–E, Meg3 overexpression increased the mRNA levels of autophagy-associated genes such as ATG3, ATG5, BECLIN-1, LAMP1 and LC3B. Similarly, an increase in the protein levels of LC3B, BECLIN-1, LAMP1 and decrease in SQSTM1/P62 were observed in the Meg3-transfected group compared with the vector group (Figure 3F and G). However, no significant change in expression of ATG3 and ATG5 proteins was observed (Figure S1D). Overall, the results confirm that Meg3 could induce autophagy in glioma cells.
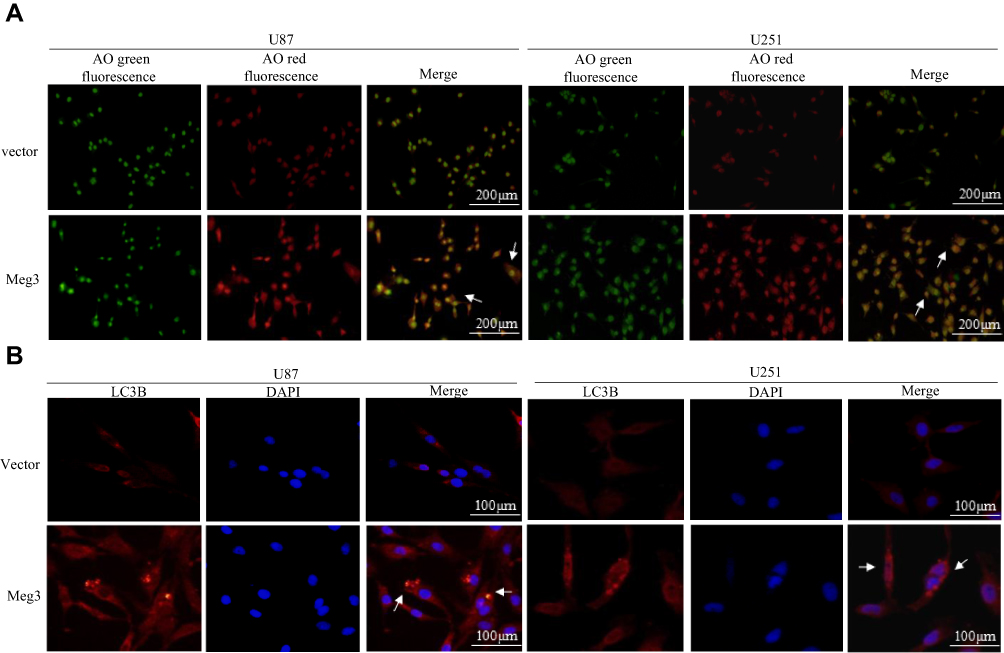 |
Figure 2 Meg3-induced autophagy in glioma cells. (A) The effect of Meg3 on the formation of acidic vacuoles was shown. Yellow to orange dots in the cytoplasm represented acidic vacuoles. Representative images were shown. (B) Immunofluorescence assay detected an elevated signal of LC3B in Meg3-transfected U87 and U251 glioma cells. |
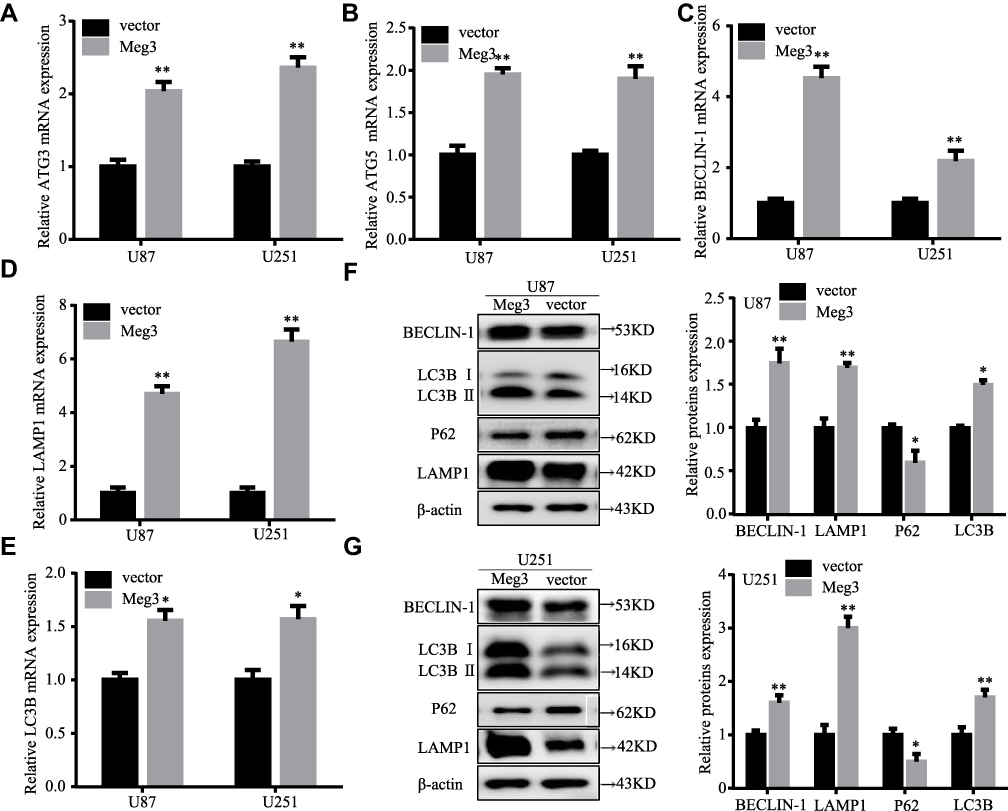 |
Figure 3 Autophagy-associated gene expression in Meg3-induced glioma cells. (A–E) The mRNAs expression levels of autophagy-associated genes (ATG3, ATG5, BECLIN-1, LAMP1 and LC3B) in U87 and U251 glioma cells transfected with Meg3 plasmids and vector were examined by RT-qPCR. (F and G) The protein expression levels of autophagy-associated genes (BECLIN-1, LC3B, P62 and LAMP1) in U87 and U251 glioma cells transfected with Meg3 plasmids and vector were tested by Western blotting. *P<0.05, **P<0.01 vs vector. |
Autophagy Inhibitors Suppress EMT of Glioma Cells
To explore the role of autophagy in EMT, U87 and U251 glioma cells were treated with CQ or Lys05, known as autophagy inhibitors, in indicated concentration for 24 h. CQ and Lys05 produced equivalent dose-dependent increases in the LC3BII/LC3BI ratio, accumulation of the autophagy cargo protein p62, indicating that CQ and Lys05 could suppress cell autophagy in glioma cells (Figure S1I–L). As shown in Figure 4A, U87 and U251 glioma cells were changed from mesenchymal morphology to epithelial morphology treated with CQ or Lys05. In addition, the protein levels of ZEB1 and ZEB2 were reduced in CQ- and Lys05-treated glioma cells for 24h in a dose-dependent manner (Figure 4B–E). These results indicated that autophagy inhibitors suppress glioma EMT.
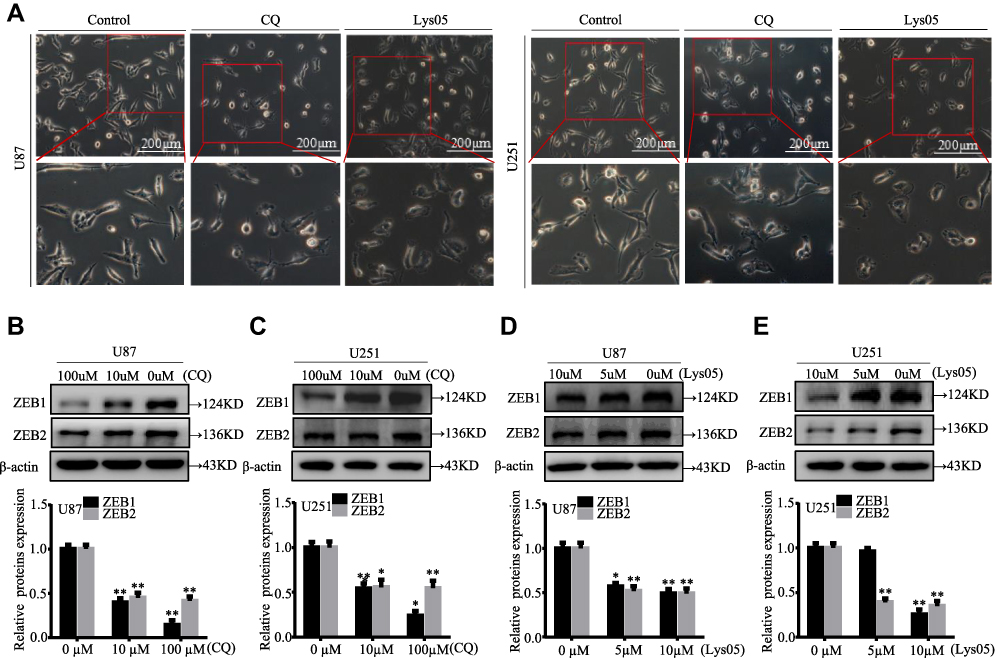 |
Figure 4 Autophagy inhibitors suppress the EMT of glioma cells. (A) The U87 and U251 glioma cells were cultured with or without CQ or Lys05 for 3 weeks, morphological changes were observed. (B–E) The protein levels of EMT markers ZEB1 and ZEB2 were examined in glioma cells treated with CQ or Lys05 for 24 h. *P<0.05, **P<0.01 vs control. |
Suppressing Autophagy Reversed Meg3-Induced EMT in Glioma Cells
To investigate whether Meg3 induced EMT in U87 and U251 glioma cells by autophagy, first, we selected CQ to detect whether autophagy inhibitors have an effect on Meg3 expression in glioma cells. The result showed that the expression of Meg3 in U87 and U251 glioma cells treated with CQ (100μm) for 24h had little changed compared with the control group (Figure S1C). Afterwards, glioma cells were transfected by Meg3 plasmid and then added CQ (100μm) or Lys05 (10μm). As shown in Figure 5A–D, Meg3 overexpression increased the mRNA expression levels of ZEB1 and ZEB2, whereas CQ treatment partially abolished Meg3-induced ZEB1 and ZEB2 mRNA expression in glioma cells. Similarly, Lys05 treatment partially abolished Meg3-induced ZEB1 and ZEB2 mRNA expression in glioma cells (Figure 5E–H). In addition, Meg3 overexpression increased the protein expression levels of ZEB1 and ZEB2, whereas CQ or Lys05 treatment partially reversed Meg3-induced ZEB1 and ZEB2 protein expression in glioma cells (Figure 5I–L) (Figure S1E–H). These results suggested that Meg3-induced EMT could be blocked by suppressing autophagy in glioma cells.
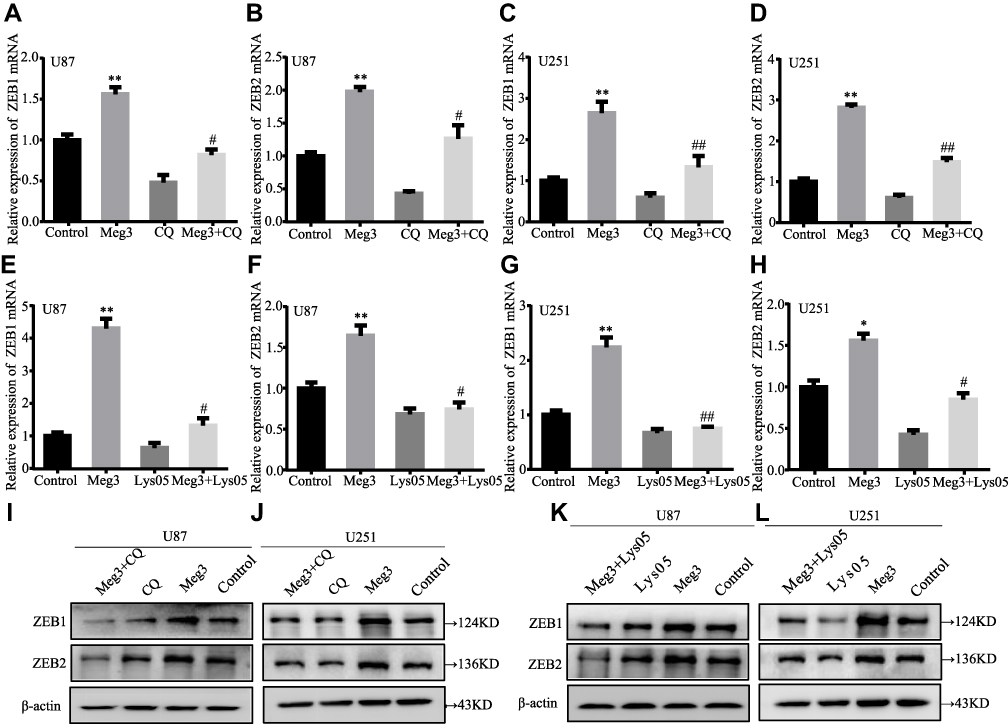 |
Figure 5 Inhibition of autophagy reverses Meg3-induced mRNA and protein expression of EMT-associated genes in glioma cells. (A–H) The mRNA expression levels of EMT markers (ZEB1 and ZEB2) in glioma cells transfected with Meg3 plasmids and vector with or without CQ or Lys05 treatment were tested by RT-qPCR. *P<0.05, **P<0.01 vs control; #P<0.05, ##P<0.01 vs Meg3. (I–L) The protein expression levels of EMT markers (ZEB1 and ZEB2) in glioma cells transfected with Meg3 plasmids and vector with or without CQ or Lys053 treatment were tested by Western blotting. |
The Inhibition of Autophagy Blocked Meg3-Induced Migration and Invasion in Glioma Cells
To determine whether Meg3 induced the migration and invasion of U87 and U251 glioma cells by autophagy, glioma cells were transfected with Meg3 plasmid and then treated with CQ (100μm) or Lys05 (10μm). The overexpression of Meg3 increased the number of migrating and invading cells compared with the control group, CQ treatment significantly decreased the migrating and invading cells induced by Meg3 (Figure 6A and B). Similarly, Lys05 treatment reversed the increase in migrating and invading cells induced by Meg3 (Figure 6C and D). These data indicate that Meg3-induced migration and invasion could be blocked by suppressing autophagy in glioma cells.
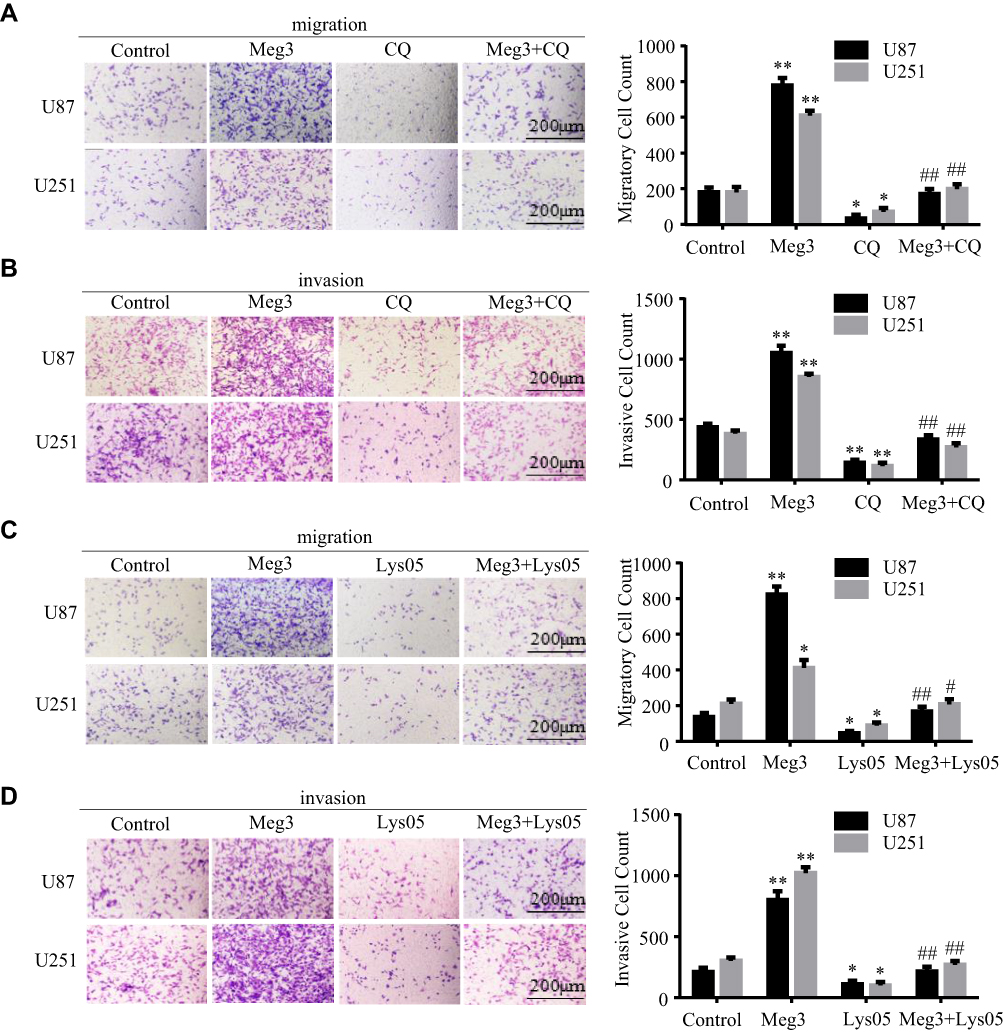 |
Figure 6 Meg3-induced migration and invasion is blocked by suppressing autophagy in glioma cells. (A–D) The migration of glioma cells transfected with Meg3 plasmids and vector with or without CQ or Lys053 treatment were tested. The invasion of glioma cells transfected with Meg3 plasmids and vector with or without CQ or Lys053 treatment was tested. *P<0.05, **P<0.01 vs control; #P<0.05, ##P<0.01 vs Meg3. |
Discussion
Recent studies have shown that lncRNA is imbalanced in several tumors and plays a key role in the biological processes of tumor, such as migration and invasion.26–28 Some studies have reported the role of lncRNAs in EMT of glioma cells. For instance, lncRNA UCA1 is reported to be necessary for EMT of glioma cells induced by TGF-β.29 It was demonstrated that the knockdown of lncRNA SPRY4-IT1 inhibits EMT in glioma cells.30 Meg3 is continually imbalanced in human cancer, and is associated with the EMT of cancer cells.31 In our previous study, we detected the expression of Meg3 in glioma samples and found downregulation of Meg3 in GBM samples compared to nonneoplastic brain tissue. Furthermore, the loss of Meg3 expression caused by hypermethylation induced proliferation and repress apoptosis in gliomas.32 But our later study found that overexpression of Meg3 induced EMT and invasion in glioma cells. These may be due to the complexity of biological processes in the occurrence and development of cancer and need further research. The findings of the present study showed that Meg3 changed the morphology of glioma cells to be a spindle-shaped morphology. In addition, Meg3 increased the expression of EMT-associated genes, such as ZEB1, ZEB2. Consistent with a previous study, Meg3 regulates EMT by epigenetic repression of the E-cadherin regulatory region in lung cancer cell lines.33
In addition, abnormal expression of the ZEB family has been reported in many cancer types, which is involved in tumor invasion and metastasis.34–36 Orellana-Serradell et al reported that the overexpression of ZEB family induced EMT and promoted invasion in prostate cancer cells.37 In glioblastoma, members of the SNAI family, such as ZEB1 and ZEB2, act as EMT activators to enhance the invasiveness of GBM cells in vitro and in vivo.38,39 Siebzehnrubl et al reported that the knockdown of ZEB family can inhibit cell invasion in glioblastoma (GBM).40 The present study demonstrated that the overexpression of Meg3 promoted the expression of ZEB family and the ability of glioma cells to migrate and invade. These results suggest that Meg3 may promote the migration and invasion of glioma cells by inducing EMT.
Generally, autophagy is a multistage process regulated by a variety of autophagy-associated proteins. In the present study, it was found that overexpression of Meg3 increased the number of acidic vacuoles consistent with autophagosomes. As for the expression of autophagy-associated genes, the immunofluorescence signal of LC3B in glioma cells transfected with Meg3 was increased. In addition, the expression of LC3B, Beclin-1 and LAMP1 was increased, whereas the expression of P62 was decreased in glioma cells transfected with Meg3. These results suggest that Meg3 induces autophagy in human glioma cells. In addition, the upregulation of Meg3 induced autophagy in epithelial ovarian cancer.41 However, the downregulation of Meg3 activates autophagy in bladder cancer.42 The effect of Meg3 on autophagy was found to be different in different tumors; this is speculated to be due to the specificity of different tumors.
CQ, a known inhibitor of autophagosome-to-lysosome fusion, is widely used to monitor autophagic flux. CQ, traditionally used as an antimalarial drug, is the only autophagy inhibitor approved by the Food and Drug Administration of the United States.43 In addition, CQ showed a concentration-dependent anti-tumor effect in glioma cells.44 Lu et al found that CQ blocked autophagy-induced cell migration and GBM infiltration.45 Furthermore, some relevant clinical studies of CQ in the treatment of brain tumor have been reported. In a double-blinded clinical trial involving 30 patients with glioblastoma multiforme (NCT00224978) (a Phase III clinical trial), randomizing eligible patients with surgically confirmed glioblastoma to receive conventional chemotherapy and radiotherapy plus placebo or 150 mg/day CQ per os, the result showed CQ-receiving patients exhibited an improved mid-term survival as compared with their control counterparts.46 CQ has also been evaluated for its ability to boost the therapeutic activity of whole-brain radiation therapy (WBRT) in 20 patients bearing intracranial metastases of various histological derivation (NCT01894633). In the context of this single-cohort Phase II clinical study, the results showed that WBRT with concurrent, short-course CQ is well tolerated in patients with brain metastases and the high intracranial disease control rate warrants additional study.47 Lys05, another autophagy inhibitor, accumulates more effectively in lysosomes and deacidifies them, resulting in damage to autophagy.48 Furthermore, Lys05 is an effective autophagy inhibitor with single-drug anti-tumor activity.49 It is reported that Lys05 can effectively inhibit autophagy of clear cell ovarian cancer and hepatocellular carcinoma.50,51 The present study demonstrated an equivalent dose-dependent increase in the LC3BII/LC3BI ratio and the accumulation of autophagic cargo protein p62 in glioma cells treated with CQ or Lys05. Thus, CQ and Lys05 could effectively inhibit autophagy in glioma cells. Following several weeks of culture, glioma cells treated with CQ or Lys05 converted from mesenchymal morphology to epithelial morphology. In addition, the expression of ZEB1 and ZEB2 decreased in a dose-dependent manner in CQ- or Lys05-treated glioma cells, suggesting that autophagy inhibitors inhibit EMT in glioma cells.
Subsequently, the data and information obtained during the present study indicate that autophagy flux is critical for the EMT, migration and invasion of Meg3-induced glioma cells. It was found that the expression of ZEB1 and ZEB2 in glioma cells transfected with Meg3 was increased, whereas the expression of ZEB1 and ZEB2 was inhibited in glioma cells transfected with Meg3, following treatment with autophagy inhibitor CQ. Furthermore, overexpression of Meg3 resulted in the promotion of glioma cell migration and invasion, which was inhibited following treatment of Meg3-transfected glioma cells with CQ. To further validate the results, another autophagy inhibitor, Lys05, was used and the results were consistent with CQ. Thus, the present study demonstrated that blocking autophagy resulted in decreased expression of EMT-associated proteins and decreased the ability of glioma cells to migrate and invade. It is noteworthy that other studies showed that glioma cells require the autophagic process to migrate, and the downregulation of some autophagy genes limits the ability of cells to migrate and invade.19,52 Other report has also indicated that autophagy-associated gene SQSTM1 is associated with EMT.53 Moreover, TGF-β2 can induce autophagy to promote the invasion of glioma cells through Smad and non-Smad pathways.54 But other than that, some evidence indicates that the activation of the autophagy acts to prevent EMT in cancer cells. Myriam Catalano et al demonstrated that silencing of autophagy-related genes 5 (ATG5), ATG7, or Beclin-1 could enhance the EMT process and increase cell motility and invasiveness by overexpression of SNAIL and SLUG, two of the major transcription factors of the EMT process in glioblastoma cells, and nutrient deprivation or treatment with mTOR inhibitors would attenuate cell migration and invasion by inducing autophagy.55 As a“double-edged sword”, autophagy might exact the opposite effects on tumor development in different tissue or development stages of cancer.56 In a word, these findings provide a new opportunity to discuss the association between autophagy and EMT, migration and invasion. However, the exact mechanism of autophagy affecting EMT is unclear, and further exploration is required in the future.
We explored the effect of Meg3 on the invasion of glioma cells and its potential mechanism in vitro. For future work, we will conduct in vivo experiments to test whether the effect of Meg3 on invasion and its potential mechanism are consistent with the results of in vitro experiments. Overall, the present study suggests that Meg3 induced EMT, migration and invasion of U87 and U251 glioma cells, and provides a novel role for autophagy in these processes. In addition, the findings of the present study may aid in guiding the use of CQ and Lys05 for clinical application. Furthermore, the present study demonstrates the potential of identifying novel options to treat glioma and to inspire new ideas to update the current knowledge on cancer.
Abbreviations
Meg3, maternal expression gene 3; lncRNA, long non-coding RNA; EMT, epithelial-mesenchymal transition; GBM, glioblastoma; CQ, chloroquine.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Strowd RE
2. Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(5):97–109. doi:10.1007/s00401-007-0243-4
3. Yuan J, Liu L, Hu Q. Mathematical modeling of brain glioma growth using modified reaction-diffusion equation on brain MR images. Comput Biol Med. 2013;43(12):2007–2013. doi:10.1016/j.compbiomed.2013.09.023
4. Yu SQ, Wang JS, Chen SY, et al. Diagnostic significance of intraoperative ultrasound contrast in evaluating the resection degree of brain glioma by transmission electron microscopic examination. Chin Med J (Engl). 2015;128(2):186–190. doi:10.4103/0366-6999.149194
5. Arnab C, Abhijit C, Delaney MA, Latham DE, Loeffler JS. The epidermal growth factor receptor pathway mediates resistance to sequential administration of radiation and chemotherapy in primary human glioblastoma cells in a RAS-dependent manner. Cancer Res. 2002;62(15):4307–4315.
6. Frosina G. DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol Cancer Res. 2009;7(7):989–999. doi:10.1158/1541-7786.MCR-09-0030
7. Lomonaco SL, Finniss S, Xiang C, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer. 2009;125(3):717–722. doi:10.1002/ijc.24402
8. Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi:10.1146/annurev-biochem-051410-092902
9. Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152(6):1298–1307. doi:10.1016/j.cell.2013.02.012
10. Sarah G, Jeff CJNRMCB. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14(11):699–712. doi:10.1038/nrm3679
11. Wang ZH, Yang B, Zhang M, et al. lncRNA epigenetic landscape analysis identifies EPIC1 as an oncogenic lncRNA that interacts with MYC and promotes cell-cycle progression in cancer. Cancer Cell. 2018;33(4):706–720. doi:10.1016/j.ccell.2018.03.006
12. Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–1076. doi:10.1038/nature08975
13. Ding HD, Liu JP, Zou RY, Cheng PR, Su Y. Long non-coding RNA TPTEP1 inhibits hepatocellular carcinoma progression by suppressing STAT3 phosphorylation. J Exp Clin Cancer Res. 2019;38(1):189. doi:10.1186/s13046-019-1193-0
14. Chen X, Qu J. Long non-coding RNA MEG3 suppresses survival, migration, and invasion of cervical cancer. Onco Targets Ther. 2018;11:4999–5007. doi:10.2147/OTT.S167053
15. Ma L, Wang F, Du C, et al. Long non-coding RNA MEG3 functions as a tumour suppressor and has prognostic predictive value in human pancreatic cancer. Oncol Rep. 2018;39(3):1132–1140. doi:10.3892/or.2018.6178
16. Zhang Z, Wang S, Liu W. EMT-related long non-coding RNA in hepatocellular carcinoma: a study with TCGA database. Biochem Biophys Res Commun. 2018;503(3):1530–1536. doi:10.1016/j.bbrc.2018.07.075
17. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. doi:10.1080/15548627.2015.1100356
18. Choi KS. Autophagy and cancer. Exp Mol Med. 2012;44(2):109–120. doi:10.3858/emm.2012.44.2.033
19. Macintosh RL, Timpson P, Thorburn J, Anderson KI, Thorburn A, Ryan KM. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle. 2012;11(10):2022–2029. doi:10.4161/cc.20424
20. Santamaria PG, Moreno-Bueno G, Portillo F, Cano A. EMT: present and future in clinical oncology. Mol Oncol. 2017;11(7):718–738. doi:10.1002/1878-0261.12091
21. Tabasum S, Singh RP. Fisetin suppresses migration, invasion and stem-cell-like phenotype of human non-small cell lung carcinoma cells via attenuation of epithelial to mesenchymal transition. Chem Biol Interact. 2019;303:14–21. doi:10.1016/j.cbi.2019.02.020
22. Wang S, Ni B, Zhang Z, et al. Long non-coding RNA DNM3OS promotes tumor progression and EMT in gastric cancer by associating with Snail. Biochem Biophys Res Commun. 2019;511(1):57–62. doi:10.1016/j.bbrc.2019.02.030
23. Li L, Wu MX, Wang CQ, et al. β-asarone inhibits invasion and EMT in human glioma U251 cells by suppressing splicing factor HnRNP A2/B1. Molecules. 2018;23(3):E671. doi:10.3390/molecules23030671
24. Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi:10.1016/j.ccr.2006.06.001
25. Livesey KM, Daolin T, Zeh HJ, Lotze MT. Current opinion in investigational drugs: autophagy inhibition in combination cancer treatment. Curr Opin Investig Drugs. 2009;10(12):1269–1279.
26. Wang R, Li Y, Zhu G, et al. Long noncoding RNA CASC2 predicts the prognosis of glioma patients and functions as a suppressor for gliomas by suppressing Wnt/beta-catenin signaling pathway. Neuropsychiatr Dis Treat. 2017;13:1805–1813. doi:10.2147/NDT.S137171
27. Alvarez-Dominguez JR, Lodish HF. Emerging mechanisms of long noncoding RNA function during normal and malignant hematopoiesis. Blood. 2017;130(18):1965–1975. doi:10.1182/blood-2017-06-788695
28. Kiang KM, Zhang XQ, Leung GK. Long non-coding RNAs: the key players in glioma pathogenesis. Cancers (Basel). 2015;7(3):1406–1424. doi:10.3390/cancers7030843
29. Li Z, Liu H, Zhong Q, Wu J, Tang Z. LncRNA UCA1 is necessary for TGF-beta-induced epithelial-mesenchymal transition and stemness via acting as a ceRNA for Slug in glioma cells. FEBS Open Bio. 2018;8(11):1855–1865. doi:10.1002/2211-5463.12533
30. Liu HJ, Lv ZQ, Guo EK. Knockdown of long noncoding RNA SPRY4-IT1 suppresses glioma cell proliferation, metastasis and epithelial-mesenchymal transition. Int J Clin Exp Pathol. 2015;8(8):9140–9146.
31. Zhu Y, Chen P, Gao YS, et al. MEG3 activated by vitamin D inhibits colorectal cancer cells proliferation and migration via regulating clusterin. EBioMedicine. 2018;30:148–157. doi:10.1016/j.ebiom.2018.03.032
32. Li J, Bian EB, He XJ, et al. Epigenetic repression of long non-coding RNA MEG3 mediated by DNMT1 represses the p53 pathway in gliomas. Int J Oncol. 2016;48(2):723–733. doi:10.3892/ijo.2015.3285
33. Terashima M, Tange S, Ishimura A, Suzuki T. MEG3 long noncoding RNA contributes to the epigenetic regulation of epithelial-mesenchymal transition in lung cancer cell lines. J Biol Chem. 2017;292(1):82–99. doi:10.1074/jbc.M116.750950
34. Guo C, Ma J, Deng G, et al. ZEB1 Promotes Oxaliplatin Resistance through the Induction of Epithelial - Mesenchymal Transition in Colon Cancer Cells. J Cancer. 2017;8(17):3555–3566. doi:10.7150/jca.20952
35. Caramel J, Ligier M, Puisieux A. Pleiotropic Roles for ZEB1 in Cancer. Cancer Res. 2018;78(1):30–35. doi:10.1158/0008-5472.CAN-17-2476
36. Zhang X, Zhang Z, Zhang Q, et al. ZEB1 confers chemotherapeutic resistance to breast cancer by activating ATM. Cell Death Dis. 2018;9(2):57. doi:10.1038/s41419-017-0087-3
37. Orellana-Serradell O, Herrera D, Castellon EA, et al. The transcription factor ZEB1 promotes an aggressive phenotype in prostate cancer cell lines. Asian J Androl. 2018;20(3):294–299. doi:10.4103/aja.aja_61_17
38. Han SP, Kim JH, Han ME, et al. SNAI1 is involved in the proliferation and migration of glioblastoma cells. Cell Mol Neurobiol. 2011;31(3):489–496. doi:10.1007/s10571-010-9643-4
39. Kahlert UD, Maciaczyk D, Doostkam S, et al. Activation of canonical WNT/beta-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012;325(1):42–53. doi:10.1016/j.canlet.2012.05.024
40. Siebzehnrubl FA, Silver DJ, Tugertimur B, et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med. 2013;5(8):1196–1212. doi:10.1002/emmm.201302827
41. Xiu Y, Sun K, Chen X, et al. Upregulation of the lncRNA Meg3 induces autophagy to inhibit tumorigenesis and progression of epithelial ovarian carcinoma by regulating activity of ATG3. Oncotarget. 2017;8(19):31714–31725. doi:10.18632/oncotarget.15955
42. Ying L, Huang Y, Chen H, et al. Downregulated MEG3 activates autophagy and increases cell proliferation in bladder cancer. Mol Biosyst. 2013;9(3):407–411. doi:10.1039/c2mb25386k
43. Katsuya S, Yukihiko I, Shigeki O, et al. Functional analysis of a novel glioma antigen, EFTUD1. Neuro-Oncology. 2014;16(12):1618–1629. doi:10.1093/neuonc/nou132
44. Kim EL, Wustenberg R, Rubsam A, et al. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro Oncol. 2010;12(4):389–400. doi:10.1093/neuonc/nop046
45. Lu Y, Xiao L, Liu Y, et al. MIR517C inhibits autophagy and the epithelial-to-mesenchymal (-like) transition phenotype in human glioblastoma through KPNA2-dependent disruption of TP53 nuclear translocation. Autophagy. 2015;11(12):2213–2232. doi:10.1080/15548627.2015.1108507
46. Sotelo J, Briceño E, López-González MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006;144(5):337–343. doi:10.7326/0003-4819-144-5-200603070-00008
47. Eldredge HB, Denittis A, Duhadaway JB, Chernick M, Metz R, Prendergast GC. Concurrent whole brain radiotherapy and short-course chloroquine in patients with brain metastases: a pilot trial. J Radiat Oncol. 2013;2(3):315–321. doi:10.1007/s13566-013-0111-x
48. McAfee Q, Zhang Z, Samanta A, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109(21):8253–8258. doi:10.1073/pnas.1118193109
49. Amaravadi RK, Winkler JD. Lys05: a new lysosomal autophagy inhibitor. Autophagy. 2012;8(9):1383–1384. doi:10.4161/auto.20958
50. DeVorkin L, Hattersley M, Kim P, et al. Autophagy inhibition enhances sunitinib efficacy in clear cell ovarian carcinoma. Mol Cancer Res. 2017;15(3):250–258. doi:10.1158/1541-7786.MCR-16-0132
51. Gade TP, Tucker E, Nakazawa MS, et al. Ischemia induces quiescence and autophagy dependence in hepatocellular carcinoma. Radiology. 2017;283(3):702–710. doi:10.1148/radiol.2017160728
52. Galavotti S, Bartesaghi S, Faccenda D, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013;32(6):699–712. doi:10.1038/onc.2012.111
53. Matthieu B, Valérie P, Ashish J, et al. SQSTM1/p62 regulates the expression of junctional proteins through epithelial-mesenchymal transition factors. Cell Cycle. 2015;14(3):364–374. doi:10.4161/15384101.2014.987619
54. Zhang C, Zhang X, Xu R, et al. TGF-beta2 initiates autophagy via Smad and non-Smad pathway to promote glioma cells’ invasion. J Exp Clin Cancer Res. 2017;36(1):162. doi:10.1186/s13046-017-0628-8
55. Catalano M, D’Alessandro G, Lepore F, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol. 2015;9(8):1612–1625. doi:10.1016/j.molonc.2015.04.016
56. Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol. 2012;30(7):671–678. doi:10.1038/nbt.2285
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.