")
Back to Journals » OncoTargets and Therapy » Volume 13
Medullary Thyroid Carcinoma in a Patient with MEN 1 Syndrome. Case Report and Literature Review
Authors Friziero A , Da Dalt G, Piotto A, Serafini S, Grego A , Galuppini F, Pennelli G, Sperti C
Received 24 April 2020
Accepted for publication 30 June 2020
Published 31 July 2020 Volume 2020:13 Pages 7599—7603
DOI https://doi.org/10.2147/OTT.S259656
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Alberto Friziero,1 Gianfranco Da Dalt,1 Andrea Piotto,1 Simone Serafini,1 Andrea Grego,1 Francesca Galuppini,2 Gianmaria Pennelli,3 Cosimo Sperti1
1Department of Surgery, Oncology and Gastroenterology-DISCOG, 3rd Surgical Clinic, University of Padua, Padua, Italy; 2Department of Woman’s and Children’s Health-SDB, University of Padua, Padua, Italy; 3Department of Medicine-DIMED, Pathology Unit, University of Padua, Padua, Italy
Correspondence: Cosimo Sperti
Department of Surgery, Oncology and Gastroenterology, 3 rd Surgical Clinic, University of Padua, via Giustiniani 2, Padova 35128, Italy
Tel +39 0498218845
Fax +39 0498218821
Email [email protected]
Abstract: Medullary thyroid cancer (MTC) is typically associated with multiple endocrine neoplasia type 2 syndrome (MEN 2), but not with multiple endocrine neoplasia type 1 (MEN 1). We report a very rare case of MTC in a patient with MEN 1 syndrome. A 60-year-old Caucasian woman with sporadic MEN 1 syndrome was admitted in October 2018 for recurrent hyperparathyroidism unresponsive to medical therapy. Her medical history included the diagnosis of a non-functioning pancreatic neuroendocrine tumor (NF-pNET) of the head of the pancreas 1.5 cm in size in 2001, and subtotal parathyroidectomy for uncontrolled hyperparathyroidism due to bilateral parathyroid hyperplasia in the same year. This history prompted genetic studies, and MEN 1 syndrome was confirmed. Family screening was performed in first-degree relatives, with negative results. Other typical clinical manifestations of MEN 1 syndrome were ruled out. In November 2018, the patient underwent excision of the residual left inferior parathyroid, extended to include the left thyroid lobe, for recurrent uncontrolled hyperparathyroidism. The pathologist identified MTC and adenoma of the parathyroid gland. Genetic tests were performed to identify any RET mutation, with negative results. The patient underwent total thyroidectomy about 6 months later, and the subsequent histological report showed only focal reactive C-cell hyperplasia of the thyroid. A literature review identified only three previously published cases of MTC coexisting with MEN 1 syndrome. This association may have two etiological hypotheses: either a sporadic MTC arising in a patient with MEN 1 syndrome, or a rare case of medullary cancer linked to a MEN 1 gene mutation.
Keywords: MEN 1 gene, multiple endocrine neoplasia type 1, multiple endocrine neoplasia type 2, RET gene, thyroid cancer, thyroidectomy
Introduction
Hereditary medullary thyroid cancer (MTC) is typically one of the neoplasia characterizing Multiple Endocrine Neoplasia type 2 syndromes (MEN 2), but it is not a clinical manifestations of MEN 1 syndrome. Here we report on the case of a patient affected by MEN 1 syndrome who developed an MTC. A review of previously described cases in the English Literature was performed to verify what is currently known about this unusual association. A comprehensive research was run in PubMed (Medline) and Scopus as at 31st December 2019 using the keywords: “medullary thyroid cancer”, “MEN 1”, and “MEN 2”. The “related articles” function was used to widen the search and all abstracts, studies, and citations retrieved were reviewed. To our knowledge, the present is the fourth case of this rare association reported so far.
Case Presentation
A 60-year-old Caucasian woman was admitted in October 2018 for recurrent hyperparathyroidism in MEN 1 syndrome. This condition had been diagnosed in 2001 after the discovery of a small non-functioning neuroendocrine tumor (NF-NET) of the head of the pancreas, 1.5 cm in diameter, and primary hyperparathyroidism due to bilateral parathyroid hyperplasia. A subtotal parathyroidectomy was performed in the same year, and the patient’s NF-NET was still being followed up. Genetic testing revealed a MEN 1 gene mutation. Family screening with the same DNA test confirmed a sporadic MEN 1 syndrome, as no other family members were affected. No other more or less typical manifestations of MEN 1 were identified. The patient’s remote medical history included carpal tunnel surgery and breast cancer treated with surgery and radiotherapy. She had no family history of thyroid disease, nor any risk factors for thyroid cancer apart from her age. Physical examination revealed no neck or lateral-cervical tenderness. The patient followed up routinely until 2018, when neck ultrasonography for relapsing hyperparathyroidism revealed an adenoma in the left inferior parathyroid, confirmed by technetium (99mTc) Sestamibi scintigraphy (Figure 1). Laboratory tests showed high serum parathyroid hormone (PTH) and calcium (Ca) levels: PTH: 153 ng/L (normal values 4.6–26.8); Ca: 2.67 mmol/L (normal values 2.1–2.55). The patient was therefore admitted to our Clinic and she underwent left inferior parathyroidectomy, extended to include the left thyroid lobe due to the presence of tenacious adhesions resulting from her previous cervical surgery. Intraoperative rapid assay showed that PTH levels dropped from 119 ng/L at the time of inducing anesthesia to 5 ng/L ten minutes after the removal of the mass. Macroscopically, the resected specimen comprised the left thyroid lobe (3.6 x 2.5 x 1 cm) with parathyroid tissue (maximal diameter 1.7 cm). Histological examination after fixing 4 μm sections from paraffin-embedded blocks in 10% buffered formalin and staining with hematoxylin and eosin identified showed: micro MTC (0.4 cm in diameter) associated with scant amyloid stroma in the thyroid gland and parathyroid adenoma consisting of clear cells and oxyphilic cells. On immunohistochemistry (Leica Bond system; Bond-III; Leica Microsystems, Milan, Italy), the medullary tumor showed positive reactions to Chromogranin A and carcinoembryonic antigen (CEA) and was slightly reactive to Calcitonin; Ki67 expression was low (<5%) (Figure 2A–D). RET gene study on DNA from the tumor tissue revealed no somatic mutations in exons 10, 11, 13, 14, 15 or 16. Subsequent computed tomography (CT) of the neck, chest and abdomen identified no abnormalities. About 6 months later, in April 2019, the patient underwent total thyroidectomy. The pathologist only reported focal reactive C-cell hyperplasia of the thyroid. The postoperative course was uneventful, and the patient was discharged after three days. With a follow-up of 12 months, the patient is asymptomatic and has normal serum Ca, PTH and calcitonin levels.
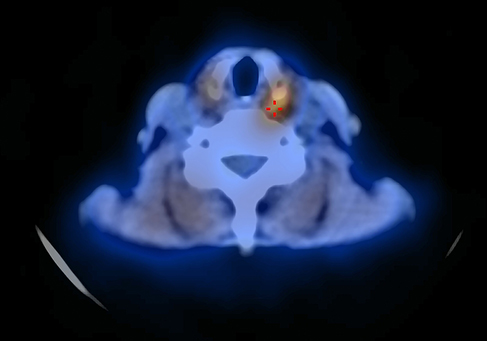 |
Figure 1 MIBI-Technetium subtraction scintigraphy, with evidence of left inferior parathyroid tracer uptake. |
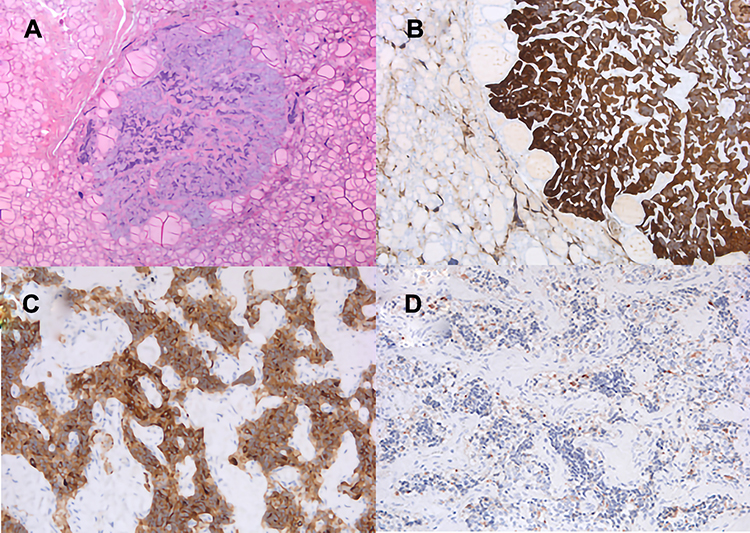 |
Figure 2 Histopathology of resected medullary thyroid cancer (MTC). Microscopically, it revealed polygonal cells with pale cytoplasm surrounded by fibrosis, collagen and amyloid tissue. H&E staining, (A). On immunohistochemistry, MTC was positive for Chromogranin (B), and CEA (C), and slightly positive for calcitonin (D). Magnification 40x (A); 100x (B); 200x (C and D). |
Discussion
Multiple endocrine neoplasia type 1 (MEN 1) is a hereditary syndrome characterized by hyperplastic and neoplastic disorders mainly affecting the parathyroid, anterior pituitary and gastro-enteric and pancreatic endocrine tissues. It is caused by a MEN 1 gene mutation in chromosome 11. A heterozygous germline mutation is identified in the MEN 1 gene of most individuals with the syndrome. Somatic mutations and large deletions of the MEN 1 locus and its surrounding region in chromosome 11, known as a loss of heterozygosity (LOH), are also found in MEN1-associated tumors.1 Medullary thyroid cancer (MTC) is a malignant tumor of the calcitonin-secreting para-follicular C-cells of the thyroid gland that is not among the variety of diseases associated with MEN 1 syndrome. Up to 25% of MTC are associated with pathogenic germinal variants on the RET proto-oncogene (locus 10q11.2) that cause familial MTC or multiple endocrine neoplasia type 2 (MEN 2).2 Consistently with the prominent role of RET, sporadic cases of MTC harbor a somatic RET mutation in a significant proportion of cases (43% to 65%), typically in exon 16 (codon 918).3 C-cell hyperplasia (CCH) should be considered a pre-neoplastic condition that leads to the development of MTC, and is commonly seen in patients with germline mutations in the RET oncogene. It is therefore important to obtain a precise pathological classification of CCH as either a pre-neoplastic or a merely reactive (physiological) process.4 MTC associated with MEN 1 is extremely rare: it has previously been reported only three times.5–7 A summary of the previously published cases is given in Table 1. There are essentially two possible explanations for this association: an inherited predisposition to MTC linked to the MEN 1 gene; or a sporadic MTC developing in a patient with MEN 1 syndrome.
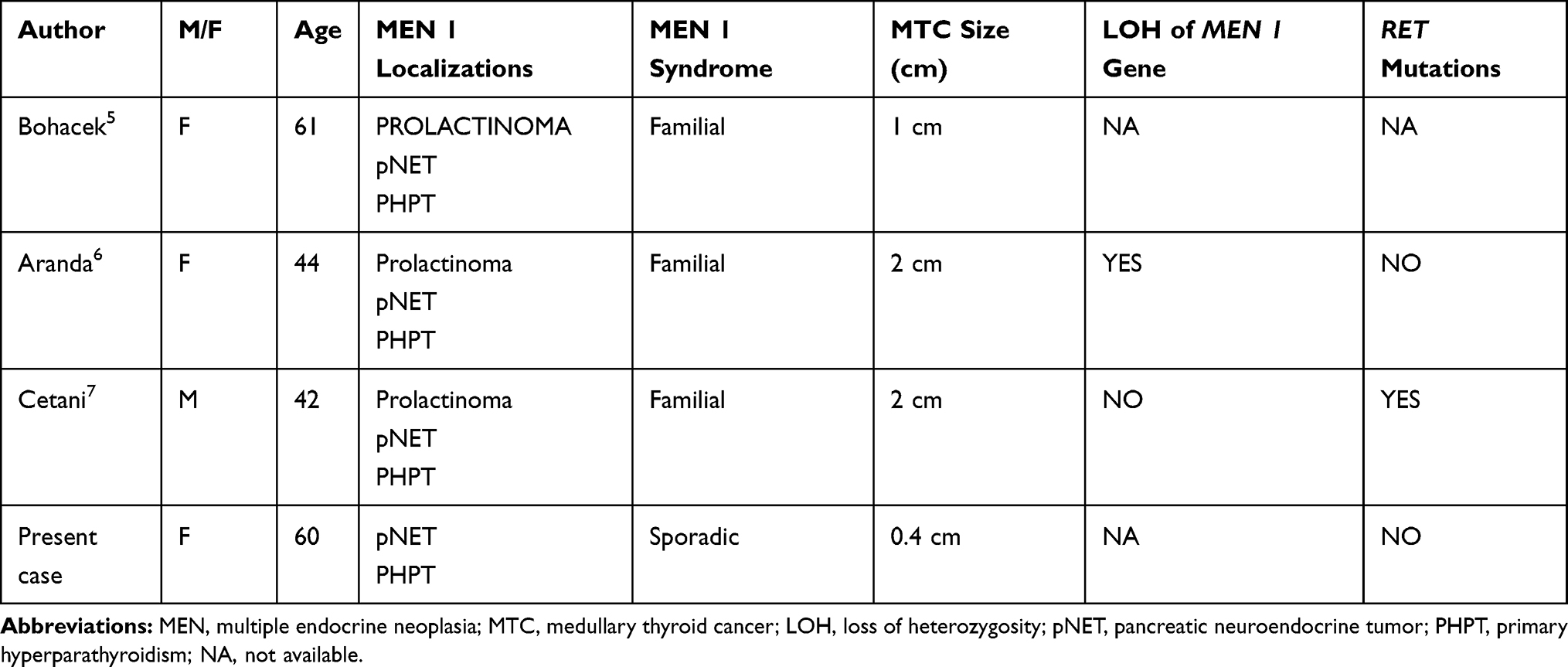 |
Table 1 Previously Published Cases, with Their Clinical and Genetic Features |
In 2014, Bohacek5 was the first to describe this combination of neoplasms in a 60-year-old Canadian woman. She was diagnosed with familial MEN 1 syndrome with a prolactinoma, a pancreatic neoplasm, and a parathyroid adenoma. She underwent total thyroidectomy for thyroid nodules many years after a first surgical procedure for uncontrolled hyperparathyroidism, and the final pathology report identified MTC. No genetic tests were carried out, so nobody knows whether this MTC was sporadic or related to MEN 1 syndrome. Aranda6 described a similar case two years later: a 44-year-old Spanish woman with familial MEN 1 syndrome and the same neuroendocrine neoplasms was incidentally found to have MTC during the histological analysis of her thyroid specimen. Genetic testing revealed LOH of the MEN 1 gene in MTC, with no evidence of any RET mutation, suggesting that the MTC occurred as part of the MEN 1 syndrome. More recently, Cetani7 described a patient with familial MEN 1 syndrome who had a prolactinoma, a pancreatic neuroendocrine tumor, hyperparathyroidism and a simultaneous metastatic MTC. Unlike the case reported by Aranda et al, genetic testing on the parathyroid and pancreatic neoplasm revealed LOH in the MEN 1 gene, but not in the MTC tissue. RET analysis on the MTC tissue revealed a point mutation in exon 16, meaning that this MTC cannot be a manifestation of MEN 1 syndrome, but an incidental, sporadic tumor.6
Our patient had sporadic MEN 1 syndrome and genetic testing on her MTC tissue identified no RET mutation. Unfortunately, no analysis of LOH of the MEN 1 gene locus was conducted, so a specific link between this patient’s MTC and her MEN 1 syndrome cannot be ruled out completely.
To date, only the patient described by Aranda et al was certainly a case of MTC as a manifestation of LOH of the MEN 1 gene.6 The other three cases, including ours, were probably incidental, sporadic cancers. That said, the thyroid gland could become a target to examine periodically in the follow-up of patients with MEN 1 syndrome. Serum calcitonin testing and neck ultrasonography could be performed at minimal expense to rule out any presence of MTC in these patients.
Abbreviations
MEN, multiple endocrine neoplasia; MTC, medullary thyroid cancer; LOH, loss of heterozygosity; pNET, pancreatic neuroendocrine tumor; NF-NET, non-functioning neuroendocrine tumor.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. No approbation was required by Ethical Committee of University of Padua to publish the case details.
Author Contributions
All Authors contributed equally to this work. In particular: in conception A.F., A.P.; in execution A.F., S.S., in acquisition of data: A.G. F.G., in interpretation: G.D., G.P, C.S. This article was drafted by A.G, G.D; written by A.F., S.S., F.G.; critically reviewed by A.P., G.P., C.S. All Authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest.
References
1. Al-Salameh A, Baudry C, Cohen R. Update on multiple endocrine neoplasia Type 1 and 2. La Presse Médicale. 2018;47(9):722–731.
2. Morrison PJ, Atkinson AB. Genetic aspects of familial thyroid cancer. Oncologist. 2009;14(6):571–577. doi:10.1634/theoncologist.2009-0046
3. Moura MM, Cavaco BM, Pinto AE, et al. Correlation of RET somatic mutations with clinicopathological features in sporadic medullary thyroid carcinomas. Br J Cancer. 2009;100:1777–1783. doi:10.1038/sj.bjc.6605056
4. Sakorafas GH, Nasikas D, Thanos D, Gantzoulas S. Incidental thyroid C cell hyperplasia: clinical significance and implications in practice. Oncol Res Treat. 2015;38:249–252. doi:10.1159/000381605
5. Bohacek L. Medullary thyroid carcinoma in a patient with MEN 1. J Surg Oncol. 2014;110(7):899–900. doi:10.1002/jso.23719
6. Aranda VG, Porta MM, Martínez D, Oriola J, Halperin Rabinovich I. Medullary thyroid carcinoma as manifestation of the loss of heterozygosity in a patient with MEN1. Endocrinol Nutr. 2016;63(7):371–373. doi:10.1016/j.endonu.2016.03.005
7. Cetani F, Pardi E, Berardi V, Romei C, Marcocci C, Elisei R. Incidental occurrence of metastatic medullary thyroid carcinoma in a patient with multiple endocrine neoplasia type 1 carrying germline MEN1 and somatic RET mutations. J Surg Oncol. 2017;116:1197–1199. doi:10.1002/jso.24793
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.