")
Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 5
MeCP2 in the regulation of neural activity: Rett syndrome pathophysiological perspectives
Authors Cuddapah V, Nwaobi S, Percy A, Olsen M
Received 28 May 2015
Accepted for publication 30 July 2015
Published 14 October 2015 Volume 2015:5 Pages 103—116
DOI https://doi.org/10.2147/DNND.S61269
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Thomas Müller
Vishnu Anand Cuddapah,1,* Sinifunanya Elvee Nwaobi,1,* Alan K Percy,2 Michelle Lynne Olsen1
1Department of Cell, Developmental, and Integrative Biology, 2Civitan International Research Center, University of Alabama at Birmingham, Birmingham, AL, USA
*These authors contributed equally to this work
Abstract: Rett syndrome (RTT), an X-linked neurodevelopment disorder, occurs in approximately one out of 10,000 females. Individuals afflicted by RTT display a constellation of signs and symptoms, affecting nearly every organ system. Most striking are the neurological manifestations, including regression of language and motor skills, increased seizure activity, autonomic dysfunction, and aberrant regulation of breathing patterns. The majority of girls with RTT have mutations in the gene encoding for methyl-CpG binding protein 2 (MeCP2). Since the discovery of this genetic cause of RTT in 1999, there has been an accelerated pace of research seeking to understand the role of MeCP2 in the brain in the hope of developing a disease-modifying therapy for RTT. In this study, we review the clinical features of RTT and then explore the latest mechanistic studies in order to explain how a mutation in MeCP2 leads to these unique features. We cover in detail studies examining the role of MeCP2 in neuronal physiology, as well as recent evidence that implicates a key role for glia in the pathogenesis of RTT. In the past 20 years, these basic and clinical studies have yielded an extraordinary understanding of RTT; as such, we end this narrative review considering the translation of these studies into clinical trials for the treatment of RTT.
Keywords: MeCP2, epigenetic regulation, Rett syndrome, neurons, glia, astrocyte, oligodendrocyte, microglia
Mutations in MeCP2 can cause Rett syndrome
MeCP2, a ~76 kb gene found on chromosome Xq28, encodes for methyl-CpG binding protein 2. MeCP2 is a transcriptional regulator that binds to methylated DNA to repress gene transcription. However, MeCP2 was recently demonstrated to bind lowly methylated promoter regions of highly transcribed genes, suggesting a role in gene activation.1 In the CNS, specifically, MeCP2 regulates expression of a variety of genes, including those involved in the neuronal differentiation and plasticity, cell adhesion, and the mitochondrial respiratory chain.2
Interest in MeCP2 greatly increased in 1999 when it was discovered that Rett Syndrome (RTT) was caused by mutations in this gene.3 Currently, MeCP2 mutations are detected in 96% of individuals diagnosed with typical RTT in the US Natural History Study (NHS), representing more than 200 unique sequence changes.4,5 In the NHS, eight point mutations representing four missense and four nonsense changes (~60%) and deletions and insertions (~20%) make up ~80% of all affected individuals. The remainder represents mutations that are found individually or in only a small number of individuals. Among the most common mutations, significant differences in clinical severity suggest phenotype–genotype correlations.4,5 Utilizing the Clinical Severity Score data for individuals enrolled in the NHS as a group of those with R133C, R294X, R306C, and 3′-truncations are associated with significantly less severity than the five other common point mutations and individuals with deletions of whole exons. However, on an individual basis, those with identical mutations may have widely differing clinical difficulties. Differences in X-chromosome inactivation (XCI) and overall genetic background may play an important role. One factor that has escaped recent attention is the clonal distribution of normal and mutant X chromosomes in the brain, where significant differences in expression patterns may lead to markedly different clinical phenotypes.6 In addition, environmental background may be critically important. Attempts at identifying variations in XCI in blood have indicated impressive skewing in more than one-third of the population, and highly skewed XCI has been noted in more than 10% of the population (unpublished data from the NHS). In a group of 150 girls, virtually equal skewing was noted in favor of the mutant or normal chromosome.
It is important to understand that a MeCP2 mutation is not synonymous with the clinical diagnosis of RTT. Individuals with MeCP2 mutations do not always demonstrate features of RTT, while up to 4% fulfill the obligate clinical criteria for RTT but do not have a MeCP2 mutation.
As an X-linked dominant disorder, RTT appears to arise spontaneously, a de-novo mutation, in rapidly dividing germinal sperm cells. In a small number, much less than 1% of the total, the mutation is transmitted by a female carrying a specific mutation, either being completely unaffected or having mild learning disability or cognitive impairment due to favorable XCI.7,8 In such families, affected females may display features of classic or typical RTT, whereas boys may have much more significant involvement resulting in infantile encephalopathy and a very much shortened life expectancy due to failure to maintain normal respiratory function.9 If the mutation is in the 3′ region, clinical involvement may include progressive dystonia.8 However, in families without an affected female, such features in a male may be difficult to associate with a MeCP2 mutation and thus go undetected.
In only a handful of instances, males with typical RTT have been reported under two different scenarios. The concurrence of Klinefelter syndrome (46 XXY) in an individual with a MeCP2 mutation or somatic mosaicism10 can result in the parallel existence of two populations, one with a normal and one with a mutant X chromosome. Additionally, shortly after the identification of MeCP2 mutations as causative in RTT, duplication of MeCP2 was identified in males producing a distinct but significantly difficult neurodevelopmental disorder.11–13 Approximately at the same time, overexpression of MeCP2 in a mouse model confirmed the existence of an abnormal phenotype.14 Subsequently, more than 100 males with MeCP2 duplications, differing in overall size, have been identified.15,16 Interestingly, the same duplication has been identified in many of the mothers of affected males. Yet, they seem to have been spared the major deleterious effects of the duplication due to favorable XCI. Close inspection of these mothers has revealed increased levels of depression and obsessive-compulsive behaviors.15,16 As with males expressing MeCP2 mutations, the recognition of MeCP2 duplication requires a high index of suspicion, resulting in likely underdiagnosis of this problem.
Finally, mutations in other genes, including CDKL5 or FOXG1, have been associated with atypical RTT. CDKL5 encodes for the cyclin-dependent kinase-like 5 protein, which promotes brain development. When mutated, CDKL5 can produce a phenotype that overlaps with RTT, but it also includes a propensity for the development of epilepsy early in life.17,18 FOXG1 may also play a role in neural development, and when mutated, it causes a congenital variant of RTT.19 The recognition of other genes associated with similar neurodevelopmental difficulties seems likely.
Clinical features of RTT
RTT was first described by Andreas Rett, a developmental pediatrician in Vienna, in 1966.20 However, the initial reports that appeared in a medical newsletter in Vienna were in German and were not widely distributed. Although Bengt Hagberg, a child neurologist in Göteborg, Sweden, noted similar clinical features at approximately the same time, it was not until 1983 that he and his colleagues in France and Portugal produced the first widely read publication.21 General awareness of this unique and puzzling disorder rapidly expanded, spurred enormously by two efforts: 1) a series of symposia organized by Rett in Vienna, and 2) the organization of patient advocacy efforts through the International Rett Syndrome Association, leading to active clinical research worldwide. The result was the elaboration of widely employed clinical diagnostic criteria first in 1984, and subsequently it was continuously updated as clinical and genetic understanding of RTT progressed.22–24 The current guidelines for diagnosis of RTT are outlined in Figure 1. The next step in progress involved a series of neuropathological studies underscoring the clinical finding of reduced brain growth and identifying specific cellular abnormalities including reduced neuronal size and simplified dendritic arborization. The absence of any suggestion of progressive neuropathological changes provided strong support for considering RTT as a neurodevelopmental disorder and conversely, opposing the notion of a neurodegenerative condition.25,26
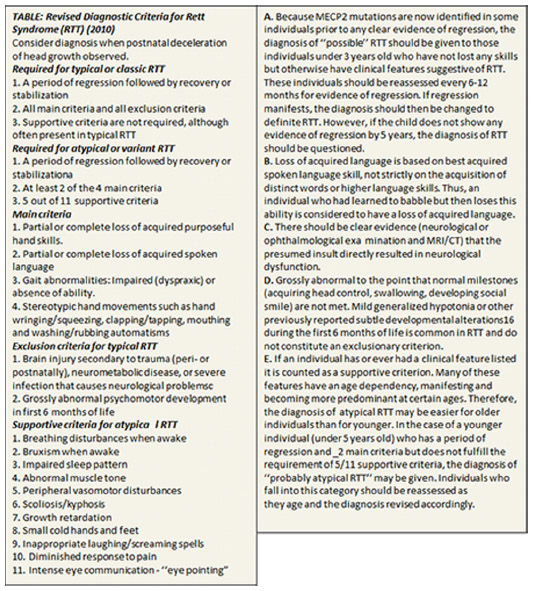 | Figure 1 Clinical criteria for the diagnosis of RTT. |
RTT affects ~1:10,000 female births27 with a broad array of quite remarkable systemic problems. RTT first appears in young females after a period of apparently normal postnatal development. Concerted study of the acquisition of early developmental skills has established that these skills are not acquired within a typical timeline.28,29 However, delayed or arrested development may be quite subtle such that it is often during the frank regression of these skills in the second year of life when concerns first emerge. In some girls, autistic-like social avoidance is noted and specific diagnosis is further delayed. The abnormal deceleration of head circumference seen as early as a few months of life,30,31 generalized reduction in muscle tone, and an overall appearance of being too quiet or good may provide important early clues to proper diagnosis. During or simultaneously with regression, periods of inappropriate screaming spells, and the appearance of stereotypic movements during wakefulness, most prominent in the hands but also seen in the oro-motor regions and the feet, also should trigger consideration of RTT. Following regression, a period of stabilization appears as early as 30 months of age marked by improved eye contact and socialization that appears to continue throughout life (NHS). This is contrasted with a gradual decline in motor performance, which accompanies an increase in muscle tone, first to normal tone and ultimately to increased tone or rigidity. This period often begins in the teenage years and can be associated with dystonic postures, particularly in the ankles and wrists (NHS). Ambulation is achieved initially in up to 80% of the population, but it is typically broad-based and nonpurposeful, often accompanied by retropulsion (first steps are backwards), and may include prominent toe-walking. Approximately, one-third of this group will stop walking such that overall ~50% maintain independent gait and another 20% are able to ambulate with some level of assistance. Numerous clinical issues arise including epilepsy, which may begin by the third year of life,32 occasionally being seen earlier; periodic breathing consisting of breath-holding or hyperventilation or both;33–35 and multiple, often-difficult to discern, gastrointestinal issues from top to bottom representing abnormal chewing and swallowing and abnormal and problematic motility concerns including gastro-esophageal reflux, delayed stomach emptying, and constipation.36 The very common and often significant decline in growth parameters involves height, weight, hands, and feet30,31 and may be crucial for maintaining the body mass index (BMI) in the normal range. Scoliosis is present in most girls by age 16 years, 13% requiring surgical rod placement to correct the severe spinal deformity and concomitant truncal rotation.37 Prior to major surgery of this type, close attention is paid to a normal BMI. Finally, heat intolerance during the summer months and cool hands and feet despite normal ambient temperature are nearly universal.
MeCP2 regulates neuronal function
In the hopes of modifying these signs and symptoms of RTT, it is first necessary to understand how mutation in MeCP2 mechanistically causes this disease. The severity of neurological deficits has drawn most research attention to the role of MeCP2 in the nervous system. As detailed later, MeCP2 plays a critical role in the development of mature neurons and the maintenance of balance between excitation and inhibition across multiple brain regions. Through the understanding of this pathophysiology, several novel molecular targets have been identified, hopefully paving the way for more effective therapeutic management.
The role of MeCP2 in neuronal differentiation and synaptic maturation
The age of onset of symptoms in girls with RTT and commonly used animal models suggest that MeCP2 is not required for neuronal cell division, neuronal migration, or early neuronal maturation. However, several structural abnormalities have been noted in neurons from girls with RTT. Cortex labels approximately one-third less than control with synaptophysin, indicating impairment in postnatal synaptic maturation.38 Additionally, dendrites extending from pyramidal neurons in multiple cortical layers are significantly shorter than control.39 MeCP2 also plays an integral role in the formation of mature synapses. Fewer mature glutamatergic synapses are found in the hippocampus of MeCP2-deficient animals in two-week-old animals, but by adulthood, the synapse number normalizes, presumably due to some undefined compensatory mechanism.40
In the cortex of adult mice lacking MeCP2, pyramidal neurons are smaller, less complex with decreased spine density, PSD95, and spontaneous excitatory post synaptic current (EPSC) amplitude as compared with wild-type animals.41–44 The decrease in EPSC amplitude can be partially explained by a decrease in mature N-methyl-D-aspartate (NMDA) receptors containing GluN1 and GluN2A42 and/or decreased quantal size and excitatory synaptic connectivity.43 This is associated with reduced brain weight and lifespan, as well as respiratory and cardiac dysfunction.
The absolute requirement for MeCP2 to maintain mature neuronal networks and normal neuronal and non-neuronal cell morphology, as well as the prevention of RTT-like symptoms, was demonstrated by ablating MeCP2 during different postnatal developmental stages.45,46 Tamoxifen-induced excision of MeCP2 in healthy mice at both 5 weeks (juvenile) and 10 weeks (adult) resulted in overall brain shrinkage, increased neuronal density, a decrease in the complexity and number of dendritic arbors and spine density, and a significant decrease in astrocyte process complexity.47 In male mice, there was 100% lethality ~20 weeks post-tamoxifen injection at both time points tested.47 A separate study using a similar paradigm demonstrated abnormal gate, reduced activity, motor abnormalities, and poor nest building following tamoxifen treatment.46 Again, median time to premature death was ~3 months post-tamoxifen treatment.46 These data reiterate the requirement for MeCP2 in maintenance of CNS functioning.
It is important to note that deficits in synaptic maturation can be reversed with a peptide fragment of insulin-like growth factor 1 (IGF-1).41 Intraperitoneal injection of a fragment of IGF-1 increased lifespan by ~50% and improved PSD95 density, brain weight, spine density, and EPSC amplitude.41 Recombinant human IGF-1 also increased lifespan, motor abilities, synaptic transmission, and spine density in a mouse model of RTT.48 However, a study utilizing PEG-IGF-1 demonstrated a narrow therapeutic window for its use – low dose (0.1 mg/kg) increased lifespan, while high dose (1.0 mg/kg) resulted in a reduction in lifespan compared with the vehicle. Contrary to previous studies utilizing IGF-1-related compounds, PEG-IGF-1 offered no improvement of breathing or motor tasks compared with vehicle, as well as aggravated an underlying metabolic syndrome in MeCP2 mutant mice.49 It may be that the addition of PEG precipitated the negative result. Therefore, despite concerns, several clinical trials using IGF in girls with RTT were initiated.
The role of MeCP2 in neuronal plasticity
Disruption of MeCP2 functions seems to have deleterious effects on both short- and long-term plasticity. In CA1 of the hippocampus, basal synaptic transmission was enhanced with impairment in long-term potentiation (LTP) in early symptomatic mice with truncated MeCP2 (MeCP2308mice).50 This decrement in LTP has been verified in several other models of RTT in male and female mice50–52 and is associated with deficits in social behavior, fear, and spatial memory. However in symptomatic MeCP2-deficient mice, while there was a deficit in short-term potentiation and LTP, basal synaptic transmission was unaltered.52 These data demonstrating deficits in hippocampal synaptic plasticity are in line with in vivo electroencephalographic (EEG) recordings, which show slower theta wave activity in the hippocampus of mutant male and female mice.53 In contrast to the hippocampus, long-term plasticity appears to be intact in the cortex. LTP is normal in L5 pyramidal neurons from MeCP2-deficient animals.43
Brain-derived neurotropic factor (BDNF) is an essential growth factor released largely by neurons to promote neuronal and glial survival, growth, and differentiation. BDNF signaling plays an important role in synaptogenesis, synaptic stability, and dendrite development, while low levels of BDNF signaling are associated with several neuropathological processes. BDNF is a direct molecular target of MeCP2,54 and reduced BDNF protein levels (~50%–70% of wild type, depending on brain region) were found in adult MeCP2 KO animals.55 Importantly, reduced levels of BDNF protein was also found in human postmortem RTT tissue relative to age-matched controls.56 Early researches utilizing in vitro techniques demonstrate that upon neuronal depolarization, BDNF expression is normally increased through at least two MeCP2-dependent mechanisms: 1) decreased CpG methylation of Bdnf, decreasing its interaction with the MeCP2-histone deacetylase-mSin3A repression complex and increasing Bdnf transcription,57 and 2) Ca2+-dependent phosphorylation of MeCP2, decreasing its repression of Bdnf expression.54 These studies indicate that Bdnf expression is controlled by MeCP2; therefore, some of the deficits observed in RTT may be due to loss of BDNF. Neurons from MeCP2-mutant hippocampus release decreased levels of BDNF after neuronal stimulation.58 As predicted, overexpression of BDNF in MeCP2-deficient mice can reverse some electrophysiological abnormalities in cortical neurons, correlating with increased lifespan and improved gait.55
MeCP2 loss causes an imbalance in excitation and inhibition
Clinically, girls with RTT experience seizures, suggestive of hyperexcitable cerebral physiology. In the laboratory, however, the data are less straightforward. Mouse models of RTT suggest features of 1) increased excitation, 2) decreased excitation, and 3) decreased inhibition, all depending on the age of the animal used and the region of the brain being studied. Studies in murine cortex suggest features of decreased spontaneous firing at the cellular level and also increased excitability at the network level as measured by EEG. Both mutant hippocampus and brainstem also demonstrate features of greater excitation than inhibition. Thus, murine models of RTT demonstrate that the imbalance in excitation and inhibition is not homogeneous across brain regions and time points and requires continued specialized study to understand this complex disease.
Changes in excitation and inhibition in the cortex
The intrinsic excitability of neurons in the cortex does not seem to be altered in early symptomatic MeCP2-deficient animals. When GABAergic and glutamatergic synaptic activity is blocked, L5 wild-type and mutant neurons fire a similar number of action potentials in response to current injection and have a similar threshold to spike and input resistance.59 However, when spontaneous firing is recorded, MeCP2-mutant L5 neurons fired less frequently.55,59 This was hypothesized to be secondary to impaired glutamatergic input and increased GABAergic input on L5 mutant neurons.60 Similarly, in L2/3, MeCP2 deficiency leads to a decrease in excitatory drive.61 In the visual cortex, voltage-sensitive dyes reveal increased inhibitory signaling in L2/3, causing deficits in visual acuity in MeCP2-deficient mice.62 In contrast, EEG recordings of the cortex are suggestive of greater excitation than inhibition in MeCP2-deficient mice. Traces from the cortex of older female mice revealed rhythmic discharges/seizure-like events, occurring spontaneously at 6–9 Hz.53,63 This abnormal electrographic activity does not occur in wild-type mice.53
There is also evidence for deficiencies in GABAergic signaling in the cortex. Using a promoter for vesicular inhibitory amino acid transporter to reduce MeCP2 expression specifically in GABAergic neurons recapitulates several deficits associated with RTT, including repetitive behaviors, gait disturbance, breathing abnormalities, and shortened lifespan.64 This impairment is associated with a reduction in total GABA, GABA quantal size in L2/3, and expression of Gad1 and Gad2 that encode for glutamic acid decarboxylase 67 and 65, respectively, manifesting as frequent epileptiform discharges on EEG.64 However, recordings from cortical L5 pyramidal neurons in mice with global MeCP2-deficiency demonstrate no change in GABA quantal size.60
Hippocampus from MeCP2-deficient mice is prone to hyperexcitability
Hippocampus from MeCP2-null mice is susceptible to hyperexcitability. While the intrinsic excitability of individual cells is unaltered, at the network level, upon 1) high-frequency stimulation, 2) exposure to GABAA receptor antagonist, 3) incubation in kainic acid, or 4) blocking of K+ channels, the hippocampus demonstrates a decreased threshold to excitation.65,66 This also correlated with decreased frequency of IPSP-based basal inhibitory rhythm.65 Unexpectedly, EPSC-like events were decreased in CA3 pyramidal neurons in MeCP2-null mice,65 possibly indicating that while there is decreased excitatory and inhibitory drive, the net effect is increased excitation in the hippocampus. This hypothesis is supported by voltage-sensitive dye imaging, which reveals that the CA3 and CA1 regions of symptomatic mice are hyperexcitable.67
Increased excitation is characteristic of brainstem from MeCP2-deficient mice
Excessive excitation has been repeatedly reported in the brainstem of MeCP2-deficient mice. The nucleus tractus solitarius contains neurons with elevated excitatory input, leading to greater firing frequencies upon stimulation.68 This excessive excitation was reversed with exogenous in situ application of BDNF.68 Thus, BDNF plays an important role not only in the survival and differentiation of neurons but also in the balance between excitation and inhibition in the brainstem. Also in brainstem, the locus ceruleus is hyperexcitable in MeCP2-null mice.69 The locus ceruleus is an important source for norepinephrine and facilitates attention and arousal. While these neurons are more excitable, there is decreased formation of norepinephrine, potentially contributing to some of the deficits seen in RTT.69 As detailed later, treatment of animals with a blocker of norepinephrine reuptake can reverse symptoms, including breathing abnormalities.70,71
Deficiencies in inhibitory signaling are also evident in the brainstem of MeCP2-deficient animals. When MeCP2 is absent, animals as young as 7 days have decreased GABAergic signaling in the ventrolateral medulla, presumably secondary to decreased GABA release and postsynaptic GABAA receptors.72 When MeCP2-null animals were given NO-711, a blocker of GABA reuptake leading to increase concentrations of GABA in the extracellular space, breathing abnormalities were mitigated.73 Using NO-711 in conjunction with a serotonin 1a agonist completely reversed several breathing abnormalities. These results indicate that MeCP2-null animals have a deficiency in inhibition in the respiratory centers of the brainstem, manifesting as breathing abnormalities.
Changes in ionic gradients?
MeCP2 deficiency may also alter the balance between excitation and inhibition by altering ionic gradients. FXYD domain-containing transport regulator 1 (FXYD1) encodes phospholemman, a protein that inhibits Na+/K+ ATPase activity, and is significantly upregulated in the brains of girls with RTT and in the frontal cortex of mice deficient in MeCP2.57 This upregulation of phospholemman, in turn, leads to decreased ouabain-sensitive Na+/K+ ATPase activity in MeCP2-deficient neurons.57 Although not specifically measured, an impairment in Na+/K+ ATPase activity may increase the extracellular concentration of K+ ([K+]o) and decrease the extracellular concentration of Na+ ([Na+]o), leading to neuronal depolarization and hyperexcitability. Of course, ~60%–85% of girls with RTT will experience at least one seizure during her lifetime.32,74,75
Restoration of global and neuronal MeCP2 function in murine models reverses deficits associated with RTT
Perhaps one of the most exciting findings in the field of RTT is the demonstration that postnatal restoration of MeCP2 function to ameliorate deficits associated with RTT. This was elegantly demonstrated by Adrian Bird’s laboratory in 2007.51 Here a floxed stop cassette was inserted between the promoter and coding sequence of MeCP2 to prevent gene transcription, essentially creating a MeCP2 null animal. Following tamoxifen treatment, the stop cassette was excised. In this way, MeCP2 expression could be turned on at any time during the development (Figure 2). These experiments demonstrated a robust activation of MeCP2 expression following tamoxifen treatment. This prevented the appearance of Rett-like symptoms in ~50 of pre-symptomatic animals. (The abrupt re-expression of MeCP2 in some animals led to neurological symptoms and death. Tamoxifen dosing schedule was adjusted to circumvent this problem). Excitingly, disease progression was prevented and symptoms were reversed by activating MeCP2 expression in mature animals. It was concluded from this study that the developmental absence of MeCP2 and its consequences can be reversed, that RTT is not just a developmental disorder, and that the MeCP2 expression is required to maintain mature neurons. This study was quickly followed by others who asked the question, Is MeCP2 re-expression in neuronal populations sufficient to rescue disease phenotype? Re-expression of MeCP2 in post-mitotic neurons under control of the Tau promoter normalized brain and body weight, as well as locomotor activity.76 It should be noted here that Tau expression at the mRNA level has been detected in other cell types including glia.77,78 Postnatal restoration of MeCP2 in neurons using the CaMKII promoter partially rescued the Rett phenotype. Although levels and patterns of MeCP2 expression did not recapitulate wild-type animals’ neuronal levels, these animals displayed enhanced survival times and a delay in the onset of RTT-like symptoms.79–81 These results are significant from a therapeutic perspective, because it suggests the possibility that restoration of MeCP2 function in girls could modify disease progression. Indeed, a recent study demonstrated that rescue of MeCP2 expression in female MeCP2-deficient mice using systemically administered viral gene therapy was able to reverse motor deficits and improve survival.82
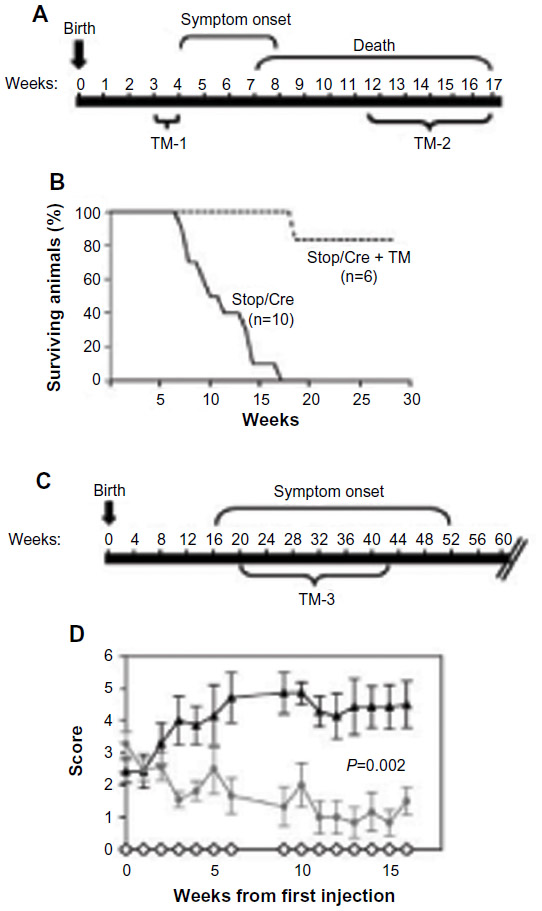 | Figure 2 Global restoration of MeCP2 reverses deficits associated with RTT. |
Glial MeCP2 plays an integral role in the pathophysiology of RTT
As mentioned earlier, the majority of research examining the role of MeCP2 in RTT has focused on neuronal, cell-autonomous mechanisms of dysfunction. And while a RTT phenotype can be induced solely by depleting MeCP2 from neurons,83,84 reversal of several key features of RTT cannot be achieved with postnatal restoration of MeCP2 in neurons alone.80 Early observations indicated that MeCP2 expression was restricted to neurons in the CNS;85 however, more recent studies demonstrate a broader expression profile of MeCP2 in the CNS, including astrocytes, microglia, and oligodendrocytes.61,86–88 Perhaps most surprising, postnatal re-expression of MeCP2 specifically in glia using a human GFAP promoter under a tamoxifen-inducible CreLoxP system resulted in longer survival rates for MeCP2-deficient mice and rescue of several key RTT phenotypes including breathing, locomotion, and neuronal structure89 (Figure 3). Such studies reveal once unappreciated non-cell autonomous mechanisms of dysfunction that alter neuronal function and survival and open the possibility for new therapeutic targets.
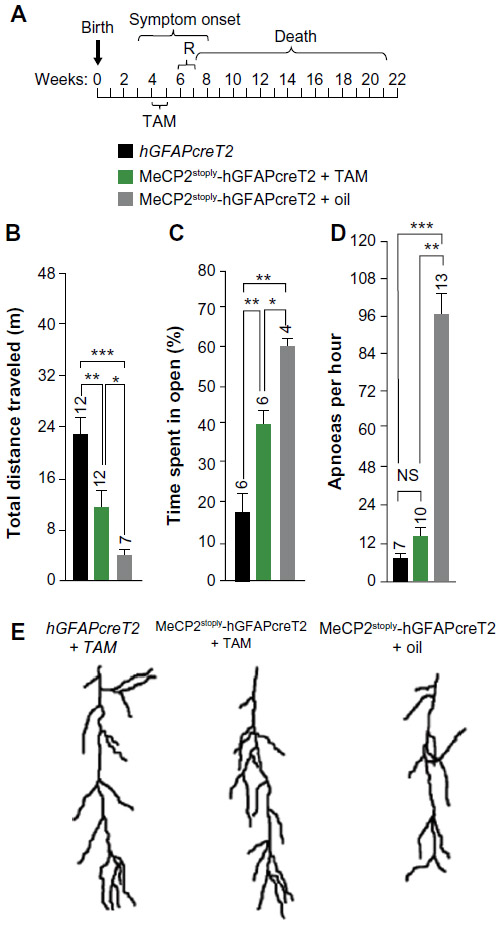 | Figure 3 Restoration of MeCP2 in astrocytes reverses deficits associated with RTT. |
Astrocytes play an important role in RTT pathophysiology
MeCP2 is highly expressed in neurons relative to other CNS cell types. The high neuronal MeCP2 expression essentially masked the expression in other cell types. The creation of a MeCP2 promoter-driven enhanced green fluorescent protein reporter mouse in 2007 enabled visualization of MeCP2 developmental, brain region and cell-type-specific MeCP2 expression.90 In this study, amplification of the enhanced green fluorescent protein signal revealed MeCP2 was indeed expressed in low levels in astrocytes. Following this study, Western blot analysis revealed MeCP2 is expressed in cultured astrocytes (881). Ballas et al confirmed MeCP2 immunoreactivity in astrocytes in culture and in situ, as well as oligodendrocytes, and MeCP2 expression of their precursor cells.61 This and other studies utilizing neuronal and astrocytic coculture systems demonstrate the inability of MeCP2-deficient astrocytes to support normal neuronal growth compared with wild-type astrocytes.61,86,91 Soluble factors released by astrocytes or the lack of uptake of ions/molecules are probable candidates in mediating abnormal neuronal growth, as neurons exposed to conditioned media from MeCP2-deficient astrocytes demonstrated a decrease in fine processes, shorter neurite outgrowth, and altered staining of MAP2.61 Conversely, culturing MeCP2-deficient neurons in conditioned media from wild-type astrocytes were sufficient to support normal neuronal growth.61 Comparable results were observed using conditioned media from astrocytes differentiated from various isogenic induced pluripotent stem cells of a patient with RTT.91 Maezawa et al also demonstrated a decrease in dendritic growth and number of branch points in primary neurons when cultured with MeCP2-deficient astrocytes.86 Interestingly, blockade of gap junctions prevented this time-dependent loss of MeCP2 in culture, suggesting gap junctions are able to mediate and spread deleterious effects from single MeCP2-deficient astrocytes to an entire culture.86 Confirming the importance of astrocytes in RTT pathogenesis, postnatal re-expression of MeCP2 in astrocytes results in the improvement of locomotion, a decrease in apneas and irregular breathing patterns, as well as the restoration of normal dendritic morphology in MeCP2 mutant animals89 (Figure 3). To re-express MeCP2 in astrocytes, the authors use the GFAP promoter, which although predominantly expressed in astrocytes is also expressed by neural precursor cells (NPCs). However, the authors minimize the possible effects of re-expression of MeCP2 in NPCs by turning on the GFAP promoter well into postnatal development when the number of NPCs is dramatically reduced. This indicates that the amelioration of deficits associated with RTT is predominantly due to re-expression of MeCP2 in astrocytes. In contrast, when MeCP2 was deleted from astrocytes, a significantly milder phenotype was observed. These mice had reduced body weight, hindlimb clasping, and irregular breathing. Several studies have demonstrated alterations in astrocytic gene expression,89–94 perhaps contributing to astrocyte dysfunction. Given many of these studies were performed using a simplified culture system composed solely of astrocytes, these data suggest that alterations in gene expression are autonomous to the astrocyte.
However, these findings are interesting when put into the larger context of both rescue and deletion experiments for other cell types. For instance, deletion of MeCP2 from GABAergic neurons recapitulated most RTT-like features, including breathing abnormalities, repetitive behaviors, hindlimb clasping, motor dysfunction, and premature death (50% at 26 weeks).64 It must be noted that disease course was significantly protracted compared with a global MeCP2 knock out. Taken at face value, these data suggest that MeCP2 expression is important to both neurons and glia. Furthermore, loss of MeCP2 from neuronal populations and subsequent neuronal dysfunction will inevitably impact other CNS cell types and vice versa. Future studies aimed at examining cell-specific gene expression patterns in disease models of RTT may help to parse out the role of different cell types to disease pathogenesis.
Mutant microglia release aberrant levels of glutamate
Several lines of evidence demonstrate a key role for microglia, the resident immune cells of the CNS, in RTT pathophysiology. Similar to astrocytes, conditioned media from MeCP2-deficient microglia resulted in abnormal neuronal structure and dendritic outgrowth.87 Specifically, a reduction of postsynaptic elements, including PSD95 and glutamate receptor units, was observed in hippocampal neurons cultured in conditioned media from MeCP2-deficient microglia compared with wild-type microglia. Excessive glutamate release from MeCP2-deficient microglia was identified as the mediator of this aberrant dendritic morphology as blockade of neuronal glutamate receptors via MK-801 and NBQX prevented toxic effects of MeCP2-deficient microglia conditioned media.87 Interestingly, blockade of Cx32 hemichannels was able to mitigate the toxic effects of conditioned media from MeCP2-deficient microglia on hippocampal neurons, once again suggesting that gap junctions are capable of spreading deleterious effects from one MeCP2-deficient cell to an entire culture.87 Whether alterations in glutamate uptake are altered in MeCP2-deficient astrocytes are yet to be examined.
Dysregulation of mitochondrial metabolism secondary to altered gene expression in MeCP2-deficient mice likely plays a central role in the overproduction of glutamate in MeCP2-deficient microglia. Jin et al demonstrated that MeCP2 functions as a transcriptional repressor of SNAT1, a glutamine transporter, which is elevated in MeCP2-deficient mice.92 Upregulation of the transporter was sufficient to increase glutamine uptake and glutamate production and induce toxicity in microglia secondary to the elevated glutamine levels. Microglia from MeCP2-deficient mice also demonstrated significantly elevated numbers of mitochondria and uncoupling of mitochondrial metabolism, possibly as a direct consequence of increased SNAT1 expression and glutamine levels.92
While these studies point to aberrations in mitochondrial bioenergetics as a mechanism for microglial toxicity in MeCP2-deficient mice, there has been a suggestion that altered microglial phagocytic activity may also play a role. Using transplantation of wild-type bone marrow into irradiated MeCP2y/- at postnatal day 28, Derecki et al was able to demonstrate arrest of the MeCP2 phenotype.93 However, a recent follow-up study published jointly by four prominent laboratories found that neither wild-type bone marrow transplantation nor transgenic re-expression of MeCP2 in microglia was sufficient to reverse these RTT-associated deficits.94 These preclinical data demonstrate that bone marrow transplantation may not be a promising therapeutic strategy for the treatment of RTT.
Oligodendrocytes also express MeCP2
As described earlier, MeCP2 is expressed in both oligodendrocytes and their precursor cells (OPCs).61,88 Studies that focus on the role of MeCP2 in oligodendrocyte lineage cells demonstrate that depletion of MeCP2 specifically in these cells results in a mild phenotype, with a predominance of hindlimb motor symptoms.88 Targeted restoration of MeCP2 in oligodendrocytes lineage cells was sufficient to provide subtle but significant increases in the lifespan of animals and improvement of motor symptoms. While MeCP2 null animals displayed alterations in myelin-associated proteins including proteolipid protein and myelin basic protein, re-expression of MeCP2 in oligodendrocytes resulted only in an increase in myelin basic protein expression, while proteolipid protein remained aberrantly high compared with wild-type animals.88 Further studies aimed at detailing the role of oligodendrocyte lineage cells, which are needed to fully understand if this cell type possesses a critical role in the pathophysiology of RTT.
Ongoing clinical trials
The therapies most likely to make a substantial impact on the clinical management of RTT in the near future are currently undergoing clinical trials. We accessed all ongoing and recently completed trials posted on ClinicalTrials.gov using the search term “Rett Syndrome” and found 28 clinical studies. Half of the studies are observational in nature, attempting to increase our knowledge of clinical, behavioral, and metabolic characteristics of disease progression. The majority of trials in which an intervention was occurring were in Phase I/II (12/28=43%), and only two trials have reached Phase III (Figure 4). Later, we highlight completed and ongoing clinical trials that will hopefully lead to improvements in our clinical management of RTT in the near future.
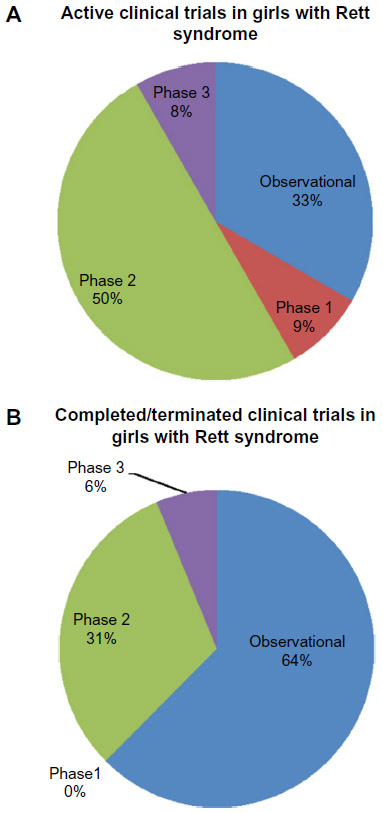 | Figure 4 All clinical trials for the treatment of RTT, as listed on ClinicalTrials.gov in May 2015. Avaliable from https://clinicaltrials.gov/. |
IGF-1
As previously alluded to, the cortex of MeCP2-deficient animals is characterized by a deficiency of mature glutamatergic synapses, which is also associated with decreased brain mass and longevity. Excitingly, treatment of mutant mice with a portion of IGF-1 can improve synaptic connections, brain mass, and longevity.41 As is the case for most drugs developed in animal models, demonstrating efficacy in humans is a major challenge. In a preliminary pilot study, six girls with RTT were given subcutaneous IGF-1 injections, twice daily, for 6 months.95 No major adverse effects were noted, paving the road for future studies to determine efficacy.
There are currently at least three trials in the USA testing the efficacy of IGF-1 for the treatment of RTT. Glycyl-L-2-Methylpropyl-L-Glutamic Acid (NNZ-2566) is a synthetic analog of the N-terminal of IGF-1 being developed by Neuren Pharmaceuticals (Camberwell, Australia). NNZ-2566 has completed a randomized, double-blind Phase II trial with primary outcomes being change in EEG activity, change in behavior, physiological changes (including autonomic, breathing, and heart function), and change in clinical severity score (ClinicalTrials.gov Identifier: NCT01703533). Recombinant Human IGF-1 is also being currently tested in a randomized, double-blind Phase II trial, and outcome measures include measurement of breathing and evaluation of cortical function using visual evoked potentials (ClinicalTrials.gov Identifier: NCT01777542).
Glatiramer acetate and fingolimod increase cortical BDNF expression
Glatiramer acetate (trade name: Copaxone) is typically used as an immunomodulatory drug approved by the Food and Drug Administration for the treatment of multiple sclerosis. Its putative mechanism of action is to act as a decoy polymer, as its amino acid sequence is similar to that of myelin basic protein, thereby diverting the immune response away from myelin. Alternatively, it may also induce the infiltration of T-cell helper type 2 cells into the brain and thereby increase the expression of anti-inflammatory cytokines and growth factors, including BDNF.96,97 Indeed, in a mouse model of multiple sclerosis, subcutaneous injections of glatiramer acetate led to a sustained elevation of BDNF levels, similar to that of control mice.97 As detailed earlier, BDNF plays a critical role in neuronal survival and synaptic maturation. Given the disturbances in BDNF expression also seen in RTT, a Phase II trial has been initiated (ClinicalTrials.gov Identifier: NCT02153723) to test the efficacy of glatiramer acetate on gait, respiratory function, visual attention, EEG activity, and quality of life in girls with RTT. Preliminary data from preclinical models support the use of glatiramer acetate in MeCP2-deficient animals.98
Similar to glatiramer acetate, fingolimod (trade name: Gilenya) is an immunomodulatory drug used for the treatment of multiple sclerosis that also increases cortical BDNF levels. Fingolimod is a sphingosine-1 phosphate receptor modulator that increases the retention of lymphocytes to lymph nodes, decreasing autoimmune reactions. Intraperitoneal injection of fingolimod into MeCP2-mutant mice increased BDNF expression in cortical neurons, corresponding with an improvement in locomotion and survival.99 These results led to the initiation of a Phase I/II trial for the treatment of girls with RTT with oral fingolimod (ClinicalTrials.gov Identifier: NCT02061137). Primary outcome measures include BDNF levels as measured by blood and cerebrospinal fluid samples before and during treatment.
Use of a tricyclic antidepressant to improve respiratory function
Breathing abnormalities are a source of considerable distress for both girls who have RTT and their families. Excitingly, pre-clinical models demonstrate that treatment of MeCP2-deficient mice with desipramine (trade name: Norpramin), a tricyclic antidepressant that inhibits the reuptake of norepinephrine (and serotonin to a lesser degree), can alleviate many breathing anomalies associated with RTT. Oral treatment of MeCP2-deficient mice with desipramine improved respiratory function and lengthened lifespan.70 Similarly, intraperitoneal injection of desipramine improved breathing and lifespan, which has been hypothesized to occur because of an increase in tyrosine hydroxylase-expressing neurons in the brainstem.71 Indeed, respiratory dysfunction appears to be secondary to a disturbed central respiratory rhythm in MeCP2-deficient mice.100 This was corrected by exogenous application of norepinephrine to in situ medullary slices.100 Based on these studies, a Phase II clinical trial has been initiated to determine if girls with RTT can benefit from desipramine (ClinicalTrials.gov Identifier: NCT00990691).
Dextromethorphan to decrease glutamatergic signaling for treatment of neurocognitive deficits
Dextromethorphan, most commonly used as an antitussive medication, is also a low-affinity NMDA-receptor antagonist.101 Therefore, dextromethorphan has been evaluated in preclinical studies to protect against glutamate-induced neurotoxicity, including in models of ischemia, seizure, and trauma.102 Given the preponderance of seizure and abnormal electrographic activity in girls with RTT, a placebo-controlled Phase II clinical trial is underway to determine if dextromethorphan can reverse some of the neurocognitive deficits associated with RTT (ClinicalTrials.gov Identifier: NCT01520363).
EPI-743 improves head growth in girls with RTT
EPI-743 (trade name: Vincerinone), marketed by Edison Pharmaceuticals Inc., Mountain View, CA, USA, is a small molecule capable of reducing oxidative stress and modulating energy metabolism in the cortex. A randomized, double-blind Phase II trial demonstrated that EPI-743 significantly increased head circumference as compared with placebo in nine out of twelve girls who received the drug (ClinicalTrials.gov Identifier: NCT01822249). However, patients did not experience a significant decrease in overall disease severity. Additionally, EPI-743 was well tolerated with no major adverse side effects.
Creatine supplementation to increase DNA methylation
In humans, most of the labile methyl groups generated are used for the synthesis of creatine. Therefore, supplementation of creatine may allow for a shift toward methylation of other substrates, including DNA. This may be valuable in RTT because an increase in methylated DNA could increase MeCP2 activity. To test this hypothesis, the diets of 18 girls with RTT were supplemented with creatine, resulting in a significant increase in global methylation103 (ClinicalTrials.gov Identifier: NCT01147575). However, there was no significant improvement in behavioral or motor outcomes, which could be attributable to the small sample size. Thus, further testing in greater cohorts is warranted.
Challenges and future directions in the treatment of RTT
The design of effective clinical trials requires the elaboration of measurable endpoints. The multiple neurological and systemic features of RTT provide several accessible clinical endpoints for which suitable outcome measures exist or are under development. These include communication, epilepsy, motor function, growth, GI dysfunction, and periodic breathing, either breath-holding or hyperventilation. Eye-tracking technology coupled with evoked response potentials present excellent potential measures of communication skills. Video-EEG acquisition provides both spectral analysis of the critical waveforms and time-locked assessments of epileptiform features that can be coupled with ECG and plethysmographic recordings to assess cardiac variability and periodic breathing. Gait analysis paradigms allow the acquisition of objective evidence regarding improvement in motor performance, which would be relevant for individuals who are ambulatory or whose ambulation skills demonstrate improvement. In addition, improvements in muscle tone or dystonia can also be assessed objectively, and actigraphic recordings provide both purposeful and stereotypic movement measures. Determination of GI function is more challenging due to the multiple levels of dysfunction. However, assessment of chewing and swallowing, gastric emptying, and level of constipation could be important areas of measurements.
One major deficiency is the absence of a relevant laboratory biomarker. As part of the recently refunded NHS, a metabolomics pilot study will be conducted. Even so, results are not anticipated before 2016. Clinical outcome measures are also critical to the conduct of future trials. Improvement of acute responsiveness of clinical severity measures, communication assessment, and neurophysiologic parameters are likely to be achieved within the near future. What is lacking is an appropriate behavioral measure, and while efforts are ongoing to provide this missing tool, nothing is currently available. Quality of life using standardized instruments not specific to RTT has been achieved in both those with RTT104 and their caregivers (analysis in progress). For individuals with RTT, it has been demonstrated that greater clinical severity is linked to more severe motor impairment, whereas lower clinical severity is associated with fewer motor impairments. On the contrary, individuals with greater clinical severity have better behavioral function, and individuals with lower clinical severity have worse behavioral function. These results suggest that a modest improvement in motor function could have an adverse effect on behavior, which would be an important aspect in the conduct of a trial. Assessment of quality of life data in the primary caregiver, generally the mother, is currently undergoing analysis. With specific attention to RTT, a specific caregiver inventory is currently being gathered to provide a standardized measure in families managing an individual with RTT. These results should be available within the next 6 months.
Conclusion
Since the initial description by Andreas Rett in 1966, we have made tremendous progress in our understanding of both the clinical and mechanistic characteristics of RTT. Federally funded programs such as the NHS continue to reveal the natural course of disease progression, paving the way for better diagnosis (Figure 1) and treatment of RTT. Additionally, the discovery that RTT is caused by a mutation in MeCP2 not only led to the demonstration of strong genotype–phenotype correlations, but also spurred the development of robust murine models of RTT. These models in turn have allowed us to better appreciate the complex pathophysiology of RTT, which not only involves deficits in neuronal maturation and signaling, but also glial dysfunction. Excitingly, several studies have now demonstrated that postnatal restoration of MeCP2 function in neurons or astrocytes can reverse deficits in mouse models of RTT (Figures 2 and 3). Whether or not we will be able to capitalize on these findings to provide better clinical care remains to be seen. Indeed several promising clinical trials approaching RTT from different angles are currently underway, with results for several trials expected in the near future.
Acknowledgments
This work was supported by NIH Grant R01 NS075062 to MLO and U54 HD061222 to AKP.
Disclosure
The authors report no conflicts of interest in this work.
References
Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. | |
Zachariah RM, Rastegar M. Linking epigenetics to human disease and Rett syndrome: the emerging novel and challenging concepts in MeCP2 research. Neural Plast. 2012;2012:415825. | |
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MeCP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. | |
Neul JL, Fang P, Barrish J, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology. 2008;70:1313–1321. | |
Cuddapah VA, Pillai RB, Shekar KV, et al. Methyl-CpG-binding protein 2 (MeCP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet. 2014;51:152–158. | |
Gibson JH, Williamson SL, Arbuckle S, Christodoulou J. X chromosome inactivation patterns in brain in Rett syndrome: implications for the disease phenotype. Brain Dev. 2005;27:266–270. | |
Schanen C, Houwink EJ, Dorrani N, et al. Phenotypic manifestations of MeCP2 mutations in classical and atypical Rett syndrome. Am J Med Genet A. 2004;126A:129–140. | |
Augenstein K, Lane JB, Horton A, Schanen C, Percy AK. Variable phenotypic expression of a MeCP2 mutation in a family. J Neurodev Disord. 2009;1:313. | |
Kankirawatana P, Leonard H, Ellaway C, et al. Early progressive encephalopathy in boys and MeCP2 mutations. Neurology. 2006;67: 164–166. | |
Schwartzman JS, Bernardino A, Nishimura A, Gomes RR, Zatz M. Rett syndrome in a boy with a 47,XXY karyotype confirmed by a rare mutation in the MeCP2 gene. Neuropediatrics. 2001;32:162–164. | |
Friez MJ, Jones JR, Clarkson K, et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MeCP2 and adjacent region in Xq28. Pediatrics. 2006;118:e1687–e1695. | |
Van Esch H, Bauters M, Ignatius J, et al. Duplication of the MeCP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77: 442–453. | |
del Gaudio D, Fang P, Scaglia F, et al. Increased MeCP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med. 2006;8:784–792. | |
Collins AL, Levenson JM, Vilaythong AP, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–2689. | |
Ramocki MB, Peters SU, Tavyev YJ, et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol. 2009;66:771–782. | |
Ramocki MB, Tavyev YJ, Peters SU. The MeCP2 duplication syndrome. Am J Med Genet A. 2010;152A:1079–1088. | |
Weaving LS, Christodoulou J, Williamson SL, et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet. 2004;75:1079–1093. | |
Mari F, Azimonti S, Bertani I, et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet. 2005;14:1935–1946. | |
Ariani F, Hayek G, Rondinella D, et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet. 2008;83:89–93. | |
Rett A. Uber ein eigenartiges hirnatrophisches Syndrom bei Hyperammonamie im Kindesalter [On a unusual brain atrophy syndrome in hyperammonemia in childhood]. Wien Med Wochenschr. 1966;116:723–726. | |
Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol. 1983;14:471–479. | |
Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:61–65. | |
Trevathan E, Naidu S. The clinical recognition and differential diagnosis of Rett syndrome. J Child Neurol. 1988;3(Suppl):S6–S16. | |
Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68:944–950. | |
Jellinger K, Armstrong D, Zoghbi HY, Percy AK. Neuropathology of Rett syndrome. Acta Neuropathol. 1988;76:142–158. | |
Armstrong DD. Neuropathology of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:72–76. | |
Laurvick CL, de Klerk N, Bower C. Rett syndrome in Australia: a review of the epidemiology. J Pediatr. 2006;148:347–352. | |
Marschik PB, Kaufmann WE, Sigafoos J, et al. Changing the perspective on early development of Rett syndrome. Res Dev Disabil. 2013;34:1236–1239. | |
Neul JL, Lane JB, Lee HS, et al. Developmental delay in Rett syndrome: data from the natural history study. J Neurodev Disord. 2014;6:20. | |
Schultz RJ, Glaze DG, Motil KJ, et al. The pattern of growth failure in Rett syndrome. Am J Dis Child. 1993;147:633–637. | |
Tarquinio DC, Motil KJ, Hou W, et al. Growth failure and outcome in Rett syndrome: specific growth references. Neurology. 2012;79:1653–1661. | |
Glaze DG, Percy AK, Skinner S, et al. Epilepsy and the natural history of Rett syndrome. Neurology. 2010;74:909–912. | |
Lugaresi E, Cirignotta F, Montagna P. Abnormal breathing in the Rett syndrome. Brain Dev. 1985;7:329–333. | |
Percy AK, Glaze DG, Schultz RJ, et al. Rett syndrome: controlled study of an oral opiate antagonist, naltrexone. Ann Neurol. 1994;35:464–470. | |
Julu PO, Kerr AM, Apartopoulos F, et al. Characterisation of breathing and associated central autonomic dysfunction in the Rett disorder. Arch Dis Child. 2001;85:29–37. | |
Motil KJ, Caeg E, Barrish JO, et al. Gastrointestinal and nutritional problems occur frequently throughout life in girls and women with Rett syndrome. J Pediatr Gastroenterol Nutr. 2012;55:292–298. | |
Percy AK, Lee HS, Neul JL, et al. Profiling scoliosis in Rett syndrome. Pediatr Res. 2010;67:435–439. | |
Belichenko PV, Hagberg B, Dahlstrom A. Morphological study of neocortical areas in Rett syndrome. Acta Neuropathol. 1997;93:50–61. | |
Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54:195–201. | |
Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. | |
Tropea D, Giacometti E, Wilson NR, et al. Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2029–2034. | |
Maliszewska-Cyna E, Bawa D, Eubanks JH. Diminished prevalence but preserved synaptic distribution of N-methyl-D-aspartate receptor subunits in the methyl CpG binding protein 2 (MeCP2)-null mouse brain. Neuroscience. 2010;168:624–632. | |
Dani VS, Nelson SB. Intact long-term potentiation but reduced connectivity between neocortical layer 5 pyramidal neurons in a mouse model of Rett syndrome. J Neurosci. 2009;29:11263–11270. | |
Kishi N, Macklis JD. MeCP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27:306–321. | |
Nguyen MV, Du F, Felice CA, et al. MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J Neurosci. 2012;32:10021–10034. | |
McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333:186. | |
Ebert DH, Gabel HW, Robinson ND, et al. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature. 2013;499:341–345. | |
Castro J, Garcia RI, Kwok S, et al. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc Natl Acad Sci U S A. 2014;111:9941–9946. | |
Pitcher MR, Ward CS, Arvide EM, et al. Insulinotropic treatments exacerbate metabolic syndrome in mice lacking MeCP2 function. Hum Mol Genet. 2013;22:2626–2633. | |
Moretti P, Levenson JM, Battaglia F, et al. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. | |
Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315: 1143–1147. | |
Asaka Y, Jugloff DGM, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21:217–227. | |
D’Cruz JA, Wu C, Zahid T, El-Hayek Y, Zhang L, Eubanks JH. Alterations of cortical and hippocampal EEG activity in MeCP2-deficient mice. Neurobiol Dis. 2010;38:8–16. | |
Chen WG, Chang Q, Lin Y, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. | |
Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. | |
Deng V, Matagne V, Banine F, et al. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum Mol Genet. 2007;16:640–650. | |
Martinowich K, Hattori D, Wu H, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. | |
Li W, Calfa G, Larimore J, Pozzo-Miller L. Activity-dependent BDNF release and TRPC signaling is impaired in hippocampal neurons of Mecp2 mutant mice. Proc Natl Acad Sci U S A. 2012;109: 17087–17092. | |
Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. | |
Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–317. | |
Wood L, Gray NW, Zhou Z, Greenberg ME, Shepherd GM. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in an RNA interference model of methyl-CpG-binding protein 2 deficiency. J Neurosci. 2009;29:12440–12448. | |
Durand S, Patrizi A, Quast KB, et al. NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron. 2012;76:1078–1090. | |
Colic S, Wither R, Eubanks JH, Zhang L, Bardakjian BL. EEG analysis for estimation of duration and inter-event intervals of seizure-like events recorded in vivo from mice. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:2570–2573. | |
Chao HT, Chen H, Samaco RC, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. | |
Zhang L, He J, Jugloff DGM, Eubanks JH. The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus. 2008;18:294–309. | |
McLeod F, Ganley R, Williams L, Selfridge J, Bird A, Cobb SR. Reduced seizure threshold and altered network oscillatory properties in a mouse model of Rett syndrome. Neuroscience. 2013;231: 195–205. | |
Calfa G, Hablitz JJ, Pozzo-Miller L. Network hyperexcitability in hippocampal slices from Mecp2 mutant mice revealed by voltage-sensitive dye imaging. J Neurophysiol. 2011;105:1768–1784. | |
Kline DD, Ogier M, Kunze DL, Katz DM. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J Neurosci. 2010;30:5303–5310. | |
Taneja P, Ogier M, Brooks-Harris G, Schmid DA, Katz DM, Nelson SB. Pathophysiology of locus ceruleus neurons in a mouse model of Rett syndrome. J Neurosci. 2009;29:12187–12195. | |
Zanella S, Mebarek S, Lajard AM, Picard N, Dutschmann M, Hilaire G. Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respir Physiol Neurobiol. 2008;160: 116–121. | |
Roux JC, Dura E, Moncla A, Mancini J, Villard L. Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur J Neurosci. 2007;25:1915–1922. | |
Medrihan L, Tantalaki E, Aramuni G, et al. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol. 2008;99:112–121. | |
Abdala APL, Dutschmann M, Bissonnette JM, Paton JFR. Correction of respiratory disorders in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2010;107:18208–18213. | |
Jian L, Nagarajan L, de Klerk N, Ravine D, Christodoulou J, Leonard H. Seizures in Rett syndrome: an overview from a one-year calendar study. Eur J Paediatr Neurol. 2007;11:310–317. | |
Bahi-Buisson N, Guellec I, Nabbout R, et al. Parental view of epilepsy in Rett syndrome. Brain Dev. 2008;30:126–130. | |
Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101:6033–6038. | |
Klein C, Kramer EM, Cardine AM, Schraven B, Brandt R, Trotter J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci. 2002;22: 698–707. | |
Zhang Y, Chen K, Sloan SA, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. | |
Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci U S A. 2007;104:1931–1936. | |
Alvarez-Saavedra M, Saez MA, Kang D, Zoghbi HY, Young JI. Cell-specific expression of wild-type MeCP2 in mouse models of Rett syndrome yields insight about pathogenesis. Hum Mol Genet. 2007;16:2315–2325. | |
Jugloff DG, Vandamme K, Logan R, Visanji NP, Brotchie JM, Eubanks JH. Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. Hum Mol Genet. 2008;17:1386–1396. | |
Garg SK, Lioy DT, Cheval H, et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci. 2013;33a:13612–13620. | |
Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. | |
Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol Psychiatry. 2006;59:468–476. | |
Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. | |
Maezawa I, Swanberg S, Harvey D, LaSalle JM, Jin LW. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci. 2009;29:5051–5061. | |
Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30: 5346–5356. | |
Nguyen MV, Felice CA, Du F, et al. Oligodendrocyte lineage cells contribute unique features to Rett syndrome neuropathology. J Neurosci. 2013;33:18764–18774. | |
Lioy DT, Garg SK, Monaghan CE, et al. A role for glia in the progression of Rett’s syndrome. Nature. 2011;475:497–500. | |
Schmid RS, Tsujimoto N, Qu Q, et al. A methyl-CpG-binding protein 2-enhanced green fluorescent protein reporter mouse model provides a new tool for studying the neuronal basis of Rett syndrome. Neuroreport. 2008;19:393–398. | |
Williams EC, Zhong X, Mohamed A, et al. Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum Mol Genet. 2014;23:2968–2980. | |
Jin LW, Horiuchi M, Wulff H, et al. Dysregulation of glutamine transporter SNAT1 in Rett syndrome microglia: a mechanism for mitochondrial dysfunction and neurotoxicity. J Neurosci. 2015;35:2516–2529. | |
Derecki NC, Cronk JC, Lu Z, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. | |
Wang J, Wegener JE, Huang TW, et al. Wild-type microglia do not reverse pathology in mouse models of Rett syndrome. Nature. 2015;521:E1–E4. | |
Pini G, Scusa MF, Congiu L, et al. IGF1 as a potential treatment for rett syndrome: safety assessment in six Rett patients. Autism Res Treat. 2012;2012:679801. | |
Aharoni R, Teitelbaum D, Sela M, Arnon R. Copolymer 1 induces T cells of the T helper type 2 that crossreact with myelin basic protein and suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 1997;94:10821–10826. | |
Aharoni R, Eilam R, Domev H, Labunskay G, Sela M, Arnon R. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc Natl Acad Sci U S A. 2005;102: 19045–19050. | |
Ben-Zeev B, Aharoni R, Nissenkorn A, Arnon R. Glatiramer acetate (GA, Copolymer-1) an hypothetical treatment option for Rett syndrome. Med Hypotheses. 2011;76:190–193. | |
Deogracias R, Yazdani M, Dekkers MP, et al. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2012;109:14230–14235. | |
Viemari JC, Roux JC, Tryba AK, et al. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. 2005;25:11521–11530. | |
Jaffe DB, Marks SS, Greenberg DA. Antagonist drug selectivity for radioligand binding sites on voltage-gated and N-methyl-D-aspartate receptor-gated Ca2+ channels. Neurosci Lett. 1989;105:227–232. | |
Werling LL, Lauterbach EC, Calef U. Dextromethorphan as a potential neuroprotective agent with unique mechanisms of action. Neurologist. 2007;13:272–293. | |
Freilinger M, Dunkler D, Lanator I, et al. Effects of creatine supplementation in Rett syndrome: a randomized, placebo-controlled trial. J Dev Behav Pediatr. 2011;32:454–460. | |
Lane JB, Lee HS, Smith LW, et al. Clinical severity and quality of life in children and adolescents with Rett syndrome. Neurology. 2011;77: 1812–1818. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.