")
Back to Journals » Journal of Inflammation Research » Volume 11
Mechanisms of NF-κB p65 and strategies for therapeutic manipulation
Authors Giridharan S, Srinivasan M
Received 6 March 2018
Accepted for publication 29 August 2018
Published 30 October 2018 Volume 2018:11 Pages 407—419
DOI https://doi.org/10.2147/JIR.S140188
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Sivagami Giridharan,1 Mythily Srinivasan2,3
1Department of Oral Medicine, Madha Dental College, Kundrathur, Chennai, TN, India; 2Department of Oral Pathology, Medicine and Radiology, Indiana University School of Dentistry, Indiana University Purdue University at Indianapolis, Indianapolis, IN, USA; 3Provaidya LLC, Indianapolis, IN, USA
Abstract: The transcription factor NF-κB is a critical regulator of immune and inflammatory responses. In mammals, the NF-κB/Rel family comprises five members: p50, p52, p65 (Rel-A), c-Rel, and Rel-B proteins, which form homo- or heterodimers and remain as an inactive complex with the inhibitory molecules called IκB proteins in resting cells. Two distinct NF-κB signaling pathways have been described: 1) the canonical pathway primarily activated by pathogens and inflammatory mediators, and 2) the noncanonical pathway mostly activated by developmental cues. The most abundant form of NF-κB activated by pathologic stimuli via the canonical pathway is the p65:p50 heterodimer. Disproportionate increase in activated p65 and subsequent transactivation of effector molecules is integral to the pathogenesis of many chronic diseases such as the rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis, and even neurodegenerative pathologies. Hence, the NF-κB p65 signaling pathway has been a pivotal point for intense drug discovery and development. This review begins with an overview of p65-mediated signaling followed by discussion of strategies that directly target NF-κB p65 in the context of chronic inflammation.
Keywords: NF-κB, inflammation, therapy
Introduction
The NF-κB signaling system is a highly dynamic protein interaction network made up of components that regulate each other. The system includes five transcriptional monomers, two precursor proteins, three ankyrin repeat containing inhibitory IκB proteins, three stimulus-responsive inhibitory kinases (IKK complex: IKK-α and IKK-β, and IKK-γ or NF-κB essential modulator/NEMO), and ankyrin repeat containing regulatory proteins.1 While all NF-κΒ translational monomers that function as homo- or heterodimers share conserved amino terminal dimerizing rel homology domain (RHD), only RelA/p65, C-rel, and RelB possess the transactivation domain (TAD) necessary for transcriptional activity. The TAD lacking monomers p50 or p52 function as trans-repressors as homodimers but can stimulate transcription when heterodimerized with a transactivating Rel subunit.2
In resting state, the NF-κB dimers remain in the cytoplasm as an inactive complex with the IκΒ inhibitory proteins that mask the nuclear localization signals (NLSs) in the RHD and block their nuclear import.2,3 Activation of NF-κB occurs through canonical or noncanonical pathways and depends on phosphorylation-induced ubiquitination of IκB proteins. The canonical NF-κB pathway is activated within minutes of exposure to proinflammatory signals such as cytokines, pathogens, and danger-associated molecular patterns. It is mediated by activation of the NEMO-containing IKK complex, which in turn designates the classical IκBs (IκBα, IκBβ, or IκBε) for degradation with consequent release of the NF-κB dimers for nuclear translocation.2–4 The noncanonical NF-κB pathway is triggered by developmental cues that signal via a select group of receptors such as a subset of tumor necrosis factor family of receptors (TNFRs), B cell-activating factor receptor, lymphotoxin β receptor, and receptor activator of NF-κB and occurs at slower kinetics.5,6 It is mediated by NF-κB inhibitory kinase (NIK) independent of NEMO. The NIK in turn phosphorylates and activates IKKα, which in turn directly acts on the non-IκB substrates of the NF-κB subunits and modulates the transcriptional responses.6–8 Interestingly suppression of canonical NF-κB activation by mutations or loss of NEMO increases NIK accumulation with aberrant noncanonical NF-κB signaling.9 This suggests that the cross talk between the two pathways and a rate-limiting canonical NF-κB signaling perhaps determine the basal pool of NIK and the constitutive NF-κB activity.2,7,8
Induced activation of NF-κB p65:p50
The most abundant form of NF-κB activated by the canonical pathway is the heterodimer of p50 and p65.2 As stated above typically triggered by an NEMO-dependent activation of IKK, phosphorylation of IκB proteins followed by ubiquitination and degradation by proteasomes releases the NF-κB p65:p50 dimers from the inhibitory complex.4,9,10 This exposes the arginine- and lysine-rich NLS of the p65 and the p50 subunits for interaction with the importin α/β heterodimers and nuclear translocation11 (Figure 1). Mechanistically, most often the importin α proteins initially bind the NLS and then recruit importin β to transport the cargo into the nucleus through the nuclear pore complexes (NPCs). However, importin β has been shown to directly bind the NLS of p65 and promote its nuclear translocation.11,12 Within the nucleus, the Ran-GTP protein binds and dissociates the importin-β from the complex with p65. The free p65 then binds specific nucleotide sequence of the target genes and mediates tightly controlled transcriptional programs that exhibit a wide degree of tissue- and context-specificity.13–16 Sequence-specific DNA-binding of p65 is greatly stimulated by the interaction with various components of the basic transcription machinery such as the TFIIB, TBP-associated factors and several coactivators including p300 and Tip60.15,17,18 Export of p65 out of the nucleus is facilitated by the exportin proteins that bind the nuclear export sequence of p65 and the Ran GTP. The complex then passes through the NPC into the cytoplasm, wherein hydrolysis of Ran GTP to Ran GDP by Ran GTPase-activating protein results in the dissociation of p65.7,8 Structural analyses of the NF-κB:IκB complex showed that the IκB proteins mask the NLS of p65 but not that of the p50 subunit.19–21 Hence, in physiologic conditions the importin α/β dimers can bind the NLS of p50 and facilitate the nuclear import of p50:p65 dimers. Exportin proteins bind the nuclear export sequence of IκBα in complex with the p65 in the nucleus and facilitate export of the complex out of the nucleus (Figure 1). Continuous shuttling of the NF-κB:importin:IκBα complex between the nucleus and cytoplasm maintains a low level of basal p65 activity.4,19,22
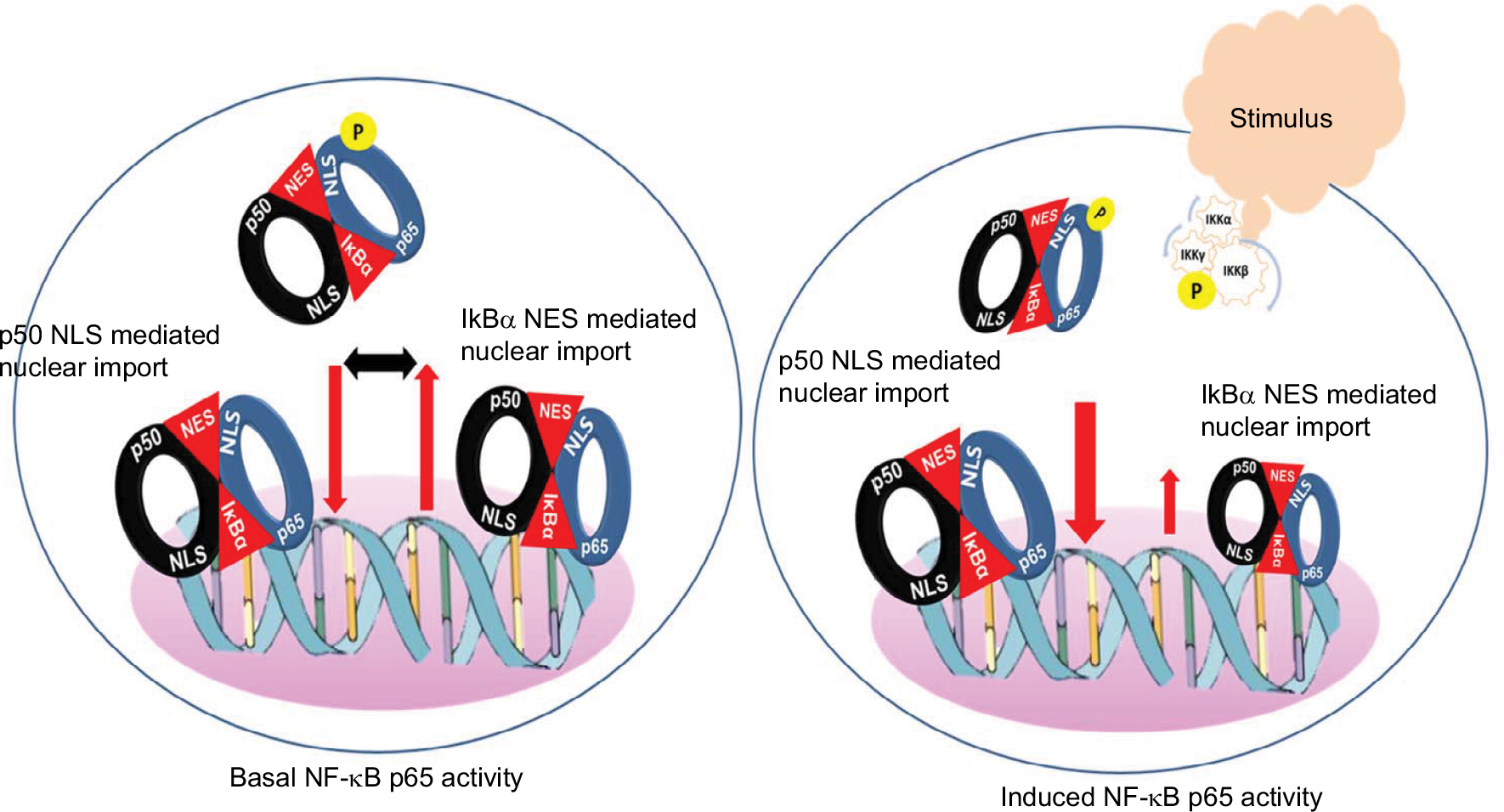 | Figure 1 Model for basal and induced NF-κB p65 activity. Notes: Basal NF-κB activity is potentially maintained by a balanced shuttling of the NF-κB p65:IκB complex between the cytoplasm and nucleus. In a steady-state condition, IκBα masks entirely the nuclear localization signal of p65 but not that of p50. This allows shuttling of the NF-κB:IκB complex by passive nuclear import to support basal activity and reciprocal export by the nuclear export signals in the terminal ankyrin repeat of IκBα. Stimulus induced phosphorylation of IKKβ followed by protein kinase-A-mediated phosphorylation of IκBα and rapid release and import of p65:p50 dimers. Postinduction, the newly synthesized IκBα not only sequesters DNA bound p65 but also shuttles to the cytoplasm to quench the free p50:p65 dimers. |
Mechanisms and modulations of the p65-mediated transactivation
Induced activation of p65 in response to a variety of stimuli is typically transient but sufficient to upregulate transactivation of target genes of diverse activities such as the cellular proliferation, inflammatory cytokines, chemokines, and mediators of apoptosis.1,23,24 It is tightly controlled by postinduction termination via negative feedback loops. Different importins including the importin α3, α4, α5, and β1 exhibit variable affinities for binding the p65 NLS and hence compete with or substitute for each other in facilitating its nuclear import.11,25 Furthermore, the expression levels of importin α and importin β have been shown to vary widely in different cells and tissues.26 Hence, the combined effects of the expression levels of the importin proteins and the differential p65 affinities determine the extent of nuclear translocation of p65 and subsequent transactivation. Efficient and controlled transactivation of the target genes is facilitated by cross talk with other transcription factors with either synergistic (eg, AP-1) or antagonistic (eg, cEBP/β) effects.2,27 In addition, a variety of posttranslational modifications including degradation and regulatory ubiquitination, acetylation, methylation, and phosphorylation play key roles in the fine-tuning and specification of the NF-κB p65 activation outcomes. These modifications often occur in a hierarchical manner with phosphorylation or acetylation as typical starting points and degradative ubiquitination as characteristic irreversible endpoints.4,28
p65 phosphorylation
Inducible p65 phosphorylation can occur both in the cytoplasm and in the nucleus in response to a variety of stimuli.4,28 Phosphorylation is mediated by protein kinases that transfer the γ-phosphate of adenosine triphosphate to the hydroxyl group of a serine (Ser), threonine (Thr), or tyrosine (Tyr) residue of the target protein.29 The NF-κΒ p65 possesses many Ser and Thr residues distributed in the RHD and the TAD. Phosphorylation of individual Ser/Thr/Tyr by multiple kinases generates a heterogeneous pool of modified NF-κΒ p65 that transactivates genes and mediates context-dependent functional responses.4,28
Ser-276 in p65 RHD is predominantly phosphorylated by protein kinase A (PKAc). In resting cells, the catalytic subunit of PKAc is masked by the cytosolic IκΒ:p65 complex. Activation of the IKK complex releases the PKAc, which in turn phosphorylates Ser-276 inducing a structural change in p65 that facilitates interaction with the cofactors p300/CBP (cAMP response element binding [CREB] protein) and enhances transcriptional activity.30 The mitogen and stress-activated protein kinase-1 (MSK-1)/MSK-2 phosphorylates Ser-276 in the nucleus and enhances inflammatory gene expression.4,9,31 This MSK1/2-mediated phosphorylation depends on the nature of stimuli and may require additional kinases such as casein kinase 2 and ribosomal s6 kinase (RSK1).9,28
Phosphorylation of p65 at Thr 254 leads to a phosphorylated Thr–proline motif that is recognized by the peptidyl-prolyl isomerase Pin1. Pin1-mediated prolyl isomerization of p65 results in reduced IκBα binding, increased p65 stability, and nuclear accumulation mediating enhanced transcription.4,32 Mutation of Thr 254 dramatically reduced the half-life of p65 protein and inhibited p65 transcriptional activity.32
Phosphorylation of Ser-468 in p65 T mediated by IKKε or glycogen synthase kinase 3β in unstimulated cells has been shown to negatively regulate basal NF-κB activity.33,34 Phosphorylation of Thr-505 by checkpoint kinase 1 has been identified as a means of cross talk between DNA damage and the activation of NF-κB.4,35
Ser-536 in the p65-TAD is also phosphorylated by multiple kinases including IKK kinases, RSK1, and NF-κB activating kinase/TNFR-associated factor NF-κB activator (TANK)-binding kinase 1 (TBK1).36–38 While phosphorylation of Ser536 in the cytosolic p65 by IKK or RSK1 promotes its nuclear translocation,37 cyclin-dependent kinase-6 (CDK-6) phosphorylates Ser-536 in the nucleus and facilitates p65 binding to specific promoter sequences. Inhibition of CDK-6 catalytic activity suppressed NF-κB p65-mediated inflammatory gene expressions.39,40 Interestingly Ser-536 has also been shown to be phosphorylated by TBK1 (TANK-binding kinase 1), a Ser-Thr kinase, belonging to the noncanonical IκB kinases, suggesting that the two NF-κB pathways are interrelated at multiple levels.41 Taken together, modulation of the Ser-536 phosphorylation status is another mechanism for self-limited activation of NF-κB.
Acetylation
Lysine acetylation is a reversible event regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs).42 Seven acetylation sites are identified in p65: Lys122, Lys123, Lys218, Lys221, Lys310, Lys314, and Lys315. The HATs p300 and p300/CBP-associated factor (PCAF) acetylate lysines 122 and 123 of p65. The p300 has also been described to acetylate lysines 310, 314, and 315.28,43 Acetylation of specific lysine residue has been shown to regulate NF-κB p65 activation or deactivation. While acetylation on Lys122 and Lys123 decreases DNA binding, Lys221 and Lys218 acetylation enhances the DNA binding of NF-κB p65 by impairing its association with IκBα.44,45 Although acetylation at Lys310 is critical for full transcriptional activity of p65, it does not affect the DNA binding or assembly with IκΒα.43,46 Lys314 and Lys315 acetylation increases promoter selectivity without affecting the NF-κB p65 shuttling or the DNA binding.43,47 The acetylated Lys310 is specifically recognized by the two bromodomains of Brd4, which recruits activated CDK9 to phosphorylate RNA polymerase II for the transcription of a subset of NF-κB target genes.28,48
Tip60 (HIV Tat-interacting protein, 60 kDa), originally identified as a binding partner for the HIV-1 Tat protein, has been shown to function as a coactivator of NF-κB p65, enhance acetylation on Lys310, and upregulate p65 transcriptional activity through a protein–protein interaction.49 The Tip60 has been shown to bind DNA prior to p65 and potentially modulate other cofactor:p65 interactions. This suggests that the site-specific early association of Tip60 could represent another mechanism that regulates p65-mediated gene expression.
Methylation
The protein methyltransferases include two enzymes, the protein lysine methyltransferases and the protein arginine methyltransferases (PRMTs). Both enzymes use small-molecule cofactor, S-adenosyl-l-methionine, as the universal methyl donor for the enzymatic methylation of lysine and arginine side chains.50,51 Methylations of both lysine and arginine on NF-κB p65 has been shown to regulate its transactivation potential.51–53
It has been suggested that the methylation of p65 in the nucleus potentially facilitates binding to specific promoters when the local chromatin remodeling machinery is active.50 Set9, a histone methyltransferase, has been shown to methylate p65 on Lys314 and Lys315 and negatively regulate the NF-κB function by inducing the ubiquitination and degradation of DNA-bound p65.54 In contrast, methylation of Lys37 by Set9 or that of Lys218 and Lys221 by nuclear receptor-binding SET domain-containing protein 1, a histone H3K36 methyltransferase, enhances the transcriptional activity by stabilizing binding of p65 to its enhancers.52,55 Apart from lysine residues, demethylation of Arg 30 by protein arginine methyltransferase 5 (PRMT5) has been shown to activate p65 suggesting that the methylation of both lysine and arginine residues regulate NF-κB-mediated transcriptional activity.50,53 Molecular modeling suggests that methylated R30 can mediate Van der Waals contacts and increase the affinity of p65 for DNA and consequent transactivation of target gene expressions.53 Hence, differential methylation could represent another regulatory mechanism for differential activation of individual genes based on the cell type, stimulus, and other factors.50,51
In summary, the sequential post-translational modifications modulate the strength and duration of NF-κB activity, contribute to the target gene specificity, and thereby fine-tune the biologic responses.28,38,56 Indeed, it has been observed that the phosphorylation of Ser276 followed by acetylation of Lys310 of p65 facilitated recruitment of specific elongation factors Brd4 and P-TEFb to proinflammatory target genes promoting efficient mRNA processing.57 Furthermore, cross-talk mechanisms link post-translational modification-mediated regulations. For example, phosphorylation at Ser-276 and Ser-536 enhances the p300- and PCAF-mediated acetylation of Lys122, Lys123, and Lys310 and upregulates transcriptional activation by p65.58 Thus, by virtue of modifying the interaction of p65 with select cofactors, the post-translational modifications at specific sites contribute to specific biologic responses.54,57
The varied and differential outcomes following individual site phosphorylation or acetylation have been attributed to the induced conformational changes in p65 that direct subsequent cofactor interactions required for transcriptional activation.4,59 In this context it is of interest to note that the p65-TAD critical for its transcriptional ability hosts multiple phosphorylation sites and is largely unstructured.4 The intrinsic disorder of the unstructured p65-TAD confers intramolecular flexibility that allows it to adopt an “induced fit” secondary structure stabilized by phosphorylation of specific residue in the context of an interacting cofactor.28,59 Such intrinsically unstructured protein regions could serve as potential targets for rational drug design based on the transition from disordered to ordered conformation through intermolecular interaction.60 Interestingly synthetic peptides derived from the p65-TAD have been shown to inhibit NF-kB p65-mediated cellular responses.61
Termination of activated p65
To avoid excessive transactivation and deleterious cellular responses, the activated NF-κΒ p65 orchestrates a highly coordinated termination program that is rapid and self-limiting.
Role of IκBα
As stated above, NF-κB activity is predominantly controlled by association with IκBα. Following translocation, the nuclear p65 binds on the κB sequence in IκBα promoter and increases its transcription. The increased nuclear IκBα in turn removes the NF-κB p65 complex from the DNA terminating its transcriptional activity.19,22 As nuclear p65:DNA interaction exhibits fast dissociation kinetics, the newly synthesized IκBα may not only reverse the binding of p65 to DNA, but also capture p65 molecules released from DNA and transport them back to the cytoplasm.20,38 The iterative cycles of IκBα degradation and replenishment by p65-mediated transactivation provide sustained supply of p65:p50 dimers to the nucleus at a basal level.3,19 In addition, strong oscillations of the negative feedback regulation between the activated p65 and IκBα are monitored by the IκBβ and IκBγ, the inhibitory proteins that undergo slow degradation, exhibit limited activation potential, and prevent abundance of activated p65 in the nucleus.2,62
Dephosphorylation of p65: It is another mechanism that facilitates termination of NF-κB signaling after stimulant removal or target gene expression and reestablish normal responsiveness. Dephosphorylation of Ser-536 by protein phosphatase 2A or by wild-type p53-induced phosphatase 1 has been shown to reduce the interaction of p65 with p300 and thereby inhibit gene expression.4,63,64 However, much like phosphorylation, dephosphorylation of p65 by phosphatases regulates the functions of NF-κB p65 in a context- and signal-dependent manner. For example, following cisplatin stimulation, phosphatase 4-mediated dephosphorylation of Thr-435 upregulated NF-κB p65 activation rather than suppression.65
Deacetylation of p65: Multiple HDACs deacetylate p65 and regulate NF-κB functions depending on the specific acetylation site. While HDAC3-mediated deacetylation of p65 at Lys122, Lys123, Lys314, and Lys315 upregulated inflammatory gene expression, deacetylation at Lys218 and Lys221 promotes the binding of p65 with IκBα leading to the nuclear export and downregulation of NF-κB.43
Demethylation of p65: Similar to phosphorylation and acetylation, methylation is also a reversible event mediated by demethylases. Demethylation of methylated Lys-218 and Lys-221 by F-box and leucine-rich repeat protein 11, an H3K36 demethylase, has been shown to negatively regulate the transcriptional activity of NF-κB p65 and decrease cell proliferation.55,66
Ubiquitination and degradation: They have been suggested as mechanisms that control the strength and duration of NF-κΒ activation.28,36 Nuclear p65 is degraded by the proteasome in a DNA-binding-dependent manner. Copper metabolism domain protein, a physiologic inhibitor of NF-κΒ, binds to an E3 ligase SOCS1 (suppressor of cytokine signaling 1) and p65, stabilizing the p65:SOCS1 interaction and thereby promote ubiquitination and degradation of p65.4,67 PDLIM2, a nuclear LIM domain containing ligase, mediates polyubiquitination and proteosomal degradation of nuclear p65 and contributes to the termination of p65-mediated transactivation.10 Interestingly, point mutations in p65 that inhibit DNA binding impaired degradation, suggesting that the sequence-specific interactions with DNA may be critical for transcriptional termination.
In conclusion, while cytoplasmic NF-κB p65 in complex with the IκBs are stable and do not undergo much turnover, the stability of nuclear p65 proteins is short-lived. Several interrelated not mutually exclusive mechanisms regulate the stability and termination of nuclear p65. In general, the postinduction repression of NF-κB p65 by newly synthesized IκBα can be considered upstream events that assist in maintaining the homeostasis between the cytoplasmic and nuclear p65. The nucleocytoplasmic shuttling mediated by the differential binding kinetics of p65 with different importin/exportin proteins or modulations by dephosphorylation, demethylation, or deacetylation are intermediate mechanisms that contribute to effective p65 activation. For example, in conditions of continuous stimulation, oscillations in Ser-536 phosphorylation have been shown to reflect oscillations in NF-κB cytosolic–nuclear translocation, suggesting potential dephosphorylation of Ser-536 in the nucleus.37,68 Ubiquitination and proteosomal degradation of DNA-bound p65 constitute downstream events that play major roles in limiting the intensity and duration of NF-κB p65 activity. In addition, the nature and extent of regulation by post-translational modification can vary with different stimulators and even the same modifications can facilitate different effects.50
Therapeutic targeting of NF-κΒ p65 in inflammation
The literature is replete with a vast number of candidate therapeutics designed to target the NF-κB pathway. These compounds belong to different chemical classes such as naturetics, peptidomimetics, small molecules, small interfering RNAs, and microbial products (Table 1).69–72 This review is restricted to therapeutic approaches that specifically target the interactions of the p65 subunit of NF-κB with a focus on inflammation, discussing representative compounds and illustrating emerging strategies to overcome the limitations of conventional NF-κB therapeutics (Figure 2).
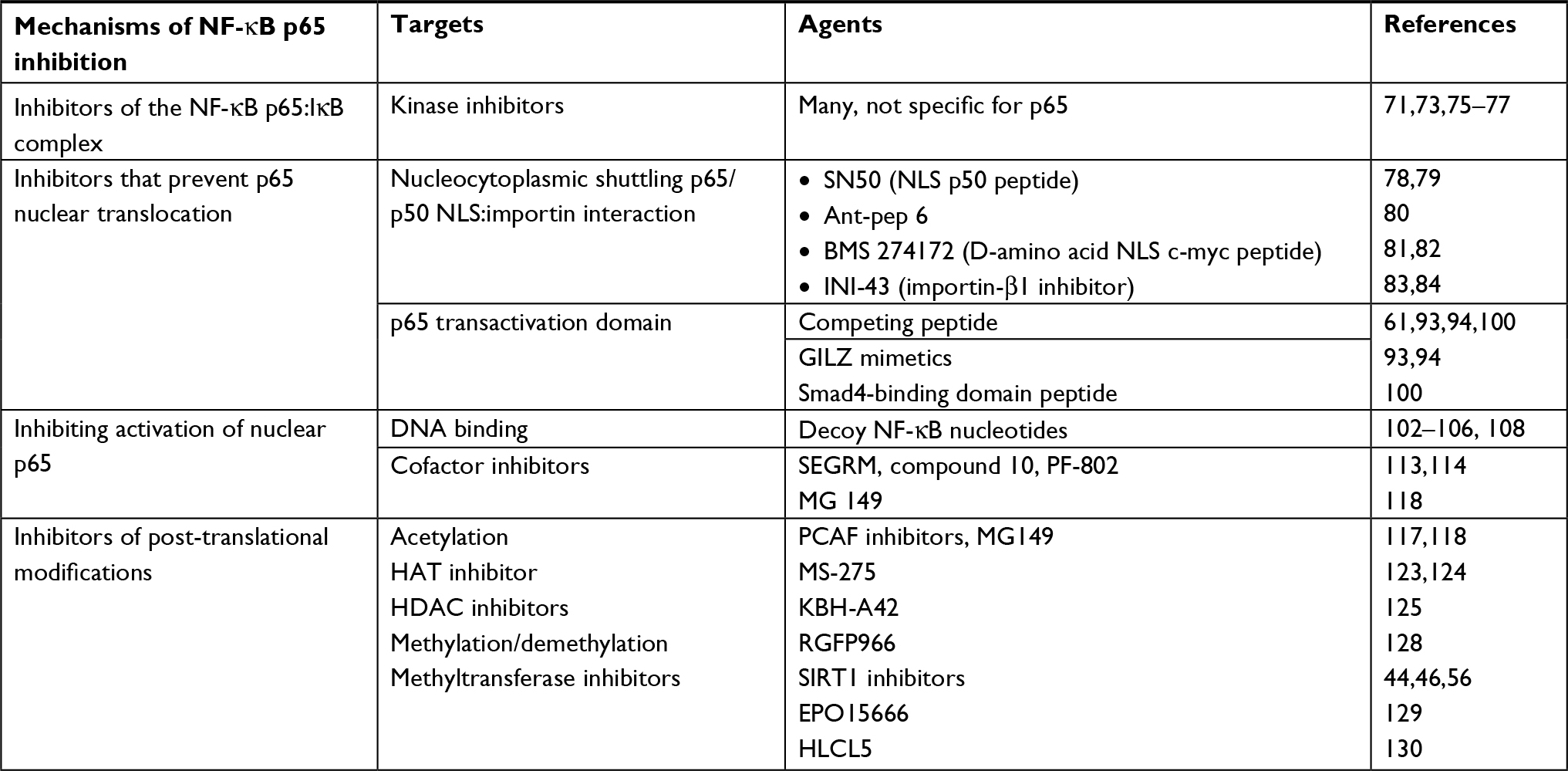 | Table 1 Strategies and agents developed and evaluated for selective targeting of NF-kB p65 Notes: The table includes representative examples of agents evaluated for selective targeting of p65 in the context of chronic inflammation. Abbreviations: HAT, histone acetyl transferase; HDAC, histone deacetylase; INI-43, inhibitor of nuclear import-43; NLS, nuclear localization signal; PCAF, p300/CBP-associated factor; SEGRM, selective glucocorticoid receptor analog or modulator. |
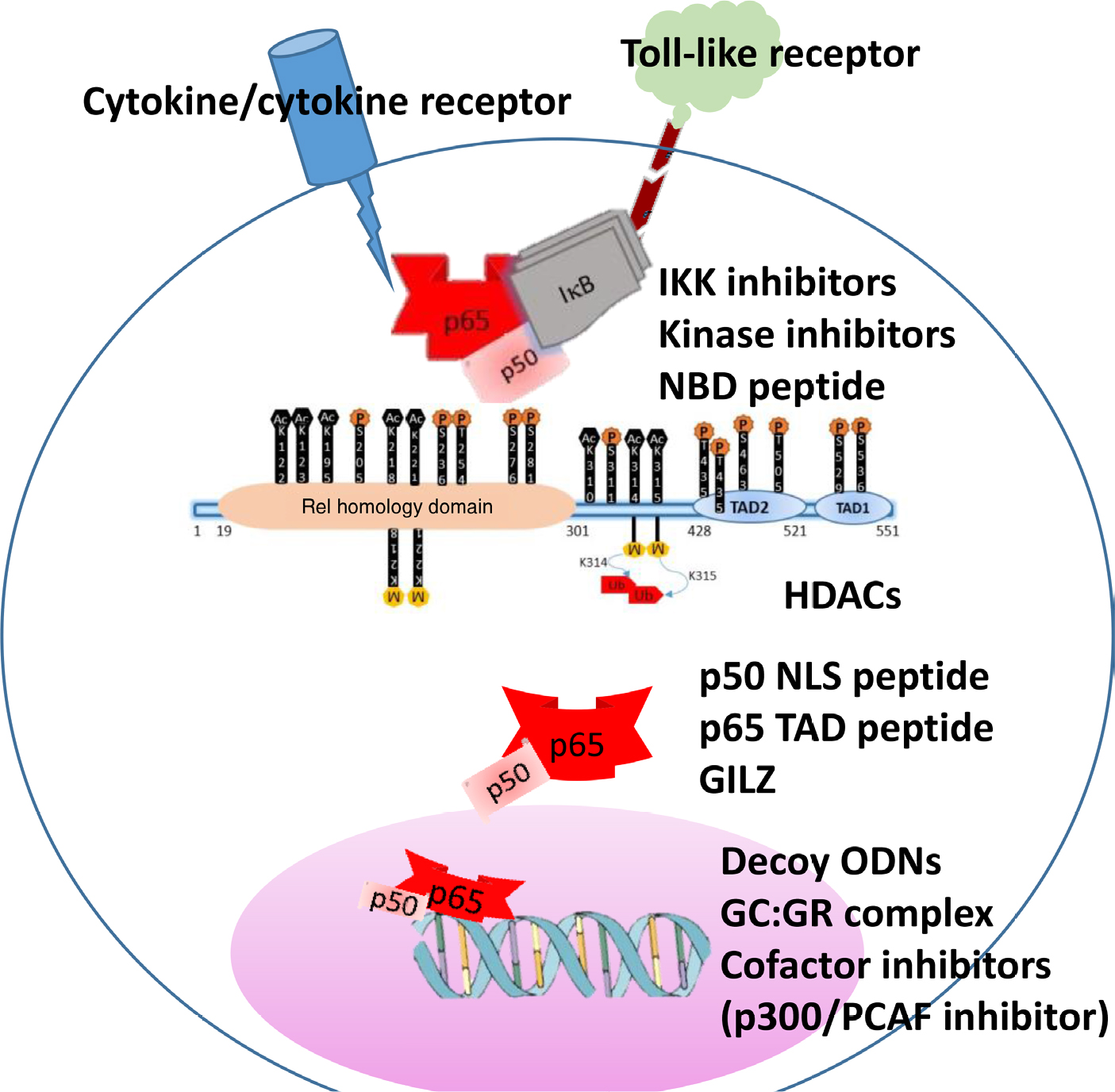 | Figure 2 Schematic representation of NF-κB p65 activation and potential inhibitors along the signaling pathway. Notes: The inset shows phosphorylation and acetylation sites of p65 as sites of kinase inhibitors and HDACs. Representative inhibitory strategies along the pathway are shown. Abbreviations: NBD, NEMO binding peptide; GC, glucocorticoid; GILZ, glucocorticoid-induced leucine zipper; GR, glucocorticoid receptor; HDAC, histone deacytelases; NLS, nuclear localization signal; ODN, oligodeoxynucleotide; PCAF, p300/CBP-associated factor; TAD, transactivation domain. |
Inhibitors of the NF-κB p65:IκΒ complex
As stated above, canonical activation of NF-κB p65 involves intracellular signaling networks that utilize adapter protein interactions, and phosphorylation and ubiquitination processes that hinge on signal-induced proximity and appropriate orientation of the multimeric subunits. Several chronic inflammatory pathologies are associated with dysregulated phosphorylation/activation of the NF-kB:IkB complex. Hence, preventing phosphorylation by using kinase inhibitors constitutes attractive therapeutic strategy. A vast number of kinase inhibitors target either the IKK complex or the adapter proteins in the NF-kB signal transduction pathway (please refer to excellent reviews3,71,73,74). The therapeutic potential of these kinase inhibitors has been predominantly evaluated in multiple cancers.3,74 While none of these have been shown to specifically target p65, a few affect the post-translational modifications in p65 by inhibiting the associated cofactors. For example, p38 mitogen-activated protein kinase (MAPK) inhibitor has been shown to inhibit phosphorylation of the coactivator p300 and preclude acetylation on Lys310 of p65, thereby preventing DNA binding and transcriptional activity.75 Second-generation p38 MAPK inhibitors are being evaluated in clinical trials for chronic inflammatory diseases such as COPD, Crohn’s disease, and Alzheimer’s disease.76,77
Inhibitors that prevent p65 nuclear translocation
a. NLS inhibitors: As stated above, nucleocytoplasmic shuttling of p65 is critical for transcriptional regulation of target genes. Binding of the p65 NLSs with the adapter proteins importins enables its translocation through the NPC.11,25 Within the nucleus, importin is dissociated by GTPases and the activated p65 is released to initiate transcription.62 This suggests that inhibitors of the p65:importin adapter interactions may be selective NF-κB inhibitors. It was proposed that rationally designed peptides derived from the NLS of the NF-κB subunits would suppress nuclear translocation of p50:p65 dimers by competitively inhibiting their interaction with importins. A linear peptide of the p50 NLS sequence (SN50) covalently synthesized with a cell-permeable compound inhibited the nuclear import of NF-κΒ p65 in human monocytic cells and murine endothelial cells stimulated with the proinflammatory agonists such as lipopolysaccharide (LPS) or the cytokine TNF-α.78,79 Interestingly a short peptide mimic (RLRWR) of the DNA binding motif of p50 designated as anti-inflammatory peptide-6 exhibited cell-penetrating ability, interacted directly with the p65 thereby inhibiting DNA binding and transactivation of inflammatory mediators.80 A cell-permeable peptide made up of NLS of c-myc has also been shown to inhibit nuclear translocation of p65 and suppress T-cell responses in models of inflammation.81,82
b. Many inhibitors that directly target select importin-α, improtin-β, or exportin proteins have been developed to inhibit nuclear translocation of p65 and subsequent cellular responses. Examples of this class of inhibitors include small molecules such as inhibitor of nuclear import-43 and selinexor, a selective inhibitor of nuclear export compound that inhibits the exportin XPO1.83,84 The efficacy of these agents has been largely investigated in tumor models and is yet to be evaluated in inflammatory pathologies.
c. Blocking p65 TAD: This includes strategies that inhibit p65 independent of other rel proteins.
i. Competing p65 peptide: The p65 nuclear translocation and consequent transactivation of target genes is facilitated by interactions of p65-TAD with other transcription factors and cofactors. A conceptually simple strategy for development of inhibitors of interprotein interactions is to take advantage of the evolutionary selection of residues for individual protein:protein interaction and design interface peptides derived from the primary sequence of one of the binding partners.85,86 As stated above, phosphorylation on Ser276, Ser529, Ser536, and Ser471 induces conformational changes in p65 and facilitates its binding with other transcriptional cofactors such as p300 and PCAF.59 Synthetic peptides encompassing these phosphorylation sites in p65 fused with a protein transduction domain have been shown to suppress NF-κΒ activation induced by a variety of stimuli such as LPS, interleukin-1, okadaic acid, phorbol 12-myristate 13-acetate, and cigarette smoke condensate inflammatory stimuli.61
ii. Glucocorticoids-mediated cytoplasmic sequestration of p65: The classical model for the therapeutic potential of glucocorticoids suggests that the direct binding of the glucocorticoid receptor on specific glucocorticoid receptor elements (GREs) in the promoter region upregulates transactivation of anti-inflammatory genes.87,88 With six GRE in its promoter, glucocorticoid-induced leucine zipper is strongly upregulated by glucocorticoids.89 GILZ has been shown to physically bind the p65 TAD via its proline-rich region and inhibit NF-κΒ signaling.90,91 In protein interfaces, proline-rich regions that adopt extended polyproline type II helical conformation constitute excellent drug templates with the specificity of the interaction determined by the residues adjacent to the interacting moieties in each partner.60,92 Synthetic peptide mimics of the p65 binding motif of GILZ suppressed nuclear translocation of p65, inflammation, and apoptosis.93,94
iii. The expanding network of NF-κB interactors has increased the potential for identifying newer targets for specific inhibition. Among the proteins that bind p65, a subset directly binds the TAD such as the p300,95,96 silencing mediator of retinoic acid and thyroid hormone receptors97 and the Smad proteins. Smads are a family of structurally similar proteins belonging to the TGF-β superfamily involved in regulating cell development and growth. In humans, eight Smads are known and are classified into three subtypes: five receptor-regulated Smads (R-Smads), one common partner Smad (co-Smad), and two inhibitory Smads (I-Smad). While the R-Smads mediate signaling from the TGF-β receptor and I-Smads suppress R-Smads, the co-Smad recruits coregulators to the transcriptional factors and modules gene expressions. Structurally, the Smad proteins have two conserved globular domains called the mad homology (MH) 1 and MH2 domains.98 Recently, using co-immunoprecipitation assays, it has been reported that the TA2 domain of p65 physically interacted with the MH1 domain of Smad4.99 A peptide derived from the amino-terminal region of TA2 that associates with the MH1 domain of Smad4 covalently synthesized with a cell-permeable peptide prevented NF-κB signaling-mediated inhibition of osteoblast differentiation by blocking the association of p65 with Smad4 and allowing increased bone morphogenetic protein-2 signaling.100
Inhibitors of nuclear p65
i. Decoy oligodeoxynucleotides (decoy ODNs): They constitute a type of gene therapy and are made up of double-stranded DNA fragments of the same sequence as the binding site of the transcription factor on DNA.101 Decoy ODNs possessing κB consensus sequence bind NF-κB p65, prevent it from interacting with the cis-element of specific target gene, and thereby inhibit initiation of the transcription process.102 Transfection of NF-κΒ decoy ODNs to the arteries by hemagglutinating virus of Japan liposomes prevented ischemia-induced myocardial infarction by suppressing expression of cytokines and adhesion molecules.103 Similar observation of protection against inflammation following decoy NF-κΒ ODNs has been reported in models of arthritis, colitis, cystic fibrosis, and atopic dermatitis.104–106 The relative ease of design and construction is a significant advantage of decoy ODNs as therapeutic agents. However, limited cellular uptake due to the negative charge and large size as well as severe toxicities due to lack of cellular restrictions is a potential drawback. Recently, several strategies have been developed for successful and targeted delivery of decoy ODNs such as the use of nanoparticles and ultrasound-targeted microbubbles.101,107 NF-κΒ decoy ODNs in such formulation are currently in clinical trials for atopic dermatitis and discogenic low back pain, and they appear to be well tolerated, nontoxic, and potentially efficacious108 (www.anges.co.jp).
ii. Selective glucocorticoid receptor agonists (SEGRA): Cross talk between the NF-κB and the GR signaling occurs at multiple levels such as the physical interaction between the DNA binding domain of GR and the nuclear p65, direct interference of GR with p65 and associated basal transcription machinery, and competitive binding of co-activators such as PKAc or CBP.87,109,110 Many SEGRA or dissociated agonists of glucocorticoid receptor based on steroidal scaffold or nonsteroidal modulators of the glucocorticoid receptor (SEGRM) developed to selectively facilitate that transrepressive actions are enhanced have been shown to inhibit NF-κB-mediated inflammatory responses.111–113 In this context it has been reported that coactivation of GR and p65 results in their association at binding sites that cluster with p65 target genes suggesting competition between GR and p65 for binding specific response elements.87 Interestingly, SEGRM of the family of diabenzoxepane or dibenzosuperane (compound 10, PF-802, Fosdagocorat) that bind GR at sites other than the ligand (glucocorticoid)-binding domain induce conformational changes precluding GR binding the GRE.113,114 Together with the enhanced potential for the nuclear GR to compete for the co-activators, the anti-inflammatory effects of such SEGRM could be attributed to specific inhibition of NF-κB p65.113,115,116 Elucidation of these mechanisms would also assist in adopting the information from the GR:p65 interactome in developing agents that provide beneficial (predominantly transrepressive) effects of glucocorticoids while avoiding the serious adverse responses.
Inhibitors of post-translational modifications
The dynamic nature of the PTMs in regulating gene expression patterns makes this system particularly amenable for epigenetic drug discovery. Considerable evidence suggests that the NF-κB p65 PTMs are deregulated in conditions of chronic inflammation and autoimmune diseases.28,58 Targeting the p65 PTM sites or the protein:protein interactions represents powerful approaches for amelioration of these conditions. Although all PTMs are important components of the epigenome, acetylation network consists of a large number of druggable targets.
i. HAT inhibitors: As stated above, HATs are epigenetic enzymes that add acetyl groups onto lysine residues of proteins including NF-κB p65. The following include few examples of HAT inhibitors that selectively target lysine residues on p65.
PCAF inhibitors: The PCAF inhibitors specifically acetylate only Lys-122 of p65 and blocking of this acetylation could diminish the nuclear retention and transcriptional activity of p65.42,45 Specific inhibitors of PCAF developed using structure-based design and molecular docking have been shown to inhibit p65-induced transactivation of inflammatory mediators in stimulated macrophages and glial cells.117
Tip60, a member of the MYST family of co-activators, has been shown to activate p65-mediated transcription by maintaining the Lys310 in the acetylated state.49 Analogs of the naturally occurring anacardic acid (6-pentadecylsalicylic acid/MG 149) that inhibit Tip60 have been shown to reduce proinflammatory gene expression in murine precision cut lung slices (PCLS).42,118
ii. HDAC inhibitors: Several studies have reported the potential of small-molecule HDAC inhibitors to regulate p65 acetylation.43,119 The functional efficacy of pan-HDAC inhibitors in multiple cancers has been reviewed extensively.120–122 The following are few examples of HADC inhibitors that have been shown to modulate inflammation.
NF-κB p65 acetylation on Lys 310 by a HDAC1–3 inhibitor, MS-275, has been shown to exert mixed effects depending on the cell type and the nature of the stimuli. In LPS-stimulated macrophages and murine PCLS, treatment with MS-275 upregulated both anti-inflammatory and proinflammatory cytokines.123 Similarly, in poly(I-C)-induced dendritic cells, MS-275 reduced the release of both the proinflammatory TNF-α, IL-6, and IL-12 cytokines and the anti-inflammatory IL-10.124 In contrast, in a model of cigarette smoke-exposed lung inflammation that mimics the human COPD, MS-275 treatment robustly attenuated the expression of inflammatory chemokines and decreased neutrophil influx in the lungs.123 Another HDAC 1–2 inhibitor, KBH-A42, reduced LPS-induced endotoxemia.125 With both NLS and nuclear export sequence, HDAC-3 exerts proinflammatory effects.126 Silencing HDAC-3 reduced vascular cell adhesion molecule 1, monocyte recruitment, proinflammatory transcriptional activity, and disease progression in an allergic skin inflammation model.126 In human macrophages, HDAC3 siRNA inhibited cytokine response to LPS.127 In contrast to the multiple HDAC inhibitor MS 275, treatment with RGFP966, a selective and potent inhibitor of HDAC-3, upregulated IL-10 and exhibited anti-inflammatory effects in response to LPS/IFNγ in macrophages and in mouse PCLS.128 Deacetylation of Lys-310 by the HDAC SIRT1 has been shown to inhibit p65-mediated transactivation of cytokines and apoptosis mediators.46 Inhibiting the activity of SIRT1 markedly increased the acetylation of Lys-310 and the transcriptional activity of NF-κB.44,56
The arginine methyltransferase, PRMT5, that demethylates R30 of p65 plays a key role in regulating endothelial cell inflammation, proliferation, and differentiation.66,123 Increased expression of PRMT5 that correlates with the enhanced inflammatory cytokines has been observed in the synovial tissues in rheumatoid arthritis. Inhibition of PRMT5 with a specific inhibitor EP015666, has been shown to decrease phosphorylation of IKK and IKBα, reduce transactivation of inflammatory cytokines, and suppress fibroblast migration potentially ameliorating disease progression in arthritis.129 Another PRMT5 inhibitor, HLCL65, has been shown to selectively inhibit pathogenic T cells in a mouse model of multiple sclerosis.130 Taken together these observations suggest that the manipulation of specific acetylation sites in p65 can lead to resultant proinflammatory or anti-inflammatory effects.
Conclusion
Overwhelming evidence supports a critical role of increased NF-κΒ p65-mediated transactivation in the pathogenesis of multiple chronic inflammatory diseases. The beneficial effects of many natural compounds such as the curcumin and that of the synthetic inhibitors of upstream signaling molecules such as the cytokine and cytokine receptor antagonists in chronic inflammatory diseases have been attributed to reduced NF-κΒ p65 signaling.3,69–71,101 Hence, targeting p65 activation directly for better clinical efficacy has been a long-sought-after goal. As discussed earlier, several strategies to block p65 at each level along the path of signaling have been assessed and novel strategies are being developed. However, a critical hurdle is selective targeting of activated p65 without the off-target effects. In this context, steric blockade of p65-TAD using GILZ mimetics or mimics of the p65:Smad4 interface appears to be a promising strategy to selectively inhibit activated p65.91,100,131 Targeted intracellular delivery in a context- and tissue-specific manner is another drawback that impedes development of effective p65 inhibitors. It is anticipated that advances in nanotechnology-based delivery systems will assist in not only improving the pharmacokinetic profile but also facilitate contextual delivery of drugs and drug-like agents. Recently, a nanocarrier delivery system consisting of a functional cell-penetrating stearoyl-oligopeptide was used for effective delivery of two RNAi agents that silenced p65 mRNA in macrophages and exerted therapeutic effects in a model of atopic dermatitis.132 The properties of biocompatibility and stability in body fluids and lack of immunogenicity make nanocarriers as ideal portals for drug delivery. In future, development of engineered nanocarriers will not only facilitate targeted delivery but also accommodate conformational changes needed for efficacious intracellular p65 inhibitors.1,133
Acknowledgment
The authors sincerely appreciate funding from the National Institutes of Health (1R41 AG053117) and from Provaidya LLC to MS.
Disclosure
The authors report no conflicts of interest in this work.
References
Basak S, Behar M, Hoffmann A. Lessons from mathematically modeling the NF-κB pathway. Immunol Rev. 2012;246(1):221–238. | ||
Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034. | ||
Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov. 2004;3(1):17–26. | ||
Christian F, Smith E, Carmody R. The regulation of NF-κB subunits by phosphorylation. Cells. 2016;5(1):12. | ||
Razani B, Zarnegar B, Ytterberg AJ, et al. Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKalpha-mediated phosphorylation. Sci Signal. 2010;3(123):ra41. | ||
Gray CM, Remouchamps C, Mccorkell KA, et al. Noncanonical NF-κB signaling is limited by classical NF-κB activity. Sci Signal. 2014;7(311):ra13. | ||
Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12(8):695–708. | ||
Shih VF-S, Tsui R, Caldwell A, Hoffmann A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011;21(1):86–102. | ||
Chen L-F, Greene WC. Shaping the nuclear action of NF-κB. Nat Rev Mol Cell Biol. 2004;5(5):392–401. | ||
Collins P, Mitxitorena I, Carmody R. The ubiquitination of NF-κB subunits in the control of transcription. Cells. 2016;5(2):23. | ||
Liang P, Zhang H, Wang G, et al. KPNB1, XPO7 and IPO8 mediate the translocation ofNF-kappaB/p65 into the nucleus. Traffic. 2013;14:1132–1143. | ||
Bayliss R, Corbett AH, Stewart M. The molecular mechanism of transport of macromolecules through nuclear pore complexes. Traffic. 2000;1(6):448–456. | ||
Korwek Z, Tudelska K, Nałęcz-Jawecki P, et al. Importins promote high-frequency NF-κB oscillations increasing information channel capacity. Biol Direct. 2016;11(1):61. | ||
Phelps CB, Sengchanthalangsy LL, Malek S, Ghosh G. Mechanism of kappa B DNA binding by Rel/NF-kappa B dimers. J Biol Chem. 2000;275:24392–24399. | ||
Schmitz ML, Baeuerle PA. The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. Embo J. 1991;10(12):3805–3817. | ||
van Essen D, Engist B, Natoli G, Saccani S. Two modes of transcriptional activation at native promoters by NF-kappaB p65. PLoS Biol. 2009;7(3):e73. | ||
Koyanagi M, Hijikata M, Watashi K, Masui O, Shimotohno K. Centrosomal P4.1-associated protein is a new member of transcriptional coactivators for nuclear factor-kappaB. J Biol Chem. 2005;280(13):12430–12437. | ||
Uranishi H, Tetsuka T, Yamashita M, et al. Involvement of the pro-oncoprotein TLS (translocated in liposarcoma) in nuclear factor-kappa B p65-mediated transcription as a coactivator. J Biol Chem. 2001;276(16):13395–13401. | ||
Huang TT, Kudo N, Yoshida M, Miyamoto S. A nuclear export signal in the N-terminal regulatory domain of Ikappa Balpha controls cytoplasmic localization of inactive NF-kappa B/Ikappa Balpha complexes. Proc Natl Acad Sci. 2000;97(3):1014–1019. | ||
Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell. 1998;95(6):759–770. | ||
Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95:749–758. | ||
Arenzana-Seisdedos F, Thompson J, Rodriguez MS, Bachelerie F, Thomas D, Hay RT. Inducible nuclear expression of newly synthesized I kappa B alpha negatively regulates DNA-binding and transcriptional activities of NF-kappa B. Mol Cell Biol. 1995;15(5):2689–2696. | ||
Baldwin AS. Regulation of cell death and autophagy by IKK and NF-κB: critical mechanisms in immune function and cancer. Immunol Rev. 2012;246(1):327–345. | ||
Yde P, Mengel B, Jensen MH, Krishna S, Trusina A. Modeling the NF-κB mediated inflammatory response predicts cytokine waves in tissue. BMC Syst Biol. 2011;5(1):115. | ||
Fagerlund R, Kinnunen L, Kohler M, Julkunen I, Melen K. NF-{kappa}B is transported into the nucleus by importin {alpha}3 and importin {alpha}4. J Biol Chem. 2005;280:15942–15951. | ||
Köhler M, Speck C, Christiansen M, et al. Evidence for distinct substrate specificities of importin alpha family members in nuclear protein import. Mol Cell Biol. 1999;19(11):7782–7791. | ||
Stein B, Cogswell PC, Baldwin AS. Functional and physical associations between NF-kappa B and C/EBP family members: a Rel domain-bZIP interaction. Mol Cell Biol. 1993;13(7):3964–3974. | ||
Huang B, Yang X-D, Lamb A, Chen L-F. Posttranslational modifications of NF-κB: another layer of regulation for NF-κB signaling pathway. Cell Signal. 2010;22(9):1282–1290. | ||
Barnes PJ. Kinases as novel therapeutic targets in asthma and chronic obstructive pulmonary disease. Pharmacol Rev. 2016;68(3):788–815. | ||
Zhong H, Suyang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89(3):413–424. | ||
Reber L, Vermeulen L, Haegeman G, Frossard N. Ser276 phosphorylation of NF-kB p65 by MSK1 controls SCF expression in inflammation. PLoS One. 2009;4(2):e4393. | ||
Ryo A, Suizu F, Yoshida Y, et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12(6):1413–1426. | ||
Buss H, Dörrie A, Schmitz ML, et al. Phosphorylation of serine 468 by GSK-3beta negatively regulates basal p65 NF-kappaB activity. J Biol Chem. 2004;279(48):49571–49574. | ||
Mattioli I, Geng H, Sebald A, et al. Inducible phosphorylation of NF-kappa B p65 at serine 468 by T cell costimulation is mediated by IKK epsilon. J Biol Chem. 2006;281(10):6175–6183. | ||
Msaki A, Sánchez AM, Koh LF, et al. The role of RelA (p65) threonine 505 phosphorylation in the regulation of cell growth, survival, and migration. Mol Biol Cell. 2011;22(17):3032–3040. | ||
Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol. 2000;18(1):621–663. | ||
Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274(43):30353–30356. | ||
Viatour P, Merville M-P, Bours V, Chariot A. Phosphorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30(1):43–52. | ||
Buss H, Handschick K, Jurrmann N, et al. Cyclin-dependent kinase 6 phosphorylates NF-κB P65 at serine 536 and contributes to the regulation of inflammatory gene expression. PLoS One. 2012;7(12):e51847. | ||
Handschick K, Beuerlein K, Jurida L, et al. Cyclin-dependent kinase 6 is a chromatin-bound cofactor for NF-κB-dependent gene expression. Mol Cell. 2014;53(2):193–208. | ||
Shanmugasundaram K, Nayak B, Shim EH, Livi CB, Block K, Sudarshan S. The oncometabolite fumarate promotes pseudohypoxia through noncanonical activation of NF-κB signaling. J Biol Chem. 2014;289(35):24691–24699. | ||
Dekker FJ, van den Bosch T, Martin NI. Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov Today. 2014;19(5):654–660. | ||
Calao M, Burny A, Quivy V, Dekoninck A, van Lint C. A pervasive role of histone acetyltransferases and deacetylases in an NF-κB-signaling code. Trends Biochem Sci. 2008;33(7):339–349. | ||
Chen L-F, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. Embo J. 2002;21(23):6539–6548. | ||
Kiernan R, Brès V, Ng RWM, et al. Post-activation turn-off of NF-κB-dependent transcription is regulated by acetylation of p65. J Biol Chem. 2003;278(4):2758–2766. | ||
Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 2004;23(12):2369–2380. | ||
Buerki C, Rothgiesser KM, Valovka T, et al. Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of RelA/p65. Nucleic Acids Res. 2008;36(5):1665–1680. | ||
Huang B, Yang X-D, Zhou M-M, Ozato K, Chen L-F. Brd4 coactivates transcriptional activation of NF- B via specific binding to acetylated RelA. Mol Cell Biol. 2009;29(5):1375–1387. | ||
Kim J-W, Jang S-M, Kim C-H, An J-H, Kang E-J, Choi K-H. New molecular bridge between RelA/p65 and NF-κB target genes via histone acetyltransferase TIP60 cofactor. J Biol Chem. 2012;287(10):7780–7791. | ||
Lu T, Stark GR. NF-κB: regulation by methylation. Cancer Res. 2015;75(18):3692–3695. | ||
Lu T, Stark GR. Using sequential immunoprecipitation and mass spectrometry to identify methylation of NF-kappaB. Methods Mol Biol. 2015;1280:383–393. | ||
Ck E, Baltimore D. Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc Natl Acad Sci U S A. 2009;106:18972–18977. | ||
Wei H, Wang B, Miyagi M, et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF- B. Proc Natl Acad Sci U S A. 2013;110(33):13516–13521. | ||
Yang X-D, Huang B, Li M, Lamb A, Kelleher NL, Chen L-F. Negative regulation of NF-κB action by Set9-mediated lysine methylation of the RelA subunit. Embo J. 2009;28(8):1055–1066. | ||
Lu T, Jackson MW, Wang B, et al. Regulation of NF- B by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc Natl Acad Sci U S A. 2010;107(1):46–51. | ||
Quivy V, van Lint C. Regulation at multiple levels of NF-κB-mediated transactivation by protein acetylation. Biochem Pharmacol. 2004;68(6):1221–1229. | ||
Bhatt D, Ghosh S. Regulation of the NF-κB-mediated transcription of inflammatory genes. Front Immunol. 2014;5:71. | ||
Chen L-F, Williams SA, Mu Y, et al. NF- B RelA phosphorylation regulates RelA acetylation. Mol Cell Biol. 2005;25(18):7966–7975. | ||
Gao J, Xu D. Correlation between posttranslational modification and intrinsic disorder in protein. Pac Symp Biocomput. 2012:94–103. | ||
Srinivasan M, Dunker AK. Proline rich motifs as drug targets in immune mediated disorders. Int J Pept. 2012;2012(7):Article ID 634769;14pp. | ||
Takada Y, Singh S, Aggarwal BB. Identification of a p65 peptide that selectively inhibits NF-kappa B activation induced by various inflammatory stimuli and its role in down-regulation of NF-kappaB-mediated gene expression and up-regulation of apoptosis. J Biol Chem. 2004;279(15):15096–15104. | ||
Macara IG. Transport into and out of the nucleus. Microbiol Mol Biol Rev. 2001;65(4):570–594. | ||
Chew J, Biswas S, Shreeram S, et al. WIP1 phosphatase is a negative regulator of NF-κB signalling. Nat Cell Biol. 2009;11(5):659–666. | ||
Yang J, Fan GH, Wadzinski BE, Sakurai H, Richmond A. Protein phosphatase 2A interacts with and directly dephosphorylates RelA. J Biol Chem. 2001;276(51):47828–47833. | ||
Yeh PY, Yeh KH, Chuang SE, Song YC, Cheng AL. Suppression of MEK/ERK signaling pathway enhances cisplatin-induced NF-kappaB activation by protein phosphatase 4-mediated NF-kappaB p65 Thr dephosphorylation. J Biol Chem. 2004;279(25):26143–26148. | ||
Lu T, Yang M, Huang DB, et al. Role of lysine methylation of NF-κB in differential gene regulation. Proc Natl Acad Sci U S A. 2013;110(33):13510–13515. | ||
Geng H, Wittwer T, Dittrich-Breiholz O, Kracht M, Schmitz ML. Phosphorylation of NF-κB p65 at Ser468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep. 2009;10(4):381–386. | ||
Buss H, Dörrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem. 2004;279(53):55633–55643. | ||
Calzado M, Bacher S, Schmitz ML. NF-kappaB inhibitors for the treatment of inflammatory diseases and cancer. Curr Med Chem. 2007;14(3):367–376. | ||
Gasparini C, Feldmann M. NF-kappaB as a target for modulating inflammatory responses. Curr Pharm Des. 2012;18(35):5735–5745. | ||
Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010;1799:775–787. | ||
Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. | ||
Uwe S. Anti-inflammatory interventions of NF-kappaB signaling: potential applications and risks. Biochem Pharmacol. 2008;75(8):1567–1579. | ||
Uehling DE, Harris PA. Recent progress on MAP kinase pathway inhibitors. Bioorg Med Chem Lett. 2015;25(19):4047–4056. | ||
Saha RN, Jana M, Pahan K. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J Immunol. 2007;179(10):7101–7109. | ||
Alam J, Blackburn K, Patrick D. Neflamapimod: clinical phase 2b-ready oral small molecule inhibitor of p38alpha to reverse synaptic dysfunction in early Alzheimer’s disease. J Prev Alzheimers Dis. 2017;4:273–278. | ||
Bühler S, Laufer SA. p38 MAPK inhibitors: a patent review (2012 – 2013. Expert Opin Ther Pat. 2014;24(5):535–554. | ||
Fujihara SM, Cleaveland JS, Grosmaire LS, et al. A D-amino acid peptide inhibitor of NF-kappa B nuclear localization is efficacious in models of inflammatory disease. J Immunol. 2000;165(2):1004–1012. | ||
Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem. 1995;270(24):14255–14258. | ||
Wang YF, Xu X, Fan X, et al. A cell-penetrating peptide suppresses inflammation by inhibiting NF-κB signaling. Mol Ther. 2011;19(10):1849–1857. | ||
Nadler SG, Tritschler D, Haffar OK, Blake J, Bruce AG, Cleaveland JS. Differential expression and sequence-specific interaction of karyopherin alpha with nuclear localization sequences. J Biol Chem. 1997;272(7):4310–4315. | ||
van der Watt PJ, Chi A, Stelma T, et al. Targeting the nuclear import receptor Kpn 1 as an anticancer therapeutic. Mol Cancer Ther. 2016;15(4):560–573. | ||
Kashyap T, Argueta C, Aboukameel A, et al. Selinexor, a selective inhibitor of nuclear export (SINE) compound, acts through NF-kappaB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death. Oncotarget. 2016;7(48):78883–78895. | ||
Stelma T, Leaner VD. KPNB1-mediated nuclear import is required for motility and inflammatory transcription factor activity in cervical cancer cells. Oncotarget. 2017;8(20):32833–32847. | ||
Oliva B, Fernandez-Fuentes N. Knowledge-based modeling of peptides at protein interfaces: PiPreD. Bioinformatics. 2015;31(9):1405–1410. | ||
Srinivasan M. Interface peptide mimetics: rationale and applications as therapeutic agents. Med Chem (Los Angeles). 2016;6(3):189–194. | ||
Rao NAS, Mccalman MT, Moulos P, et al. Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res. 2011;21(9):1404–1416. | ||
Yamamoto KR. Steroid receptor regulated transcription of specific genes and gene networks. Annu Rev Genet. 1985;19(1):209–252. | ||
Riccardi C. GILZ (glucocorticoid-induced leucine zipper), a mediator of the anti-inflammatory and immunosuppressive activity of glucocorticoids. Ann Ig. 2010;22:53–59. | ||
Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. Faseb J. 2009;23(11):3649–3658. | ||
Srinivasan M, Bayon B, Chopra N, Lahiri DK. Novel nuclear factor-kappaB targeting peptide suppresses β-amyloid induced inflammatory and apoptotic responses in neuronal cells. PLoS One. 2016;11(10):e0160314. | ||
Cubellis MV, Caillez F, Blundell TL, Lovell SC. Properties of polyproline II, a secondary structure element implicated in protein-protein interactions. Proteins. 2005;58(4):880–892. | ||
Srinivasan M, Blackburn C, Lahiri DK. Functional characterization of a competitive peptide antagonist of p65 in human macrophage-like cells suggests therapeutic potential for chronic inflammation. Drug Des Devel Ther. 2014;8:2409–2421. | ||
Srinivasan M, Janardhanam S. Novel p65 binding glucocorticoid-induced leucine zipper peptide suppresses experimental autoimmune encephalomyelitis. J Biol Chem. 2011;286(52):44799–44810. | ||
O’Shea JM, Perkins ND. Regulation of the RelA (p65) transactivation domain. Biochem Soc Trans. 2008;36(4):603–608. | ||
Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci U S A. 1997;94(7):2927–2932. | ||
Lee SK, Kim JH, Lee YC, Cheong J, Lee JW. Silencing mediator of retinoic acid and thyroid hormone receptors, as a novel transcriptional corepressor molecule of activating protein-1, nuclear factor-kappaB, and serum response factor. J Biol Chem. 2000;275(17):12470–12474. | ||
Dennler S, Dijke T. Smad proteins in TGF-beta signaling. In: Schwab M, editor. Encyclopedia of Cancer. Berlin: Springer; 2011. | ||
Hirata-Tsuchiya S, Fukushima H, Katagiri T, et al. Inhibition of BMP2-induced bone formation by the p65 subunit of NF-κB via an interaction with Smad4. Mol Endocrinol. 2014;28(9):1460–1470. | ||
Urata M, Kokabu S, Matsubara T, et al. A peptide that blocks the interaction of NF-κB p65 subunit with Smad4 enhances BMP2-induced osteogenesis. J Cell Physiol. 2018;233(9):7356–7366. | ||
Hecker M, Wagner AH. Transcription factor decoy technology: a therapeutic update. Biochem Pharmacol. 2017;144:29–34. | ||
Morishita R, Sugimoto T, Aoki M, et al. In vivo transfection of cis element “decoy” against nuclear factor-kappaB binding site prevents myocardial infarction. Nat Med. 1997;3(8):894–899. | ||
Sawa Y, Morishita R, Suzuki K, et al. A novel strategy for myocardial protection using in vivo transfection of cis element ‘decoy’ against NFkappaB binding site: evidence for a role of NFkappaB in ischemia-reperfusion injury. Circulation. 1997;96(9 Suppl):280–284. | ||
Dajee M, Muchamuel T, Schryver B, et al. Blockade of experimental atopic dermatitis via topical NF-kappaB decoy oligonucleotide. J Invest Dermatol. 2006;126(8):1792–1803. | ||
de Stefano D. Oligonucleotides decoy to NF-kappaB: becoming a reality? Discov Med. 2011;12(63):97–105. | ||
Tahara K, Samura S, Tsuji K, et al. Oral nuclear factor-κB decoy oligonucleotides delivery system with chitosan modified poly(D,L-lactide-co-glycolide) nanospheres for inflammatory bowel disease. Biomaterials. 2011;32(3):870–878. | ||
Farahmand L, Darvishi B, Majidzadeh-A K. Suppression of chronic inflammation with engineered nanomaterials delivering nuclear factor κB transcription factor decoy oligodeoxynucleotides. Drug Deliv. 2017;24(1):1249–1261. | ||
Tamai K, Kaneda Y, Morishita R, Katayama I. Development of NF-kappa B decoy ointment and clinical trial for atopic dermatitis. Dermatitis. 2008;19:293–294. | ||
Doucas V, Shi Y, Miyamoto S, West A, Verma I, Evans RM. Cytoplasmic catalytic subunit of protein kinase A mediates cross-repression by NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci U S A. 2000;97(22):11893–11898. | ||
Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci U S A. 1994;91(2):752–756. | ||
Lesovaya E, Yemelyanov A, Swart AC, Swart P, Haegeman G, Budunova I. Discovery of compound A--a selective activator of the glucocorticoid receptor with anti-inflammatory and anti-cancer activity. Oncotarget. 2015;6(31):30730–30744. | ||
Schäcke H, Berger M, Rehwinkel H, Asadullah K. Selective glucocorticoid receptor agonists (SEGRAs): novel ligands with an improved therapeutic index. Mol Cell Endocrinol. 2007;275(1–2):109–117. | ||
Sundahl N, Bridelance J, Libert C, de Bosscher K, Beck IM. Selective glucocorticoid receptor modulation: new directions with non-steroidal scaffolds. Pharmacol Ther. 2015;152:28–41. | ||
Carson MW, Luz JG, Suen C, et al. Glucocorticoid receptor modulators informed by crystallography lead to a new rationale for receptor selectivity, function, and implications for structure-based design. J Med Chem. 2014;57(3):849–860. | ||
Stock T, Fleishaker D, Wang X, Mukherjee A, Mebus C. Improved disease activity with fosdagrocorat (PF-04171327), a partial agonist of the glucocorticoid receptor, in patients with rheumatoid arthritis: a Phase 2 randomized study. Int J Rheum Dis. 2017;20(8):960–970. | ||
Catley M. Dissociated steroids. ScientificWorldJournal. 2007;7:421–430. | ||
Park SY, Kim MJ, Kim YJ, et al. Selective PCAF inhibitor ameliorates cognitive and behavioral deficits by suppressing NF-κB-mediated neuroinflammation induced by Aβ in a model of Alzheimer’s disease. Int J Mol Med. 2015;35(4):1109–1118. | ||
van den Bosch T, Leus NGJ, Wapenaar H, et al. A 6-alkylsalicylate histone acetyltransferase inhibitor inhibits histone acetylation and pro-inflammatory gene expression in murine precision-cut lung slices. Pulm Pharmacol Ther. 2017;44:88–95. | ||
Legartová S, Stixová L, Strnad H, et al. Basic nuclear processes affected by histone acetyltransferases and histone deacetylase inhibitors. Epigenomics. 2013;5(4):379–396. | ||
Xu J, Sun J, Wang P, Ma X, Li S. Pendant HDAC inhibitor SAHA derivatised polymer as a novel prodrug micellar carrier for anticancer drugs. J Drug Target. 2018;26(5–6):448–457. | ||
Li X, Zhang J, Xie Y, Jiang Y, Yingjie Z, Xu W. Progress of HDAC inhibitor panobinostat in the treatment of cancer. Curr Drug Targets. 2014;15(6):622–634. | ||
Gao S, Li X, Zang J, Xu W, Zhang Y. Preclinical and clinical studies of chidamide (CS055/HBI-8000), an orally available subtype-selective HDAC inhibitor for cancer therapy. Anticancer Agents Med Chem. 2017;17(6):802–812. | ||
Leus NG, van den Bosch T, van der Wouden PE, et al. HDAC1-3 inhibitor MS-275 enhances IL10 expression in RAW264.7 macrophages and reduces cigarette smoke-induced airway inflammation in mice. Sci Rep. 2017;7:45047. | ||
Nencioni A, Beck J, Werth D, et al. Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin Cancer Res. 2007;13(13):3933–3941. | ||
Choi Y, Park SK, Kim HM, et al. Histone deacetylase inhibitor KBH-A42 inhibits cytokine production in RAW 264.7 macrophage cells and in vivo endotoxemia model. Exp Mol Med. 2008;40(5):574–581. | ||
Leus NG, Zwinderman MR, Dekker FJ. Histone deacetylase 3 (HDAC 3) as emerging drug target in NF-κB-mediated inflammation. Curr Opin Chem Biol. 2016;33:160–168. | ||
Winkler AR, Nocka KN, Williams CM. Smoke exposure of human macrophages reduces HDAC3 activity, resulting in enhanced inflammatory cytokine production. Pulm Pharmacol Ther. 2012;25(4):286–292. | ||
Leus NG, van der Wouden PE, van den Bosch T, et al. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-κB p65 transcriptional activity. Biochem Pharmacol. 2016;108:58–74. | ||
Chen D, Zeng S, Huang M, Xu H, Liang L, Yang X. Role of protein arginine methyltransferase 5 in inflammation and migration of fibroblast-like synoviocytes in rheumatoid arthritis. J Cell Mol Med. 2017;21(4):781–790. | ||
Webb LM, Amici SA, Jablonski KA, et al. PRMT5-selective inhibitors suppress inflammatory T cell responses and experimental autoimmune encephalomyelitis. J Immunol. 2017;198(4):1439–1451. | ||
Srinivasan M, Lahiri DK. Significance of NF-κB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer’s disease and multiple sclerosis. Expert Opin Ther Targets. 2015;19(4):471–487. | ||
Kanazawa T, Hamasaki T, Endo T, et al. Functional peptide nanocarriers for delivery of novel anti-RelA RNA interference agents as a topical treatment of atopic dermatitis. Int J Pharm. 2015;489(1–2):261–267. | ||
Deci MB, Liu M, Dinh QT, Nguyen J. Precision engineering of targeted nanocarriers. Wiley Interdiscip Rev Nanomed Nanobiotechnol. Epub 2018 Feb 13. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.