")
Back to Journals » OncoTargets and Therapy » Volume 11
Mechanisms, monitoring, and management of tyrosine kinase inhibitors-associated cardiovascular toxicities
Authors Chaar M , Kamta J , Ait-Oudhia S
Received 4 April 2018
Accepted for publication 19 July 2018
Published 25 September 2018 Volume 2018:11 Pages 6227—6237
DOI https://doi.org/10.2147/OTT.S170138
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Maher Chaar,* Jeff Kamta,* Sihem Ait-Oudhia
Center for Pharmacometrics and Systems Pharmacology, Department of Pharmaceutics, College of Pharmacy, University of Florida, Orlando, FL, USA
*These authors contributed equally to this work
Abstract: The tyrosine kinase inhibitor (TKI) drug class is a prominently used option in the treatment of various cancers. Safety evaluation of these drugs has shown evidence of cardiotoxicity of varying frequency and severity between agents; concern has led to updated labeling, warning prescribers of such. This review seeks to clarify the present dangers and investigate cardiotoxic mechanisms of action for each discussed TKI. Dasatinib was connected primarily with an incidence of fluid retention, edema, QT prolongation, and pulmonary hypertension in clinical studies. It is theorized that this is due to a combination of off-target kinase binding and on-target binding of Bcr-Abl, and less likely, mitochondrial induced apoptosis. Studies showed sorafenib to carry the risk of hypertension, QT prolongation, and myocardial infarction. Proposed mechanisms for these side effects include inhibition of proteins, vascular endothelium growth factor receptor, hERG potassium channels, and the RAF/MERK/ERK pro-survival pathway. Finally, lapatinib showed evidence of decreased left ventricular ejection fraction (LVEF) and QT prolongation in clinical studies. The literature attributes these as side effects of on-target ErbB2 binding leading to mitochondrial induced apoptosis. The concern warranted by these findings is in question. Pooled safety data suggest that the overall risk for cardiotoxicity is minimal in dasatinib and lapatinib. Sorafenib seems to carry a moderate concern. For the discussed agents, recommendations agree that routine monitoring via methods such as electroencephalogram, cardiac biomarkers, and blood pressure is warranted during the course of treatment, in addition to a comprehensive collection of past medical history and risk factors to identify those at heightened risk for cardiovascular events.
Keywords: cardiovascular toxicity, tyrosine kinase inhibitors, dasatinib, sorafenib, lapatinib
Introduction
Kinases are a class of enzyme that mediates phosphate transfer from adenosine triphosphate (ATP) onto certain amino acid residues to produce cell signal transduction resulting in a range of cellular processes. The discovery of their overexpression in various cancers, particularly the receptor tyrosine kinase subtype, has led to the development of several tyrosine kinase inhibitors (TKIs). Their binding to TKIs is usually via competitive inhibition at the ATP binding pocket, stopping cell proliferation signaling. The vast interplay in the resultant network of cell signaling is currently being studied in the context of homeostatic cardiac function. Limited but crucial evidence suggests that adverse effects of the TKI class include cardiotoxicity, dysfunction, or damage to cardiomyocytes which can manifest clinically as a multitude of cardiovascular complications. A high variability of selectivity between agents, and off-target kinase binding are characteristics of the drug class impeding our understanding of the significance of the threat in each agent.1 Understanding the context of the clinical use of these drugs reveals the delicacy of this issue. Risk–benefit analysis is inherently more tolerant in the chemotherapeutics setting, and the use of other cardiotoxic drugs (particularly the anthracycline class) in a given regimen is likely. Clarification of the underlying mechanism of these side effects will be needed as concern grows and the capacity to anticipate them is lacking. Preclinical studies often lack the follow-up time needed to observe the development of long-term cardiac side effects or fail to capture their presence at all.2 The interplay between these side effects and patient baseline risk factors further complicates studies. Pre-existing cardiac disease, hypertension, diabetes, and hyperlipidemia are major diagnoses that can contribute to cardiotoxicity. Factors such as family history, activity level, smoking status, and alcohol intake are more difficult to capture, not always accounted for, and can confound study results.
Proposed mechanisms of cardiotoxicity vary and appear to be drug-specific. Disruption of mitochondrial function within the cardiomyocyte has been implicated; several off-target kinases such as c-Jun N-terminal kinase, pyruvate dehydrogenase kinase, and protein kinase A are possible targets that when inhibited can interrupt oxidative phosphorylation leading to morphological abnormalities of the mitochondria and hypertrophy of the cardiomyocyte itself as the cell increases dependence on anaerobic metabolism.3 Caspase-mediated mitochondrial apoptosis seems to be an additional consequence.4 Knockout mouse models have revealed further possible targets and have taught much about the complexity of cardiac cell signaling. Knockout of platelet-derived growth factor receptor (PDGFR), ErbB2, Raf-1, and Shp2 have all shown cardiotoxicity with a common theme of cardiomyopathy and reduced contractility.1 The downstream effects of some of these kinases appear to play a role in ion channel activation. Reduced phosphorylation of the hERG potassium channel in particular has been explored;5 ion channel blockade manifests as QT prolongation in the clinic, which has been connected with the use of some TKIs. It is possible that observed cardiac adverse effects occur secondary to TKI binding in the vasculature. Inhibition of the vascular endothelium growth factor receptor (VEGFR) is a mechanism of some agents with the intention to block tumor angiogenesis, but which can also lead to the development of hypertension.
Selected TKIs
Dasatinib
Dasatinib (Sprycel®; Bristol-Myers Squibb, New York, NY, USA) is a TKI approved for the treatment of chronic myeloid leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) following imatinib treatment failure. Its range of targets include Bcr-Abl, c-Kit, PDGFR, and members of the Scr family (Table 1).6 Evidence of cardiotoxicity was seen early in clinical trials. In a Phase I dose escalation study of 84 patients, pleural effusion was seen in 15 (18%) patients and peripheral edema in 19% of patients.7 Phase II trials leading to the approval of dasatinib further indicated evidence of cardiotoxicity. A pooled analysis of these studies, all single-arm trials, examined safety outcomes in a combined 911 patients with a typical starting dose of 70 mg twice a day, for a median duration of 6 months.8 Among all trials, patients received electroencephalograms (ECG) on days 1 and 8 of treatment, and at the end. ECGs revealed statistically significant QTc prolongation of 3–6 ms.8 Clinical significance remains in question; <1% of these patients exceeded a clinically significant threshold of 500 ms, and only 2.9% experienced an elevation beyond a significant threshold of 60 ms from baseline. While 4% of patients experienced congestive heart failure or ventricular dysfunction, over half of these already had a history of cardiovascular disease.
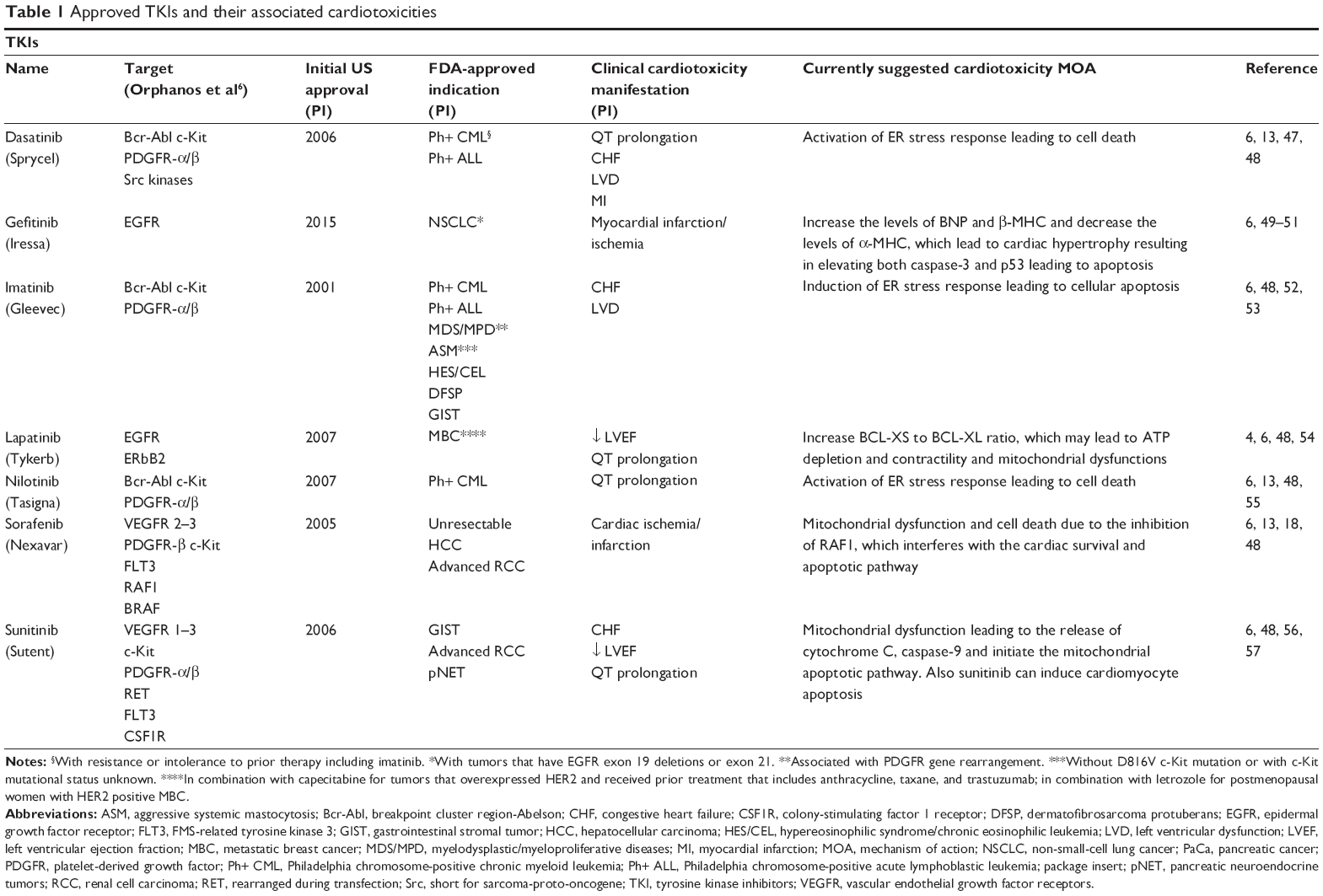 | Table 1 Approved TKIs and their associated cardiotoxicities |
A following Phase III dose optimization study of 670 patients treated with dasatinib 100 mg once daily saw no evidence of CHF and less than half the rate of pleural effusion (7% vs 16%) was seen in the pooled Phase II studies.9 Half of the patients among all Phase II trials experienced fluid retention of some kind, with 9% of these being grade 3 or 4. It is known that fluid retention can lead to edema; however, this consequence was not frequently seen; pulmonary edema and generalized edema each occurred at only 1%. The Phase III trial leading to the approval of dasatinib contrasts some of these findings and suggests that concern for cardiotoxicity is still warranted when compared to the standard of treatment.
In the DASISION trial, dasatinib 100 mg once daily and imatinib 400 mg once daily were compared, with 258 patients in each treatment arm. Both superficial edema and fluid retention were seen at higher rates in the imatinib treatment arm (36% vs 11% and 44% vs 27%, respectively). Dasatinib however did show a significantly higher rate of pleural effusion, 15% vs 0% in the imatinib arm.10 Thirty-six months into the follow-up for the DASISION trial, pulmonary arterial hypertension (PAH) was identified in 3% of patients on dasatinib and in no patients on imatinib. PAH had previously been implicated in the deaths of two patients undergoing dasatinib treatment,11 and the results from DASISION therefore prompted the US Food and Drug Administration (FDA) in 2011 to release a warning on the risk of PAH with dasatinib use. The READY trial was another head-to-head Phase III trial comparing dasatinib 100 mg once daily plus docetaxel 75 mg/m2 every 3 weeks vs docetaxel alone. Superficial edema occurred at a lower rate in the dasatinib-treated group (48% vs 62%) as did peripheral edema (44% vs 57%). Pleural effusion was seen at a higher rate in the dasatinib group (21% vs 7%) and led to treatment discontinuation in 2% of patients.12 These results seem to follow those of the DASISION trial, but the limitations must be acknowledged considering dasatinib was not used as monotherapy.
Mechanism of dasatinib cardiotoxicity
An in vitro study investigating cardiac toxicity among several TKI agents tested dasatinib treatment in H9c2 cardiac cells supplemented with either 25 mM glucose or 10 mM galactose. Cells grown in galactose were resultantly forced to subsist on oxidative phosphorylation instead of glycolysis, allowing for researchers to examine the role of mitochondrial TKR inhibition as a mechanism for dasatinib toxicity. Researchers found that the values of the inhibitory concentration leading to 50% decrease in the maximum effect (IC50) for dasatinib were equal amongst both treatment groups. Additionally, no change in mitochondrial oxygen consumption was observed in a separate experiment. The same study found dasatinib to inhibit complexes IV (cytochrome c oxidase) and V (human mitochondrial ATP synthase) of the electron transport chain, but at doses well above clinical.13 Taken together, these results suggest that adverse effects are not originating at the level of the mitochondria and are supported by a separate study comparing dasatinib and imatinib in terms of their effects on mitochondrial structure or apoptosis.5
It is likely that toxicity is due to receptor kinase binding both on- and off-target. This conclusion was reached in a study that treated neonatal rat cardiomyocytes with both tyrosine kinase and serine-threonine kinase inhibitors. Results showed that a lack of target selectivity was correlated with myocyte damage, but a correlation also existed with the strength of on-target Kd (dissociation constant).14 The study used lactate dehydrogenase (LDH) enzyme as a surrogate for myocyte damage, which is also used clinically as a marker for tissue damage in the heart. Results showed dasatinib to induce a greater percentage LDH release in both 2 and 5 μM concentrations tested compared to imatinib; this trends with the relative drug potencies as dasatinib binds about 325 times stronger than imatinib.15 This mixed view was supported by a later follow-up study using the same neonatal rat cardiomyocyte model, again identifying poor selectivity among TKIs as a driver for damage, and also pointing to the potency of on-target ABL1 inhibition as a simultaneous contributor.16 It has long been theorized that on-target inhibition of the ABL protein can be a cause of TKI-induced cardiotoxicity, when such adverse effects were seen in clinical trials with imatinib. Conferring imatinib resistance in cardiomyocytes has been shown to prevent the onset of toxicity, and the findings have therefore been extrapolated to dasatinib since it shares the Bcr-Abl target (Figure 1). However, in contrast to the above previous findings with dasatinib, imatinib-induced cardiotoxicity is thought to occur at the level of the mitochondria, resulting in apoptosis.4
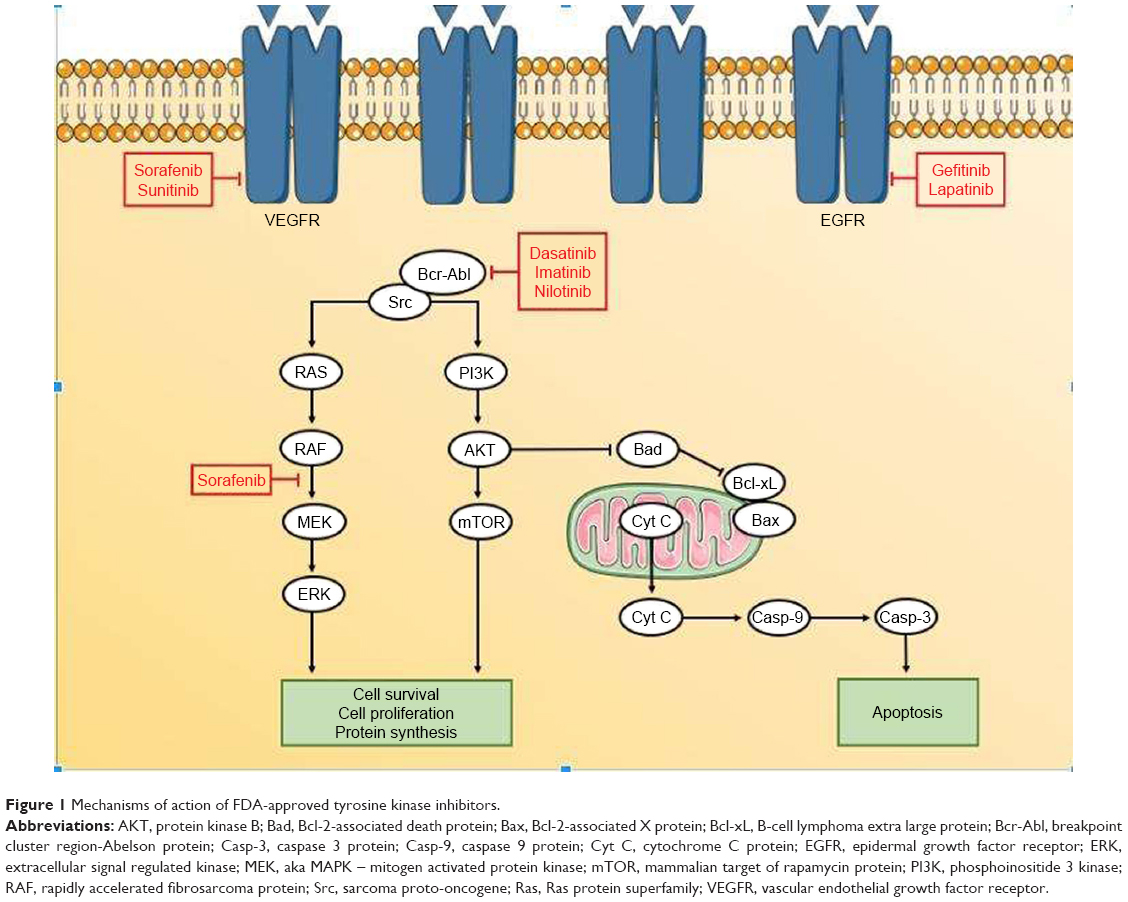 | Figure 1 Mechanisms of action of FDA-approved tyrosine kinase inhibitors. |
Management and monitoring of dasatinib cardiotoxicity
The safety outcomes from various clinical trials testing dasatinib have reported mixed results about the significance of cardiotoxicity. Hence, a conservative approach has so far been taken in the use of this drug. Product labeling currently includes warnings in reference to fluid retention, cardiac ischemia, PAH, and QT prolongation. Interestingly, a statistical evaluation of cardiovascular ischemia in dasatinib-treated patients using standardized incidence ratios suggests that dasatinib does not significantly increase the risk for cardiovascular ischemia when compared to similar patient populations. The study pooled safety outcomes from the DASISION trial, READY trial, and 11 Phase I and II trials. Researchers found the incidence of ischemia to be 2%–4%; however, the majority of these patients had a history of or risk factors for atherosclerosis. When adjusted for age and sex, dasatinib-treated patients did not have a significantly higher risk when compared to CML and prostate cancer control populations as observed in publicly available databases, and there was no cumulative risk dependent on the length of dasatinib treatment (most events occurred within the first year of treatment).17
In deciding how to manage the possibility of dasatinib-induced cardiotoxicity, Saglio et al agreed with the recommendations of many of the above-cited trials and the current FDA labeling, which at this time find it sufficient to recommend caution when using dasatinib concomitantly with cardiotoxic therapies or in patients with a history of cardiovascular disease, and do suggest monitoring of cardiac function during therapy.17 These sources do not mention the need for cardiac evaluation in the absence of the above risk factors before starting treatment. The aforementioned 2011 warning on PAH similarly advised healthcare providers to evaluate patients for underlying cardiopulmonary disease before starting treatment with dasatinib and to monitor for signs and symptoms of PAH during treatment (dyspnea, hypoxia, and fluid retention). This condition is considered reversible upon stopping treatment, and therefore the warning suggests an interruption of treatment when these symptoms manifest and PAH is suspected, and a permanent discontinuation if PAH is confirmed through right heart catheterization.
Sorafenib
Sorafenib (Nexavar®; Bayer HealthCare Pharmaceuticals, Inc, Wayne, NJ, USA) is a TKI approved for the treatment of unresectable hepatocellular carcinoma and advanced renal cell carcinoma (RCC). It is a multi-kinase inhibitor that binds VEGFR-2 and 3 kinases, PDGFR, c-Kit, RAF, and BRAF (Table 1).18 Like dasatinib, the drug labeling of sorafenib contains a warning about the potential for cardiotoxicity, based on a small incidence seen throughout several studies. Phase I and II clinical trials of sorafenib were mostly underwhelming in the occurrence of these side effects. One Phase II trial treating 34 patients with sorafenib 400 mg twice daily reported two patients who developed hypertension during the course of treatment.19 A separate Phase II trial with 137 patients under the same treatment did not report any significant cardiovascular related events,20 and a third Phase II trial of 54 patients under the same treatment reported two patients developing grade 3 hypertension and one patient developing a nonfatal myocardial infarction.21 A pooled analysis of four Phase I dose escalation clinical trials totaling 173 patients showed the incidence of hypertension ranging from 5%–11%, with up to 5% of these being grade 3 or higher.22 The propensity of sorafenib to lead to hypertension was seen in a placebo-controlled Phase III clinical trial. Seventy-six (16.8%) of 451 patients on sorafenib 400 mg twice daily developed hypertension, with 16 of these being grade 3 or 4, these rates being significantly higher than placebo. Additionally, 12 patients in this study experienced cardiac ischemia or myocardial infarction.23 As a VEGFR inhibitor, the propensity of sorafenib to induce thromboembolic events has also been studied in a meta-analysis of ten trials totaling 10,255 patients. Sorafenib therapy was found to have a relative risk of 3.03 for the development of these events when compared to placebo in patients with the same malignancies and baseline cardiovascular health.24
Another placebo-controlled Phase III clinical trial of sorafenib 400 mg twice daily reported five (1.7%) of 297 patients having developed hypertension (significant compared to placebo), with two of these being grade 3 or 4 (not at significance). Myocardial infarction and cardiac ischemia were reported in 3% of treated patients, but this was not at significance compared to placebo.25 Schmidinger et al26 conducted an observational study investigating the risk of cardiotoxicity of sunitinib and sorafenib in 74 patients and identified 13 (17.5%) patients treated with sorafenib 800 mg/day who experienced a cardiac event, defined as elevation of cardiac enzymes compared to baseline (creatine kinase-MB [CK-MB], troponin T [TnT]), symptomatic arrhythmia requiring treatment, new left ventricular dysfunction, or acute coronary syndrome. Of these 13 patients, seven required medical intervention and all the treated patients have made recoveries.26 Also, six of the 13 presented with ECG abnormalities (one of which entered the study with a pre-existing arrhythmia). Past medical histories of cardiac dysfunction or hypertension might have contributed to the toxicities seen in this study. Of importance, all symptomatic patients were effectively managed and eligible to continue treatment. This study and the accompanying clinical trials highlight the significance of monitoring patients on sorafenib therapy for any cardiovascular changes, in accordance with the product labeling, despite these events being infrequent; quickly identifying and resolving these side effects will reduce the chance of treatment being permanently discontinued.
Mechanism of sorafenib cardiotoxicity
Numerous reports of hypertension in the above clinical trials can be easily attributed to sorafenib VEGFR inhibition. Blocking the actions of VEGFR stops angiogenesis and vasodilatory processes, leads to an increase in vascular resistance, and has been observed in other chemotherapeutics using this target.27 While not doing direct damage to the heart, hypertension is a well-known precursor to more serious heart conditions and therefore its contribution to cardiotoxicity weighs heavily. Impairment of endothelial cell survival would additionally explain the findings of increased risk of thromboembolic events with sorafenib, as a consequence of vascular injury. Further explanation for the cause of hypertension is seen in a study of 57 patients treated with sorafenib or sunitinib that found significant increases in measured pulse wave velocity (PWV), a surrogate for arterial stiffness, when corrected for blood pressure. The increases found were rapid and large. Authors theorized that TKIs might interact with integrins and thereby components of arterial structure, reducing elasticity. Another suggestion was damage to the vasa-vasorum, which has been linked with PWV increases.28 Although this study was small and featured pooled results, increases in arterial stiffness have been suggested elsewhere as a class effect of anti-angiogenic medications.29
In a preclinical study on dogs, sorafenib disturbed action potentials in the hERG potassium channel of selected purkinje fibers, although without any clinical manifestation.1 Inhibition of the hERG channel has been explored in TKIs dasatinib, nilotinib, and imatinib as a cause of arrhythmias. Although sorafenib has not been tested specifically, it shares kinase targets with these agents which raises the possibility that sorafenib may affect hERG as well.5 RAF1 is a kinase mediating a major pro-survival pathway in the cell, and its inhibition by sorafenib has also been explored as a contributor to observed toxicities. Although its role in cardiac function has not been clearly defined, RAF1 knockout mouse models have demonstrated reduced contractility and fibrosis in the heart. It is theorized however that these effects are due to downstream pro-apoptotic factors ASK1 and MST2, and these factors do not seem to rely solely on RAF for normal inhibition.4 This conclusion is supported by another study in which cardiomyocytes treated with sorafenib did not show a significant change in downstream ERK (of the RAF/MEK/ERK pro-survival pathway) phosphorylation (Figure 1).14 Should RAF-1 inhibition have a role, the toxicity seen would be an end result of mitochondrial mediated apoptosis. Whether RAF inhibition itself is a cause of direct cardiomyocyte damage is yet unclear; therefore, another contributory mechanism could be considered which increases the likelihood of cardiac dysfunction or worsens conditions when they do occur. In the same manner, c-Kit, another sorafenib target, seems to have a role in repairing damage to the heart caused by ischemic injury.4 The same study to investigate ERK phosphorylation in regard to sorafenib examined caspase activity in treated cardiomyocytes and found no significant increase either, ruling out the possibility of mitochondrial mediated apoptosis as a mechanism. Supplementation with dexrazoxane also showed no change in cell death. Dexrazoxane is a drug used to combat cardiotoxicity due to doxorubicin-induced reactive oxygen species (ROS).4 This would seem to eliminate oxidative stress as a mechanism for sorafenib as well, although this conclusion is opposed by the results of a study conducting a transcriptome analysis of sorafenib-treated zebrafish. These authors observed a reduction in STC1 protein, a regulator of calcium homeostasis, accompanied by an increase in ROS generation; results were confirmed in vitro using human cardiomyocytes. It is possible that the origin of ROS generation determines whether oxidative stress contributes to sorafenib-induced cardiotoxicity. Taken together, these studies suggest that like dasatanib, sorafenib-induced cardiotoxicity cannot be pinned upon a singular mechanism. Kinase inhibition both on- and off-target is likely to blame, with an emphasis on inhibition of VEGFR leading to frequently seen hypertension. One of the earlier TKIs, sorafenib features poor selectivity within the TKI class.30 Identifying the kinases at fault for toxicities seen in the clinic among the hundreds of possibilities is a very difficult task. At the moment, it might suffice for clinicians to simply familiarize themselves with the phenotypes of the cardiotoxic mechanism of action, such as hypertension, and work to predict and manage them.
Management and monitoring of sorafenib cardiotoxicity
Patients who developed symptoms of cardiotoxicity in the previously mentioned Schmidinger et al26 study were all effectively managed and able to continue treatment. As discussed, risk factors and pre-existing conditions may have played a role in the development of these toxicities. This study serves as a good example that, like dasatinib, careful cardiac monitoring during the course of therapy is likely the current best option for prevention of adverse drug effects, in addition to a proper collection of past medical history. The Schmidinger study defined a cardiac event in such a way as to identify markers of ischemia or cardiac cell damage before a more serious pathology could present. This was achieved through monitoring of biomarkers CK-MB and TnT bi-monthly, ECG once monthly as needed, blood pressure measurements three times a day as needed, and echocardiograms at baseline and as needed.26 This vigilant monitoring likely prevented the development of serious cardiac dysfunction in patients and therefore allowed them to remain eligible for continuation of TKI therapy. While effective, these methods might not be practical to repeat in a normal clinical setting, but should serve as examples as to the laboratory tests that would be appropriate for cardiac safety monitoring during sorafenib treatment and the time intervals for their measurement.
Interestingly, the authors also discussed the utility of a prophylactic regimen of an angiotensin-converting enzyme inhibitor, beta blocker, and statin for cardioprotection before initiating the TKI therapy. This could also be considered especially in patients with risk factors. Specific agents include carvedilol and simvastatin, which have evidence that they protect the cardiomyocyte at the level of the mitochondria, in addition to their acknowledged mechanisms of action.26 The product labeling for sorafenib suggests similar precautions as seen in the Schmidinger study. It suggests weekly blood pressure monitoring during the first 6 weeks of therapy, and electrolyte and ECG monitoring for patients at risk for QT prolongation (those with pre-existing arrhythmias or on concomitant QT prolonging drugs). The development of hypertension is not an indicator to interrupt therapy as it can be appropriately managed with drug therapy, or prophylactically as discussed above. The labeling also suggests temporary or permanent discontinuation in patients who develop cardiac ischemia, myocardial infarction (MI), uncontrolled hypertension, or QT prolongation over 500 ms or over 60 ms from baseline.18 Management of thromboembolic events is less proactive. Primary prevention for thromboembolic events outside the inpatient setting is typically limited to antiplatelet therapy for high-risk patients with atherosclerotic cardiovascular disease. Increased contributions to thromboembolic risk from both TKI therapy and inherently due to malignancy could be factors that prompt clinicians to initiate primary prevention in patients who otherwise would not be considered and should be evaluated on a case-by-case basis. Studies evaluating the safety and efficacy of such therapy in this patient population would serve as valuable additions to current literature. Otherwise, thromboembolic events are managed based on symptoms. Following the occurrence of an event, prophylactic anticoagulation becomes an important part of treatment to avoid recurrence. The administration of TKI therapy can be a factor that prompts clinicians to treat these patients with extended prophylaxis (>3 months).31
Lapatinib
Lapatinib (Tykerb®; GlaxoSmithKline, Research Triangle Park, NC, USA) is approved, in combination with capecitabine,32 for the treatment of advanced or metastatic breast cancer (MBC) in overexpressed HER2 patients who received a prior treatment that included an anthracycline, a taxane, and trastuzumab (Table 1). It is also approved for postmenopausal women, in combination with letrozole, in treating HER2-positive MBC.33 Lapatinib is a dual kinase inhibitor for both endothelium growth factor receptor (EGFR or ERbB1) and HER2 (ERbB2).34 The emergence of cardiotoxicity with trastuzumab, the standard antibody therapy for HER2-positive MBC, prompted early investigation into lapatinib for the same adverse effects, although the data do not appear to support serious risk.35 A pooled analysis of 3,558 patients in 18 Phase I to Phase III trials was conducted to evaluate lapatinib for cardiac safety. Lapatinib was used as monotherapy or in combination with other chemotherapeutics, and in some patients following treatment with agents such as trastuzumab or an anthracycline. The analysis identified a decrease in left ventricular ejection fraction (LVEF) in 1.6% of patients, with only 0.2% of this being symptomatic. These rates are lower than that would be expected in trastuzumab-treated patients.36 The results suggest a niche in the therapy of lapatinib for HER2-positive patients with a risk for cardiovascular complications.
Another Phase III study evaluated lapatinib in combination with capecitabine in HER2-positive, locally advanced, or MBC patients. In this study, patients who were previously treated with an anthracycline, a taxane, and trastuzumab were randomized to receive a combination therapy of 1,250 mg/day lapatinib plus 2,000 mg/m2/day capecitabine or capecitabine monotherapy. The only cardiotoxicity seen was an asymptomatic decrease in LVEF in four women (2.5%) in the combination therapy group; this did not occur at a rate statistically greater than the monotherapy group and LVEF decreases did not require discontinuation of lapatinib.37 Similar results were seen in another Phase III trial that combined lapatinib 1,500 mg/day with paclitaxel 175 mg/m2 every 3 weeks. Six patients (2%) in the combination treatment arm experienced a decrease in LVEF, with five being asymptomatic and only three of the six being considered treatment related. None of the six patients required dosage adjustment.38
A subset analysis of the LEAP trial, an expansive global program treating patients with lapatinib 1,250 mg/day plus capecitabine 2,000 mg/m2 also agrees with the above studies. The subset involved 293 patients from 12 central and eastern European countries. Results showed 71 patients with an LVEF decrease of <20% from baseline, which was not outside institutional normal limits. Only three patients (1%) experienced a serious decline over 20% from baseline and below institutional normal limits. This low incidence was reflected in the global results, where only 21 of 4,283 (0.5%) patients met the same serious definition of LVEF decline.39 Similarity in results was seen in a Phase III trial treating 399 patients with the same aforementioned lapatinib plus capecitabine regimen; reversible LVEF decline was seen in 2% of patients.32
A more recently published Phase I dose escalation study saw no evidence of cardiotoxicity at all in the 40 patients evaluated, using measurements of ejection fraction and troponin 1.40 The recent Phase III ALTTO trial is perhaps the best evidence of the relatively low risk in lapatinib. The trial featured four study arms testing trastuzumab and lapatinib as monotherapies or in combination. The lapatinib monotherapy group followed safety events for 2,057 patients who were treated with lapatinib 1,500 mg/day for 52 weeks. Symptomatic heart failure was seen in only 2% of patients. LVEF decrease ≥10% and below the lower limit of normal was seen in 3% of patients.41 An anomaly to the trend set by these studies was seen in a small observational study treating patients with lapatinib 1,250 mg/day plus capecitabine 2,000 mg/m2. The study reported that five of 25 eligible patients experienced a cardiac event attributable to treatment, which included LVEF decline, QT prolongation, and sinus tachycardia.42
Mechanism of lapatinib cardiotoxicity
Data on the cellular mechanism of lapatinib cardiotoxicity are lacking, possibly because the threat is considered mild among the HER2 class of therapeutics. Presumptions are often drawn from studies on trastuzumab since it shares the ErbB2 target but is more widely studied and shows greater cardiotoxicity. For this reason, many studies seek to investigate why lapatinib appears safer rather than why it shows the minimal toxicity that it does. It is known that ErbB2 function is essential for cardiomyocyte survival and that its inhibition by trastuzumab in cardiomyocytes likely leads to reduced contractility and cell death via the BCL-X protein family, which causes mitochondrial induced apoptosis.43 Therefore, it is possible that lapatinib toxicity could also be a result of this mechanism.
A comparison of trastuzumab and a generic ErbB2 inhibitor with a low cardiotoxic profile observed protein expression in cardiomyocytes treated with these drugs and found trastuzumab to inhibit the AMP-activated protein kinase (AMPK) pathway while the generic inhibitor activated it instead. AMPK is needed for mitochondrial energy production, and its inhibition could explain why trastuzumab is one of the more cardiotoxic TKIs. Cardiomyocytes subsist on low stores of ATP, supplied in part by the AMPK pathway, making them particularly sensitive to inhibition.1 These findings agree with the toxicity seen in trastuzumab, as a lack of ATP would hinder cardiac muscle cell contractility. Authors theorized that these results could be related to lapatinib, where like the generic inhibitor it activates the AMPK pathway and spares cardiomyocytes in this regard.44
The low cardiac toxicity of lapatinib could also help explain by itself the mechanism of toxicity. Lapatinib is known to be one of the more selective TKIs, and therefore, it is possibly rendered safer than other agents in the class through minimal off-target binding. Researchers have suggested that the current view on lapatinib cardiotoxicity is mis-represented in one direction or another. Schools of thought include the theory that in the lapatinib treatment, there is no significant risk of cardiac dysfunction at all, pointing out that in published trials it is often employed following treatment with known cardiotoxins trastuzumab and anthracyclines. This would suggest that adverse effects only appear with lapatinib after cumulative insult to cardiomyocytes from other agents. Another observation follows that patients started on lapatinib in published trials were often those who had tolerated trastuzumab without cardiovascular complications or had a longer treatment free interval following an anthracycline regimen than those who started trastuzumab. The implication is that lapatinib-treated patients are sheltered from cumulative cardiotoxicity that would otherwise manifest adverse effects and if its place in therapy were that of trastuzumab’s, toxicity would be more prominent.45
The earlier discussed ALTTO trial could help clarify the pathology. The study arms were lapatinib monotherapy, trastuzumab monotherapy, trastuzumab followed by lapatinib in sequence, or lapatinib and trastuzumab in combination. These arms were applied in one of the three different chemotherapeutic regimens. One regimen included these drugs alongside concomitant taxane therapy following adjuvant chemo with an anthracycline; this regimen represented about 40% of treated patients for each arm. Despite a significant portion of the overall study population having undergone adjuvant anthracycline treatment, the incidence of cardiac events was low and similar among all treatment arms. Symptomatic heart failure of any grade was 2% each in lapatinib monotherapy and sequential therapy, and 3% each in trastuzumab monotherapy and combination therapy. LVEF decrease of ≥10% and below the lower limit of normal was seen at a rate of 3% each in lapatinib monotherapy and sequential therapy, and 5% each in trastuzumab monotherapy and combination therapy.41 The difference in events between arms is not strong enough to support the theory that cardiac events emerge as a result of cumulative toxicity, but would seem to support the hypothesis that the cardiotoxicity profile of these two drugs is similar when used in similar regimens. Based on these results, a final conclusion could be made that cardiotoxicity in both agents is primarily a result of ERbB2 inhibition but occurs in lapatinib with cardioprotective mechanisms not seen in trastuzumab.
Monitoring and management of lapatinib cardiotoxicity
Of the three TKIs discussed in this article, lapatinib appears to have the best cardiac safety profile. Overall, the studies above show that cardiotoxicity caused by lapatinib seems to be rare, and when it does occur, it is mild, easily managed, and does not often warrant discontinuation of treatment. Data from separate trials agree that an important monitoring parameter with this drug is LVEF. As with dasatinib and sorafenib, proper monitoring of patients during therapy is again important, but particular to lapatinib, measurement of baseline heart function is strongly recommended as a standard precaution. Lapatinib shares the ERbB2 target with monoclonal antibody trastuzumab, and literature suggests that both show LVEF declines in patients.46 Accordingly, monitoring guidelines have been proposed. The Cardiac Guidelines Consensus Committee recommends a baseline LVEF of ≥50% to initiate treatment, with follow-up evaluations via echocardiogram or multigated acquisition scan one to two times during therapy and at the end of treatment. If these follow-ups identify an asymptomatic drop in LVEF, assessment should be repeated in 3–4 weeks. Treatment should be put on hold if the LVEF is <40%, and the patient should then be referred to a cardiologist. The guidelines also strongly recommend monitoring of cardiac biomarkers such as CK, troponin, and brain natriuretic peptide. Clinically significant abnormalities in these labs would indicate a more acute problem than LVEF decline, although the guidelines do not propose a frequency for their monitoring; this will likely be left to professional judgment. An increase in these biomarkers would also warrant referral to a cardiologist.46 The product labeling includes warnings for LVEF decline and QT prolongation and suggests monitoring in line with these guidelines, with a detailed collection of past medical history.
Conclusion
Our review of the literature finds that there is indeed variability in the cardiotoxic effects of the TKI class. While commonalities exist, the predominant adverse effects are different in the agents discussed. Studies showed fluid retention to be most common in dasatinib treatment, hypertension in sorafenib, and lowered LVEF in lapatinib. This implies that cardiotoxicity is not due to a single mechanism for the TKI class but varies among agents; this is supported by the studies reviewed in this article. Notably, the threat posed by some of the discussed TKIs is not as severe as many practitioners might believe. Pooled safety data suggest a minimal risk of cardiotoxicity for dasatinib and lapatinib. An analysis of guidelines and professional recommendations identifies routine monitoring of heart function during therapy and identification of at risk patients before therapy to be the key steps in preventing cardiovascular events, regardless of the agent used. Off-target tyrosine kinase binding is thought to have a role in the toxicities seen in dasatinib and sorafenib. A complete understanding of their mechanisms of cardiotoxicity will therefore require further investigation to identify which of several possible kinases is at fault. On-target binding side effects have been implicated for each of the discussed drugs as well; targets such as Bcr-Abl, VEGFR, and ErbB2 are all thought to have a role in proper cardiovascular function. In addition, specific pro-survival or pro-apoptotic pathways are thought to be affected with each of the drugs. The result is that cardiotoxicity is likely explained as a combination of mechanisms specific to each TKI.
Author contributions
MC and JK wrote the review. SAO created and edited the review.
Disclosure
The authors report no conflicts of interest in this work.
References
Mellor HR, Bell AR, Valentin JP, Roberts RR. Cardiotoxicity associated with targeting kinase pathways in cancer. Toxicol Sci. 2011;120(1):14–32. | ||
Zuppinger C, Suter TM. Cancer therapy-associated cardiotoxicity and signaling in the myocardium. J Cardiovasc Pharmacol. 2010;56(2):141–146. | ||
Thomson M. Evidence of undiscovered cell regulatory mechanisms: phosphoproteins and protein kinases in mitochondria. Cell Mol Life Sci. 2002;59(2):213–219. | ||
Force T, Krause DS, van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. 2007;7(5):332–344. | ||
Freebern WJ, Fang HS, Slade MD. In vitro cardiotoxicity potential comparative assessments of chronic myelogenous leukemia tyrosine kinase inhibitor therapies: dasatinib, imatinib and nilotinib. Am Soc Hematol. 2007;110:4582. | ||
Orphanos GS, Ioannidis GN, Ardavanis AG. Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol. 2009;48(7):964–970. | ||
Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531–2541. | ||
Brave M, Goodman V, Kaminskas E, et al. Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin Cancer Res. 2008;14(2):352–359. | ||
Shah NP, Kantarjian HM, Kim DW, et al. Intermittent target inhibition with dasatinib 100 mg once daily preserves efficacy and improves tolerability in imatinib-resistant and -intolerant chronic-phase chronic myeloid leukemia. J Clin Oncol. 2008;26(19):3204–3212. | ||
Kantarjian HM, Shah NP, Cortes JE, et al. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood. 2012;119(5):1123–1129. | ||
Moslehi JJ, Deininger M. Tyrosine kinase inhibitor-associated cardiovascular toxicity in chronic myeloid leukemia. J Clin Oncol. 2015;33(35):4210–4218. | ||
Araujo JC, Trudel GC, Saad F, et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. Lancet Oncol. 2013;14(13):1307–1316. | ||
Will Y, Dykens JA, Nadanaciva S, et al. Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol Sci. 2008;106(1):153–161. | ||
Hasinoff BB, Patel D. Mechanisms of myocyte cytotoxicity induced by the multikinase inhibitor sorafenib. Cardiovasc Toxicol. 2010;10(1):1–8. | ||
Verstovsek S. Preclinical and clinical experience with dasatinib in Philadelphia chromosome-negative leukemias and myeloid disorders. Leuk Res. 2009;33(5):617–623. | ||
Hasinoff BB, Patel D, Wu X. The myocyte-damaging effects of the BCR-ABL1-targeted tyrosine kinase inhibitors increase with potency and decrease with specificity. Cardiovasc Toxicol. 2017;17(3):297–306. | ||
Saglio G, Le Coutre P, Cortes J, et al. Evaluation of cardiovascular ischemic event rates in dasatinib-treated patients using standardized incidence ratios. Ann Hematol. 2017;96(8):1303–1313. | ||
Nexavar (R) [package insert]; 2005. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021923s008s009lbl.pdf. Accessed December 3, 2017. | ||
Janjigian YY, Vakiani E, Ku GY, et al. Phase II Trial of Sorafenib in Patients with Chemotherapy Refractory Metastatic Esophageal and Gastroesophageal (GE) Junction Cancer. PLoS One. 2015;10(8):e0134731. | ||
Abou-Alfa GK, Schwartz L, Ricci S, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2006;24(26):4293–4300. | ||
Gridelli C, Maione P, del Gaizo F, et al. Sorafenib and sunitinib in the treatment of advanced non-small cell lung cancer. Oncologist. 2007;12(2):191–200. | ||
Strumberg D, Clark JW, Awada A, et al. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist. 2007;12(4):426–437. | ||
Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–134. | ||
Choueiri TK, Schutz FA, Je Y, Rosenberg JE, Bellmunt J. Risk of arterial thromboembolic events with sunitinib and sorafenib: a systematic review and meta-analysis of clinical trials. J Clin Oncol. 2010;28(13):2280–2285. | ||
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | ||
Schmidinger M, Zielinski CC, Vogl UM, et al. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2008;26(32):5204–5212. | ||
Syrigos KN, Karapanagiotou E, Boura P, Manegold C, Harrington K. Bevacizumab-induced hypertension: pathogenesis and management. BioDrugs. 2011;25(3):159–169. | ||
Alivon M, Giroux J, Briet M, Goldwasser F, Laurent S, Boutouyrie P. Large artery stiffness and hypertension after antiangiogenic drugs: influence on cancer progression. J Hypertens. 2015;33(6):1310–1317. | ||
Giroux J, Alivon M, Briet M, et al. Arterial stiffness to predict hypertensive response to antiangiogenic drugs. J Clin Oncol. 2013;31(15_Suppl):e13589–e13589. | ||
Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26(1):127–132. | ||
Zamorano JL, Lancellotti P, Rodriguez Muñoz D, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur Heart J. 2016;37(36):2768–2801. | ||
Ryan Q, Ibrahim A, Cohen MH, et al. FDA drug approval summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist. 2008;13(10):1114–1119. | ||
National Cancer Institute. Lapatinib ditosylate March 9, 2018. Available from: https://www.cancer.gov/about-cancer/treatment/drugs/fda-lapatinib#Anchor-Hormone1234. Accessed August 31, 2018. | ||
Paul B, Trovato JA, Thompson J. Lapatinib: a dual tyrosine kinase inhibitor for metastatic breast cancer. Am J Health Syst Pharm. 2008;65(18):1703–1710. | ||
Pondé NF, Lambertini M, de Azambuja E. Twenty years of anti-HER2 therapy-associated cardiotoxicity. ESMO Open. 2016;1(4):e000073. | ||
Moy B, Goss PE. Lapatinib-associated toxicity and practical management recommendations. Oncologist. 2007;12(7):756–765. | ||
Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–2743. | ||
di Leo A, Gomez HL, Aziz Z, et al. Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. J Clin Oncol. 2008;26(34):5544–5552. | ||
Greil R, Borštnar S, Petráková K, et al. Combination therapy of lapatinib and Capecitabine for ErbB2-positive metastatic or locally advanced breast cancer: results from the Lapatinib Expanded Access Program (LEAP) in Central and Eastern Europe. Onkologie. 2011;34(5):233–238. | ||
Chien AJ, Munster PN, Melisko ME, et al. Phase I dose-escalation study of 5-day intermittent oral lapatinib therapy in patients with human epidermal growth factor receptor 2-overexpressing breast cancer. J Clin Oncol. 2014;32(14):1472–1479. | ||
Piccart-Gebhart M, Holmes E, Baselga J, et al. Adjuvant lapatinib and trastuzumab for early human epidermal growth factor receptor 2-positive breast cancer: results from the randomized phase III adjuvant lapatinib and/or trastuzumab treatment optimization trial. J Clin Oncol. 2016;34(10):1034–1042. | ||
Dogan E, Yorgun H, Petekkaya I, Ozer N, Altundag K, Ozisik Y. Evaluation of cardiac safety of lapatinib therapy for ErbB2-positive metastatic breast cancer: a single center experience. Med Oncol. 2012;29(5):3232–3239. | ||
Yeh ET, Bickford CL. Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol. 2009;53(24):2231–2247. | ||
Shell SA, Lyass L, Trusk PB, Pry KJ, Wappel RL, Bacus SS. Activation of AMPK is necessary for killing cancer cells and sparing cardiac cells. Cell Cycle. 2008;7(12):1769–1775. | ||
de Azambuja E, Bedard PL, Suter T, Piccart-Gebhart M. Cardiac toxicity with anti-HER-2 therapies: what have we learned so far? Target Oncol. 2009;4(2):77–88. | ||
Chatsiproios D. Safety Profile and Clinical Recommendations for the Use of Lapatinib. Breast Care. 2010;5(s1):16–21. | ||
Sprycel (R) [package insert]; June 2006. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021986s7s8lbl.pdf. Accessed December 3, 2017. | ||
Brana I, Tabernero J. Cardiotoxicity. Ann Oncol. 2010;21(Suppl 7):vii173–vii179. | ||
Iressa (R) [package insert]; July 2015. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206995s000lbl.pdf. Accessed December 3, 2017. | ||
Korashy HM, Attafi IM, Ansari MA, et al. Molecular mechanisms of cardiotoxicity of gefitinib in vivo and in vitro rat cardiomyocyte: role of apoptosis and oxidative stress. Toxicol Lett. 2016;252:50–61. | ||
Yamaguchi K, Kanazawa S, Kinoshita Y, Muramatsu M, Nomura S. Acute myocardial infarction with lung cancer during treatment with gefitinib: the possibility of gefitinib-induced thrombosis. Pathophysiol Haemost Thromb. 2005;34(1):48–50. | ||
Gleevec (R) [package insert]; 2001. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021588s024lbl.pdf. Accessed December 3, 2017. | ||
Chen MH, Kerkelä R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation. 2008;118(1):84–95. | ||
Tykerb (R) [package insert]; 2007. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022059s007lbl.pdf. Accessed December 3, 2017. | ||
Tasigna (R) [package insert]; June 2010. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022068s004s005lbl.pdf. December 3, 2017. | ||
Sutent (R) [package insert]; May 2011. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021938s13s17s18lbl.pdf. Accessed December 3, 2017. | ||
Chu TF, Rupnick MA, Kerkela R, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007;370(9604):2011–2019. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.