")
Back to Journals » Cancer Management and Research » Volume 11
Mechanism of trastuzumab resistance caused by HER-2 mutation in breast carcinomas
Authors Kong X, Zhang K, Wang X, Yang X, Li Y, Zhai J, Xing Z, Qi Y , Gao R, Feng X, Wang J, Fang Y
Received 10 November 2018
Accepted for publication 23 April 2019
Published 2 July 2019 Volume 2019:11 Pages 5971—5982
DOI https://doi.org/10.2147/CMAR.S194137
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Rituraj Purohit
Xiangyi Kong,1,2,* Kai Zhang,3,* Xiangyu Wang,1,4,* Xue Yang,1,* Yalun Li,5 Jie Zhai,1 Zeyu Xing,1 Yihang Qi,1 Ran Gao,1 Xiaoli Feng,6 Jing Wang,1 Yi Fang1
1Department of Breast Surgical Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, People’s Republic of China; 2Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital, Harvard Medical School, Harvard University, Boston, MA 02114-3117, USA; 3Department of Cancer Prevention, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, People’s Republic of China; 4Department of Laboratory Medicine, Mayo Clinic, Rochester, MN 55902, USA; 5Department of Breast Surgery, Yantai Yuhuangding Hospital, Yantai 264000, People’s Republic of China; 6Department of Pathology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, People’s Republic of China
*These authors contributed equally to this work
Background: Trastuzumab is an effective drug for the treatment of Her2-positive breast cancer. But, primary or secondary resistances to trastuzumab have become an important factor influencing the curative effect. The mechanisms of trastuzumab resistance are somewhat complex. The present work aims to explore the mechanism of trastuzumab resistance caused by HER-2 mutation in breast carcinomas.
Methods: Firstly, the HER2 wild type (WT) and HER2 mutant (HER2 Q429R, HER2 Q429H and HER2 T798M are the commonest 3 types of mutations) MCF7 cell lines were established. Cell proliferation inhibition was then assessed by the Cell Counting Kit-8 assay and BrdU assay. Transwell invasion assays were also conducted to investigate the metastatic potential influenced by the HER2 mutation. Furthermore, Western blotting and co-immunoprecipitation were conducted to detect protein levels and the physical interaction of HER2 and trastuzumab.
Results: The results showed that the mutant MCF7 cells were less sensitive to trastuzumab than the WTMCF7 cells. The mutation of HER2 almost had no influence on the expression of HER2 and the interaction of HER2 and trastuzumab. Finally, the mutation of HER2 weakened the inhibition of trastuzumab in the PI3K/AKT pathways. In addition, the inhibition of PI3K/AKT signaling-pathway increased the trastuzumab-sensitivity of HER2-mutant MCF7 cells.
Conclusions: Dysregulation of the PI3K-AKT signaling-pathway was a key mechanism inducing the trastuzumab-resistance to HER2 mutant breast cancer cells.
Keywords: HER2, trastuzumab, resistance, PI3K
Introduction
The human epidermal growth factor receptor (ErbB) family of transmembrane RTKs1 has been demonstrated to play a pivotal role in the occurrence and development of various malignant tumors.2 Studies have shown that there are a few critical mutations in the tyrosine kinase domain of HER-2 proteins in a variety of cancers including the head and neck cancer, lung cancer and breast cancer.3–6 Around 25% of breast cancers have HER2-gene amplifications or protein/mRNA over-expressions. Deregulations of HER2-gene, through over-expressions or gene’s amplifications, have been demonstrated of importance in cancer’s tumorigenesis or proliferation. The over-expression of HER2 is able to confer stronger responses to specific anti-HER2 therapies for the breast cancer.
HER2 oncoprotein is one of the class-1 transmembrane-receptors with tyrosine-kinase activities and plays important roles in the oncogenic activations of lots of signaling pathways, like the phosphoinositide 3-kinase (PI3K) pathway and mitogen-activated protein kinase (MAPK) pathway.7 Trastuzumab is a monoclonal antibody that directly targets part of the HER2 tyrosine kinase receptor, which is an effective drug for the treatment of HER2-positive breast cancer.8 But, primary or secondary resistances to trastuzumab have become a key factor affecting the curative effects.9 Moreover, most Her-2 positive patients will have resistances to trastuzumab after a period of time of trastuzumab treatments. The mechanisms of trastuzumab resistance are relatively complex.10 Recently, several studies have demonstrated that PI3K/AKT/mTOR pathway played an important role in the trastuzumab resistance.7
Through transmitting signal-transduction events in response to extracellular stimulations, the PI3K/AKT/mTOR pathway plays important roles in the cell physiology, including cell proliferations, growths, survivals, motilities, and metabolisms.11 The activation of PI3K/Akt signal-pathway has been shown to be an important mechanism of trastuzumab-resistance.7,12 Assessments of PI3K-pathway activations may function as one of the key biomarkers to identify patients less likely to respond well to trastuzumab.
In the present research, the human breast carcinoma cell line MCF7 was used to establish the HER2 wild type (WT) and HER2 mutant MCF7 cell models, analyzed the characteristics of each kind of cell lines, and deeply investigated the mechanisms controlling the in vitro trastuzumab-resistance in breast cancers. Moreover, we found that the dysregulation of the PI3K-AKT signaling pathway was one of the major mechanisms leading to resistance to trastuzumab in HER2 mutant breast cancer cell line. Our results provide supporting evidence for future investigations combining inhibitors of the PI3-kinase pathway with trastuzumab-based therapy.
Materials and methods
PCR amplification and cloning in T vector
The PCR reaction was performed in 50 μL of the solution containing: 4 μL of oligo mix, 1 μL of dNTP, 2 μL of cDNA, 5 μL of 10X PCR buffer, 0.3 μL Pfu DNA polymerase, 36 μL of distilled water and 1 μL of each of primers. The sequences of the primers used in this study are shown in Table 1. The second PCR solution containing: 1 μL of PCR product, 5 μL of 10X PCR buffer, 1 μL of dNTP, 1 μL of each of primers, 41 μL of distilled water, 0.3 μL Pfu DNA polymerase. PCR product was cloned into T vector using clonExpress® Entry One Step Cloning Kit (Vazyme Biotech Co., Nanjing, China) and transformed in competent cell (E. coli DH5α, Takara Bio Inc., Otsu, Japan). The analysis of recombinant colony was done by sequencing.
 |
Table 1 The primers used in this study |
Table 1 The primers used in this study
Cell culture and cell transfection
Human breast carcinoma cell line MCF7 was obtained from the American Type Culture Collection (Manassas, VA, USA). MCF7 cell is a commonly used breast cancer cell line that has been promoted for >40 years by multiple research groups and has been considered the most representative. MCF7 cells were routinely cultured in complete DMEM (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (Gibco) with 100 units/mL penicillin and 100 μg/mL streptomycin (Sigma, St-Louis, MO, USA) in a humidified incubator at 37°C containing 5% CO2, with 95% humidity (Thermo Scientific SW-CJ-1FD incubator; Thermo Scientific, Waltham, MA, USA). Cells were separately cultured onto six-well plates and transfected with 2 μg of each plasmid in each well using 4 μL of Lipofectamine 200 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions.
Cytometry analysis
MCF7 cells were disaggregated with Trypsin-EDTA for 15 mins at 37°C. Cells were washed and counted to reach a concentration of 106 cells/mL. Cells were incubated with specific antibodies: FITC anti-human CD340 (erbB2/HER-2) antibody and FITC Mouse IgG1, κ Isotype Ctrl (FC) antibody (Biolegend, San Diego, CA, USA), and analyzed by flow cytometry (BD Biosciences, San Jose, CA, USA). Experiments were performed in triplicate.
Apoptotic cells were identified using an Annexin V/FITC kit (KGA108, Nanking, China). According to the manufacturer’s instructions, cells were then centrifuged once more, and the supernatants were discarded. The cells were resuspended in 200 μL of binding buffer at 37°C for 30 mins in the dark; 2 μL Annexin V-FITC solution and 5 μL propidium iodide (1 μg/mL) were added. Cell suspensions were transferred to flow cytometry test tubes. The percent of early apoptotic, late apoptotic and necrotic cells were detected and analyzed by the flow cytometry. Bivariate analyses were adopted for defining the cell populations, where FITC (−) and PI (−) cells were designated as viable cells, FITC (+) and PI (−) cells were designated as apoptotic cells, and FITC (+) and PI (+) cells were designated as late apoptotic or necrotic cells. Results were representative of three independent experiments with triplicate samples.
Western blot analysis and CCK8 assay
About 1×107 cells were gathered and lysed in RIPA buffer (Beyotime Biotech, Nantong, China) in the presence of protease inhibitor (PMSF) and phosphatase inhibitor (Na-ortho-vanadate, NaF). Cell Counting Kit (CCK8) assay is a one-bottle solution; no premixing of components is required. CCK8, being nonradioactive, allows sensitive colorimetric assays for the determination of the number of viable cells in cell proliferation and cytotoxicity assays. The detection sensitivity of CCK8 is higher than the other tetrazolium salts such as MTT, XTT, MTS or WST-1. For CCK8 assay, 100 μL cell suspensions (3,000 cells/well) were dispensed in 96-well plates and pre-incubated for 24 hrs to allow cell attachments (humidified atmosphere, 37°C, 5% CO2). After 24 hrs pre-incubation, cells were treated with different concentrations (1, 3.16, 10, 31.6, 100, 316, 1,000, 3,160, 10,000 nM) of trastuzumab and incubated for 4 days. This design was chosen as a compromise that allows assessing the statistical significance of treatment effects at the replicated concentrations while still allowing for the possibility to describe concentration-response trends over a broad range of test concentrations.13 Subsequently, the cells were then incubated with 10 μL of CCK8 (Beyotime Biotech) per well at 37°C for 4 hrs in the incubator. The absorbance at 460 nm (A460) was examined using a scanning multi-well spectrophotometer (Thermo Scientific). We determined the trastuzumab concentration and exposure time for CCK8 assay according to Yang et al’s reported methods.14
Proliferation (BrdU) assay and transwell invasion assays
Cell proliferation was determined using 5′-bromo-2′-deoxyuridine (BrdU) staining kit (Ebioscience, San Diego, CA, USA). Briefly, cells were labeled with BrdU (10 μM) for 2 hrs prior to be harvested, and then cells were fixed by the fixing solution at 4°C overnight. The ensuing steps were carried out according to the manufacturer’s protocol. OD values were measured by a spectrophotometer microplate reader set at a dual wavelength of 450/550 nm. Evaluations of the cell migrations and invasions were assessed using Transwell Permeable Supports (Corning Inc., Corning, NY, USA). Invasive cells at the bottom of the membrane were fixed with methanol and then stained with 0.1% crystal violet for 20 mins. The number of invasive cells was finally counted.
Co-immunoprecipitation
The cells were gathered and lysed in RIPA buffer (Beyotime Biotech) in the presence of protease inhibitor (PMSF). The lysates were centrifuged for 15 mins at 13,000×g. After centrifugation, the supernatant was incubated with the indicated antibody and subsequently with A-protein agarose beads at 4°C overnight. Then, each sample was centrifuged at 3,000 rpm at 4°C for 3 mins, and the pellet was washed three times with 1× immunoprecipitation buffer and eluted by boiling in protein sample buffer under reducing conditions. Sample proteins were then separated by 10–12% SDS-PAGE before the proteins were transferred onto PVDF membranes (Millipore). After blocking in 5% skimmed milk for 1 hrs, the membranes were rinsed and probed with 1:1,000-diluted primary antibodies against HER2 (Abcam, Cambridge, MA, USA) overnight at 4°C, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies for 2 hrs at room temperature. Next, the immune-reactive protein bands were visualized using the ECL detection reagents (Beyotime Biotech) and X-ray film. Finally, the blots were scanned, and densitometric analysis was performed on the scanned images using Image J Software.
Statistical analysis
All data were expressed in the form of mean values ± standard error. Comparisons among multiple groups were made with a one-way ANOVA followed by Dunnett’s test. P<0.05 was used for statistical significance.
Results
The generation of HER2-mutantbreast cancer cells
Recombinant plasmid was sequenced by dideoxy chain termination method and compared with HER2 gene (3,768 bp/3,768 bp) by www.ncbi.nlm.nih.gov/blast-showed high homology of 98% (as shown in Figure S1A–C). The comparison of gene sequence in the recombinant plasmid HER2 Q429R, HER2 Q429H, HER2 T798M and Her2 was shown in Figure 1D–F. Flow cytometric analysis of Her2 demonstrated that MCF7 cells and MCF7 cells transfected with control vector showed a low HER2 level, while MCF7 cells in the presence of HER2, HER2 Q429R, HER2 Q429H, HER2 T798M showed a high HER2 level (as shown in Figure 1A). The HER2 expression was also confirmed by SDS-PAGE/immunoblotting. Consistent with the flow cytometry results, the expression of Her2 in transfected MCF7 cells was detected by SDS-PAGE/immunoblotting with anti-HER2 antibodies. Conversely, there was zero expression of HER2 in MCF7+control vector cells (Figure 1B).
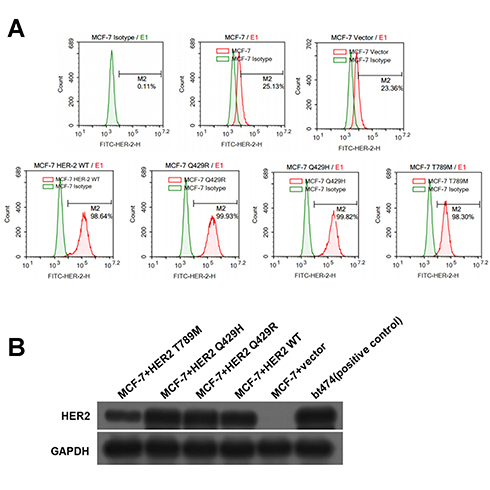 |
Figure 1 The assessment of HER2 expression in the transfected cells. The levels of HER2 in transfected MCF7 cells were quantified by flow cytometry. The mean fluorescence signal ± SD (n=3) (A). The expression of Her2 in transfected MCF7 cells was detected by SDS-PAGE/immunoblotting with anti-HER2 antibodies. Conversely, there was zero expression of HER2 in MCF7+control vector cells. GAPDH was used as internal control (B). These experiments were repeated in triplicate, quantified, and represented. Results are the average ± SD of triplicate determination. |
HER2 mutant MCF7 cells were resistant to trastuzumab
Trastuzumab undoubtedly provides a major advancement in the treatment of breast cancer alone or in combination with chemotherapy. However, the resistance to trastuzumab is common.15 Mutant-expressing MCF7 cells were used to investigate the resistance to trastuzumab after the mutation of HER2. Cells were treated with trastuzumab (1 μg/mL) for 4 days. The results showed that compared with the control vector group, MCF7+HER2, MCF7+HER2 Q429R, MCF7+HER2 Q429H and MCF7+HER2 T798M were less sensitive to the trastuzumab (as shown in Figure 2A). Moreover, the mutant MCF7 cells were less sensitive to the trastuzumab than the WTMCF7 cells. The BrdU assay was also used to evaluate the anti-proliferative activity of mutant-expressing MCF7 cells. Similarly, WTMCF7 cells were more sensitive to trastuzumab than the mutant MCF7 cells (as shown in Figure 2B and C). These results indicated that the mutations of HER2 played an important part in the resistance to trastuzumab.
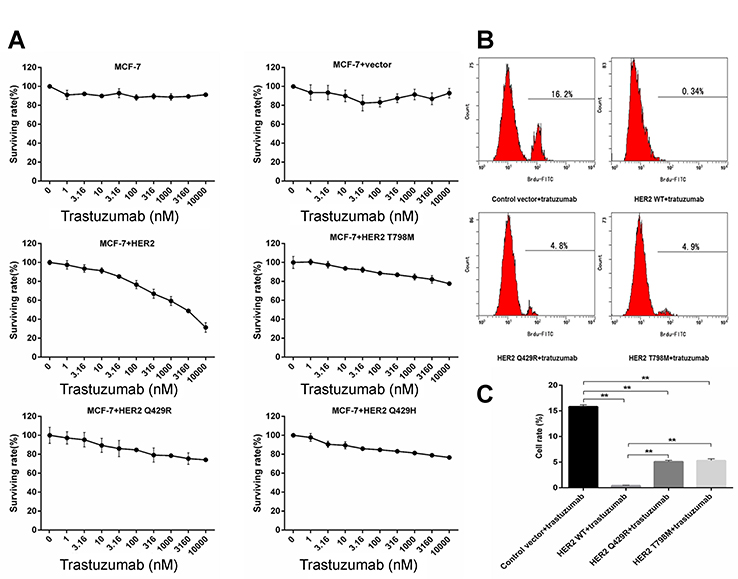 |
Figure 2 The effect of trastuzumab on the cell proliferation of HER2 Mutant MCF7 cells. (A) HER2 WT, HER2 Q429R, HER2 Q429H and HER2 T798M cells were treated with increasing concentrations of trastuzumab (1 μg/mL) for 4 days. CCK8 assay was performed at the end of treatment and dose–inhibition curves were produced by Graphpad Prism software. The experiment was performed at least three times at different times. Results are shown as percentage of control surviving rate. Error bars, SD. (B and C) Cell proliferation was determined using BrdU staining kit. Cells were treated with increasing concentrations of trastuzumab (1 μg/mL). The experiment was performed at least three times at different times. Results are shown as percentage of control surviving rate. Error bars, SD **P<0.01. |
The effect of the trastuzumab on the apoptosis of mutant-expressing MCF7 cells was then assessed by Annexin V. As shown in Figure 3A and B, following a 24 hrs trastuzumab treatment (1 μg/mL), HER2 WT cells exhibited 3.2±0.45 and 4.0±0.34 fold-changes in the number of Annexin V-positive cells, as compared with the HER2 Q429R and HER2 T798M cells, respectively. Transwell invasion assay was used to explore the metastasis potential of the mutant-expressing MCF7 cells. As shown in Figure 3C and D, following a treatment with trastuzumab (1 μg/mL), HER2 WT cells showed a reduced invaded cell number, while the HER2 Q429R and HER2 T798M cells exhibited resistance in the trastuzumab-treated invasion.
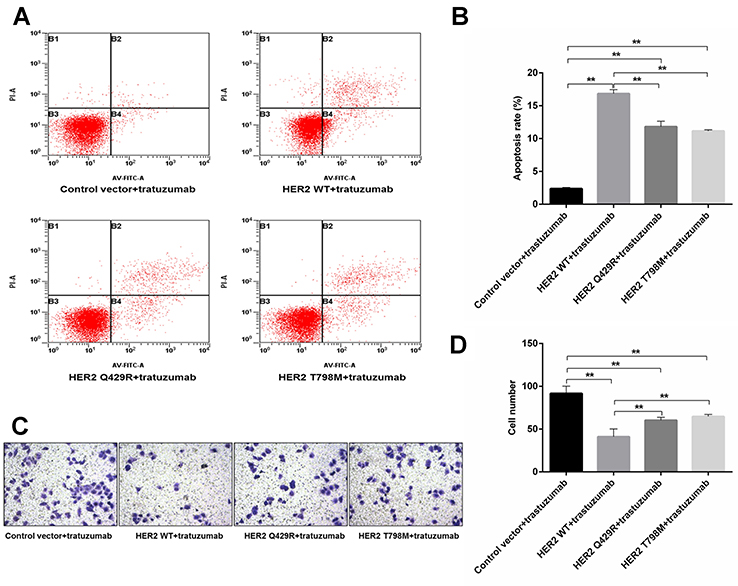 |
Figure 3 The effect of trastuzumab on the apoptosis and invasion of mutant-expressing MCF7 cells. (A) The effect of trastuzumab treatment on apoptosis was assessed by Annexin V/ FITC assay. Annexin V-positive cells were counted using flow cytometry. (B) Graphs exhibit apoptosis rate = number of Annexin V positive staining cells/number of total cells ×100%. As shown in (A) and (B), following a 24 hrs trastuzumab treatment (1 μg/mL), HER2 WT cells exhibited 3.2±0.45 and 4.0±0.34 fold-changes in the number of Annexin V-positive cells, as compared with the HER2 Q429R and HER2 T798M cells, respectively. Transwell invasion assay was used to explore the metastasis potential of the mutant-expressing MCF7 cells. (C) Representative images of the trastuzumab treatment on the invasion of mutant-expressing MCF7cells in a Transwell invasion assay. The photographs were taken at a magnification of x20. (D) Quantitative analysis of the number of invasive cells, as determined by counting cells under a microscope. As shown in (C) and (D), following a treatment with trastuzumab (1 μg/mL), HER2 WT cells showed a reduced invaded cell number, while the HER2 Q429R and HER2 T798M cells exhibited resistance in the trastuzumab-treated invasion. Data are presented as the mean ± SD; **P<0.01. Comparisons were made by one-way ANOVA followed by Dunnett’s test. |
The mutation of HER2 almost had no influence on the expression of HER2 and the interaction of HER2 and trastuzumab
To identify the difference of the expression of HER2 in the mutant-expressing cells, immunofluorescence staining was performed. As shown in Figure 4A, up-regulations of HER2 were detected in HER2 WT cells and the mutant-expressing cells when compared with the control group. However, there were no significant differences in the expression of HER2 between the HER2 WT cells and the mutant-expressing cells.
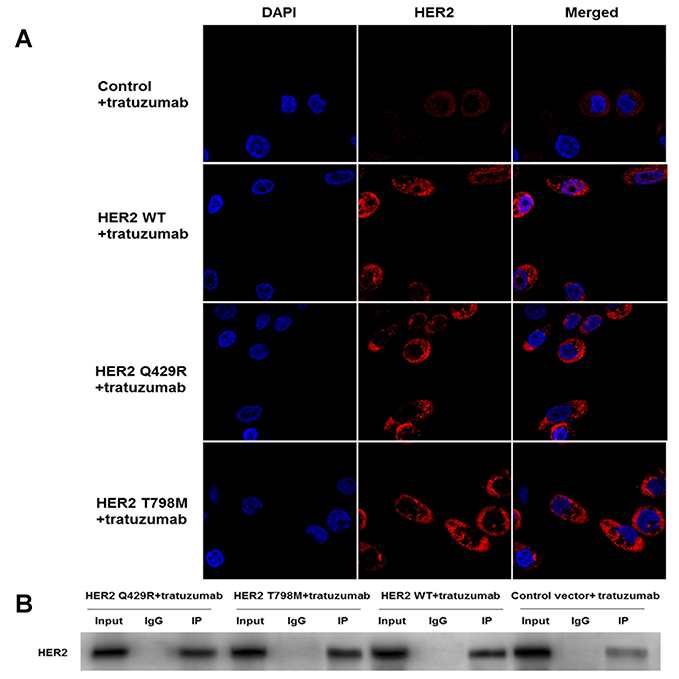 |
Figure 4 The mutation of HER2 almost had no influence on the expression of HER2 and the interaction of HER2 and trastuzumab. (A) The expression of HER2 (red) was examined by immunofluorescence analysis with a confocal microscope. Up-regulations of HER2 were detected in HER2 WT cells and the mutant-expressing cells when compared with the control group. However, there were no significant differences in the expression of HER2 between the HER2 WT cells and the mutant-expressing cells. DAPI, a blue tint to the nucleus. The photographs were taken at a magnification of x200. (B) Co-immunoprecipitation (IP) was carried out to confirm the physical interaction of HER2 and trastuzumab. Immuno-reactive signals of HER2 and trastuzumab were clearly detected in the immune-precipitates of anti-HER2. The cell lysate was immunoprecipitated and subjected to Western blot to detect HER2. The representative result is shown from three repeats with a similar pattern. |
To test the interactions between trastuzumab and HER2, immune-precipitates of trastuzumab were detected by the anti-HER2 antibody (as shown in Figure 4B). Immuno-reactive signals of HER2 and trastuzumab were clearly detected in the immune-precipitates of anti-HER2. When compared with the control group, the immune-reactive signals of the mutant-expressing cells and the HER2 WT cells were much stronger. In addition, there were no significant differences between the mutant-expressing MCF7 cells and the HER2 WT cells.
The mutation of HER2 weakened the inhibition of trastuzumab in the PI3K/AKT pathway
HER2 is involved in the oncogenic activations of numerous signaling pathways, like the PI3K pathways and the MAPK pathways.16,17 Western blot analysis was performed to analyze the roles of HER2 on the expressions of PI3K/AKT signaling pathway key proteins in MCF7 cells with incubation trastuzumab. When compared with the control group, the expression of HER3 and AKT stayed the same in the HER2 WT cells and the mutant-expressing cells following the treatment of trastuzumab for 24 hrs (as shown in Figure 5A). The expression of HER2, PTEN and P27K1P1 was up-regulated. The expression of PI3K, p-AKT and mTOR was inhibited when compared with the control group. Furthermore, the mutant-expressing cells showed a decreased expression of PTEN and P27K1P1 and an increased expression of PI3K, p-AKT and mTOR when compared with the HER2 WT cells following the treatment of trastuzumab for 24 hrs. Previous reports demonstrated that trastuzumab can inhibit the MAPK- and PI3K-pathways for cell lines overexpressing HER2, but not on HER2-overexpressing cancers that have received preoperative treatments as a monotherapy.18 In this study, PI3K pathways were inhibited by trastuzumab in HER2 WT cells, but not in the mutant-expressing cells. These results revealed that the mutation of HER2 might have changed the PI3K/AKT cascade signal transmission, which might weaken the inhibition of trastuzumab in the PI3K/AKT pathway.
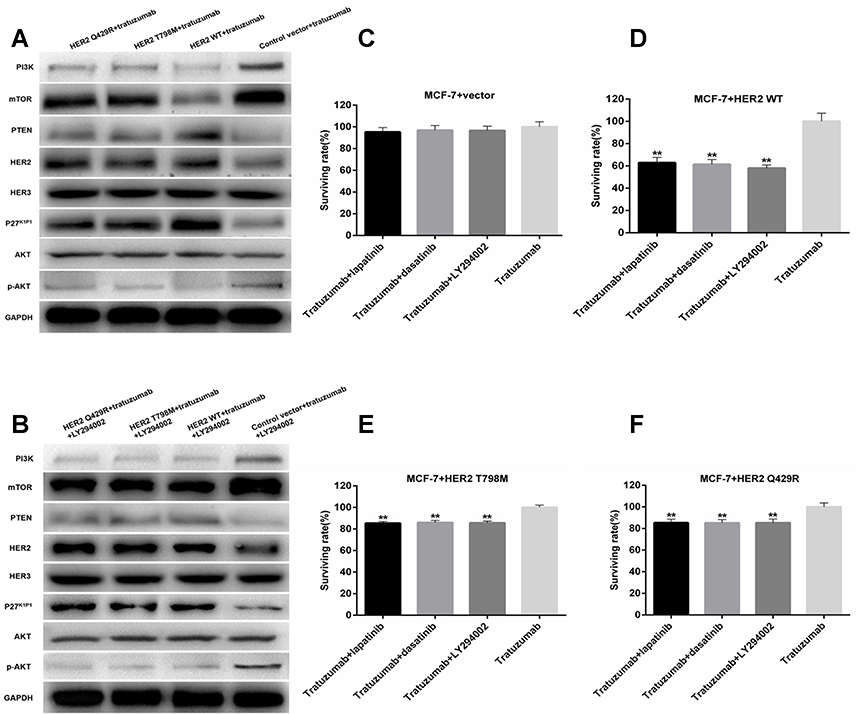 |
Figure 5 The mutation of HER2 weakened the inhibition of trastuzumab in the PI3K/AKT pathway. The combination with LY294002 and other chemotherapies could restore the inhibition of PI3K/AKT pathway. (A–B) Protein extracts from the indicated MCF7 cells were immunoblotted with the specified antibodies against PI3K, mTOR, PTEN, HER2, HER3, AKT, P27K1P1, AKT, p-AKT and GAPDH. (A) The indicated MCF7 cells were incubated with trastuzumab (1 μg/mL) for 24 hrs. (B) The indicated MCF7 cells were incubated with trastuzumab (1 μg/mL) and LY294002 (10 μmol/L) for 24 hrs. (C–F) The surviving rates were assessed by the CCK8 assay. The indicated cells were treated with trastuzumab (1 μg/mL), trastuzumab (1 μg/mL) and lapatinib (1 μmol/L), trastuzumab (1 μg/mL) and dasatinib (10 μmol/L), trastuzumab (1 μg/mL) and LY294002 (10 μmol/L). (C) MCF7 cells transfected with the control plasmid (C), the HER2-WT cells (D), HER2 T798M cells (E), and HER2 Q429R cells (F). |
The inhibition of PI3K/AKT signaling increases the trastuzumab sensitivity of HER2-mutant MCF7 cells
In the previous studies,19 the trastuzumab-resistant NCI-N87/TR cells were treated with a PI3K inhibitor (LY294002). Western blotting showed that when the cells were co-treated with LY294002 and trastuzumab, there was no difference between the HER2 WT cells and the HER2-mutant MCF7 cells with the expression of HER3, AKT, HER2, PTEN, P27K1P1, p-AKT and mTOR (as shown in Figure 5B). When compared with the trastuzumab alone, the regulations of PTEN and P27K1P1 were optimized. These results revealed that trastuzumab in combination with LY294002 re-regulated the PI3K/AKT signaling pathway of the HER2-mutant MCF7 cells, and increased the sensitivity to trastuzumab.
To further confirm the sensitivity caused by the combination treatment, the CCK8 assay was performed to determine the cell viability. As shown in Figure 5C–F, the HER2 WT cells were more sensitive when treated with trastuzumab (1 μg/mL) and lapatinib (1 μmol/L), trastuzumab (1 μg/mL) and dasatinib (10 μmol/L), trastuzumab (1 μg/mL) and LY294002 (10 μmol/L).
Discussion
HER2 is over-expressed widely in breast cancers. ErbB2 over-expressions are identified in around 25–30% of human breast cancers, and are closely related to the tumorigenesis, cancer progressions and poor prognoses.20,21 Trastuzumab is a recombinant antibody that has been approved by the FDA for the treatment of HER2-positive breast cancers.8 Although it has substantially improved the outcomes of patients with ErbB2-positive breast cancers, about 70% of ErbB2-amplified breast cancers do not respond to trastuzumab.22,23 There have been some studies that investigated the underlying mechanisms of trastuzumab-resistance. According to Pohlmann et al’s review, mechanisms for resistance are grouped into 4 main categories: a) Obstacles preventing trastuzumab binding to HER2; b) Upregulation of HER2 downstream signaling pathways; c) Signaling through alternate pathways; and d) Failure to trigger an immune-mediated mechanism to destroy tumor cells.24 In Luque-Cabal et al.’s study, the phosphatidylinositol 3’-kinase/protein kinase B/mammalian target of rapamycin pathway, cross-talk with estrogen receptors, immune response, cell cycle control mechanisms, and other tyrosine kinase receptors such as insulin-like growth factor I receptor are potential pathways involved in trastuzumab resistance.25 Gajria et al highlighted putative models of trastuzumab-resistance, including activation of the downstream PI3K-signaling pathway, accumulation of a constitutively active form of HER2, and crosstalk of HER2 with other growth factor receptors. The identification of these specific mechanisms of trastuzumab resistance has provided a rationale for the development of several novel HER2-targeted agents as the mechanisms have largely suggested a continued tumor dependence on HER2 signaling.26 Additionally, Different therapeutic interventions targeting these pathways are currently under evaluation. In Bose et al’s study, 13 HER2 mutations were functionally characterized using in vitro kinase assays, protein structure analysis, cell culture, and xenograft experiments.27 The majority of HER2 somatic mutations in breast cancer patients are activating mutations that likely drive tumorigenesis. Some patients had mutations that are resistant to the reversible HER2 inhibitor lapatinib, but are sensitive to the irreversible HER2 inhibitor, neratinib. Their results suggest that patients with HER2 mutation-positive breast cancers could benefit from existing HER2-targeted drugs.27
Despite these, the exact mode of action of trastuzumab remains partly elucidated, and several studies have suggested that the inhibition of oncogenic signaling may not be the main mode of action,28 particularly when trastuzumab is administered alone for chemotherapy-naïve patients.29,30 Thus, there is an urgent need to investigate the mechanisms of trastuzumab resistance and to develop new strategies to circumvent the trastuzumab resistance.
The purpose of the present study is to evaluate the sensitivity of the HER2 mutated cancer cell lines to the drug trastuzumab. Trastuzumab responsiveness was evaluated in breast cancer cell lines by treating the HER2 mutant and WT cell lines with trastuzumab followed by a proliferation assay. Consistent with a previous report,31 the mutant MCF7 cells were less sensitive to trastuzumab than the WTMCF7 cells in the CCK8 assay and the cytometry analysis. The Transwell invasion assay also showed that the mutant MCF7 cells exhibited resistance. These results indicated that HER2 played an important part in the resistance to trastuzumab. To explore the mechanisms of trastuzumab resistance, we firstly examined the expressions of the HER2 and the interactions between trastuzumab and HER2. It turned out that there were no significant differences between the mutant-expressing MCF7 cells and the HER2 WT cells.
Recent researches have focused on the abnormalities involved in the mechanisms of trastuzumab resistance. Hyperactivated PI3K pathway is such an important abnormality, which exists in over 70% of breast cancers.32 Berns et al showed that ectopic expressions of either WT or oncogenic mutant PIK3CA led to a trastuzumab resistance phenotype.7 Nagata et al demonstrated that PI3 kinase inhibitors restored trastuzumab sensitivity in PTEN-deficient cells.33 Therefore, the PI3K/AKT pathway was then evaluated to find out that the regulation of the PI3K/AKT pathway was weakened in the HER2-mutant MCF7 cells. These results revealed that the mutation of HER2 might have changed the PI3K/AKT cascade signal transmission, which weakened the inhibition of trastuzumab in the PI3K/AKT pathway.
A typical PI3K inhibitor LY294002 was also used to confirm the roles of HER2 in the trastuzumab resistance and to restrain the activity of PI3K/Akt. LY294002, transformed from quercetins, is a typical PI3K inhibitor. The inhibitor of targeted PI3K’s catalytic subunit p110 can effectively restrain the growth of tumor cells in vivo and in vitro, block the PI3K/Akt signaling pathway upstream in a non-specific way, and thereby induce tumor cell apoptosis.34 Our results showed that trastuzumab in combination with LY294002 re-regulated the PI3K/AKT signaling pathway of the HER2-mutant MCF7 cells, and increased the sensitivity to trastuzumab. Several drugs against PI3K, mTOR, and AKT, such as Everolimus, are in clinical development. Moreover, the mechanism of trastuzumab resistance is relatively complex. Several other hypotheses have been raised: the loss of PTEN, activation of alternative pathways, expression of ligands of the EGFR family and receptor masking or epitope inaccessibility.10 PI3K/Akt signaling pathway and continuous activation state of p-Akt are just two important target spots for drug resistance of trastuzumab, which need to be further investigated in future experiments.
Our study has some limitations. Firstly, we only used one single cell line to assess the mechanisms of resistance caused by the mutations. We did not conduct other sets of experiments in another cell line. We understand that our results would be more convincing if their findings could be replicated in a panel of cell lines with different hormone receptor statuses. However, in the present study, we mainly focused on the Her-2-related resistance using the MCF7 cell line which is a commonly used breast cancer cell line that has been promoted for >40 years by multiple research groups and has been considered the most representative, and we think that should be sufficient to draw a conclusion to some degree. Of course, in our other unpublished studies, we also used SK-BR-3, a breast cancer cell line isolated by the Memorial Sloan–Kettering Cancer Center in 1970 that is used in studies seeking to overcome trastuzumab resistance to HER2-overexpressing breast cancers. Secondly, we used the Western blot to prove that when compared with the control group, the expression of HER3 and AKT stayed the same in the HER2 WT cells and the mutant-expressing cells following treatment with trastuzumab for 24 hrs (Figure 5A and so does Figure 5B). We understand that the densitometry may better reveal this finding. However, in the present study, we mainly focused on protein expression differences, and we think that although a Western blot experiment may not be optimal, it should be sufficient to draw a conclusion. Also, when we explored if the HER2 WT cells were more sensitive when treated with trastuzumab and lapatinib, trastuzumab and dasatinib, trastuzumab and LY294002, we only choose the most commonly used drug concentrations (Figure 5C–F). Although we understand that setting a range of different concentrations may be more comprehensive, in the present study, we mainly focused on the sensitivity rather than determining an optimal concentration. The concentration-comparing work is also under investigation in our laboratory. Unfortunately, results are unavailable at this point. Similarly, the mechanism of cancer cell death induced by trastuzumab was performed only using Annexin V method rather than further TUNEL assay and PI staining, and a cell cycle arrest was not conducted in the situation that increase of the trastuzumab-sensitivity of HER2-mutant MCF7 cells by the inhibition of PI3K/AKT signaling pathway in HER2-mutant MCF7 cells was obtained. We have already started broadening the cell biological methods and molecular biological strategies, and hope these results could be presented in our next article in the near feature. Thirdly, despite considering very specific MCF7 mutations, the PI3K/Akt/mTOR pathway has been already demonstrated to be implicated in trastuzumab resistance in HER2-overexpressing breast cancer. Also, preclinical studies have indicated that inhibitors of this pathway can act synergistically with trastuzumab in resistant cells.24–26 Seeing this, we have already been broadening the experimental settings including using the xenograft mouse models of breast cancer which transplanted Her-2 mutation cell lines in vivo to improve the research significance. What is novel about this present research is that the HER2 WT and HER2 mutant MCF7 cell models were established, and the characteristics of each kind of cell lines were analyzed. The mechanism was investigated from the perspective of HER2 mutation.
Conclusions
Taken together, our findings showed that dysregulation of the PI3K-AKT signaling pathway was one of the major mechanisms leading to the resistance to trastuzumab in HER2 mutant breast cancer cells. Our study provides a theoretical basis for further improving the effect of trastuzumab targeted breast cancer treatment.
Abbreviation list
HER2, human epidermal growth factor receptor 2; PI3K, phosphatidylinositol 3-kinase; WT, wild type; MAPK, mitogen-activated protein kinase; ErbB2, receptor tyrosine-protein kinase.
Authors’ contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Acknowledgments
The authors gratefully acknowledge the support from the Department of Breast Surgical Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College. This research was supported by Beijing Municipal Natural Science Foundation (J.W., Key program No. 7191009), CAMS Initiative for Innovative Medicine (J.W., No.2016-12M-1-007), Capital Public Health Education Beijing Science and Technology Program (J.W., No. Z161100000116093), Capital Public Health Education Beijing Science and Technology Program (Y.F., No. 171100000417028), PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (Y.F., No.3332015157) and National Natural Science Foundation of China (J. W., General program No.81872160). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Troyer KL, Lee DC. Regulation of mouse mammary gland development and tumorigenesis by the ERBB signaling network. J Mammary Gland Biol Neoplasia. 2001;6:7–21.
2. Hurst HC. Update on HER-2 as a target for cancer therapy: the ERBB2 promoter and its exploitation for cancer treatment. Breast Cancer Res. 2001;3:395–398.
3. Cohen EE, Lingen MW, Martin LE, et al. Response of some head and neck cancers to epidermal growth factor receptor tyrosine kinase inhibitors may be linked to mutation of ERBB2 rather than EGFR. Clin Cancer Res. 2005;11:8105–8108. doi:10.1158/1078-0432.CCR-05-0926
4. Lee JW, Soung YH, Kim SY, et al. ERBB2 kinase domain mutation in the lung squamous cell carcinoma. Cancer Lett. 2006;237:89–94. doi:10.1016/j.canlet.2005.05.026
5. Shigematsu H, Takahashi T, Nomura M, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65:1642–1646. doi:10.1158/0008-5472.CAN-04-4235
6. Stephens P, Hunter C, Bignell G, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525–526. doi:10.1038/431525b
7. Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi:10.1016/j.ccr.2007.08.030
8. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi:10.1056/NEJM200103153441101
9. Singer CF, Kostler WJ, Hudelist G. Predicting the efficacy of trastuzumab-based therapy in breast cancer: current standards and future strategies. Biochim Biophys Acta. 2008;1786:105–113. doi:10.1016/j.bbcan.2008.02.003
10. Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol. 2007;18:977–984. doi:10.1093/annonc/mdl475
11. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi:10.1038/nrg1879
12. Park BH, Davidson NE. PI3 kinase activation and response to Trastuzumab Therapy: what’s neu with herceptin resistance? Cancer Cell. 2007;12:297–299. doi:10.1016/j.ccr.2007.10.004
13. Eriksson KM, Johansson CH, Fihlman V, et al. Long-term effects of the antibacterial agent triclosan on marine periphyton communities. Environ Toxicol Chem. 2015;34:2067–2077. doi:10.1002/etc.3030
14. Yang Y, Ren F, Tian Z, Song W, Cheng B, Feng Z. Osthole synergizes with HER2 inhibitor, trastuzumab in HER2-overexpressed N87 gastric cancer by inducing apoptosis and inhibition of AKT-MAPK pathway. Front Pharmacol. 2018;9:1392. doi:10.3389/fphar.2018.01392
15. Wang L, Yu X, Wang C, et al. The anti-ErbB2 antibody H2-18 and the pan-PI3K inhibitor GDC-0941 effectively inhibit trastuzumab-resistant ErbB2-overexpressing breast cancer. Oncotarget. 2017;8:52877–52888. doi:10.18632/oncotarget.17907
16. Hudis CA. Trastuzumab–mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39–51. doi:10.1056/NEJMra043186
17. Baselga J. Treatment of HER2-overexpressing breast cancer. Ann Oncol. 2010;21 Suppl 7:vii36–40. doi:10.1093/annonc/mdq421
18. Lion M, Harle A, Salleron J, Ramacci C, Campone M, Merlin JL. Trastuzumab as a preoperative monotherapy does not inhibit HER2 downstream signaling in HER2-positive breast cancer. Oncol Lett. 2016;12:2028–2032. doi:10.3892/ol.2016.4891
19. Zuo Q, Liu J, Zhang J, Wu M, Guo L, Liao W. Development of trastuzumab-resistant human gastric carcinoma cell lines and mechanisms of drug resistance. Sci Rep. 2015;5:11634. doi:10.1038/srep11634
20. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182.
21. Sachdev JC, Jahanzeb M. Blockade of the HER family of receptors in the treatment of HER2-positive metastatic breast cancer. Clin Breast Cancer. 2012;12:19–29. doi:10.1016/j.clbc.2011.07.001
22. Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–726. doi:10.1200/JCO.2002.20.3.719
23. Pusztai L, Esteva FJ. Continued use of trastuzumab (herceptin) after progression on prior trastuzumab therapy in HER-2-positive metastatic breast cancer. Cancer Invest. 2006;24:187–191.
24. Pohlmann PR, Mayer IA, Mernaugh R. Resistance to trastuzumab in breast cancer. Clin Cancer Res. 2009;15:7479–7491. doi:10.1158/1078-0432.CCR-09-0636
25. Luque-Cabal M, Garcia-Teijido P, Fernandez-Perez Y, Sanchez-Lorenzo L, Palacio-Vazquez I. Mechanisms behind the resistance to trastuzumab in HER2-amplified breast cancer and strategies to overcome it. Clin Med Insights Oncol. 2016;10:21–30. doi:10.4137/CMO.S34537
26. Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11:263–275. doi:10.1586/era.10.226
27. Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–237. doi:10.1158/2159-8290.CD-12-0349
28. Arnould L, Gelly M, Penault-Llorca F, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer. 2006;94:259–267. doi:10.1038/sj.bjc.6602930
29. Andre F, Dieci MV, Dubsky P, et al. Molecular pathways: involvement of immune pathways in the therapeutic response and outcome in breast cancer. Clin Cancer Res. 2013;19:28–33. doi:10.1158/1078-0432.CCR-11-2701
30. Gennari R, Menard S, Fagnoni F, et al. Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clin Cancer Res. 2004;10:5650–5655. doi:10.1158/1078-0432.CCR-04-0225
31. Chen X, Wang H, Ou-Yang XN, Xie FW, Wu JJ. Research on drug resistance mechanism of trastuzumab caused by activation of the PI3K/Akt signaling pathway. Contemp Oncol (Pozn). 2013;17:363–369. doi:10.5114/wo.2013.35292
32. Lopez-Knowles E, O’Toole SA, McNeil CM, et al. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int J Cancer. 2010;126:1121–1131. doi:10.1002/ijc.24831
33. Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi:10.1016/j.ccr.2004.06.022
34. Hu L, Zaloudek C, Mills GB, Gray J, Jaffe RB. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002). Clin Cancer Res. 2000;6:880–886.
Supplementary materials
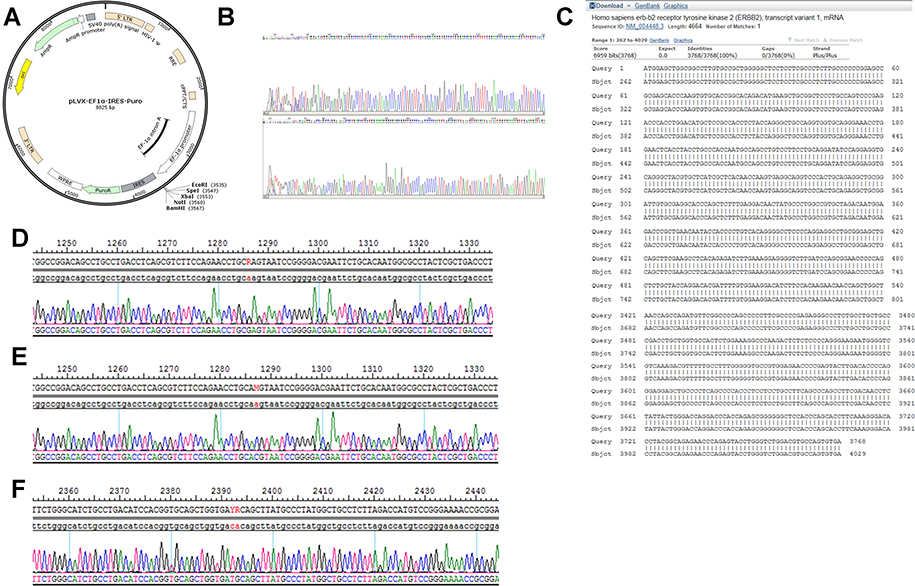 |
Figure S1 The construction and identification of HER2 Q429R, HER2 Q429H and HER2 T798M. (A) Circular map of plasmid pLVX-EF1α-IRES-Puro. (B) The sequencing of plasmid vector. (C) The comparison of gene sequence in the recombinantplasmidHER2 WT and the sequence of Homo sapiens erb-b2 receptor tyrosine kinase 2 (ERBB2) in NCBI. (D) The comparison of gene sequence in the recombinantplasmidHER2 Q429R and Her2. (E) The comparison of gene sequence in the recombinantplasmidHER2 Q429H and HER2. (F) The comparison of gene sequence in the recombinantplasmidHER2 T798M and HER2. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.