")
Back to Journals » OncoTargets and Therapy » Volume 10
Mechanism of immune evasion in breast cancer
Authors Wang M, Zhang C, Song Y , Wang Z, Wang Y, Luo F, Xu Y, Zhao Y, Wu Z, Xu Y
Received 3 November 2016
Accepted for publication 17 December 2016
Published 14 March 2017 Volume 2017:10 Pages 1561—1573
DOI https://doi.org/10.2147/OTT.S126424
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Ingrid Espinoza
Mozhi Wang,1,* Changwang Zhang,2,* Yongxi Song,2 Zhenning Wang,2 Yaojia Wang,1 Fang Luo,1 Yujie Xu,1 Yi Zhao,1 Zhonghua Wu,2 Yingying Xu1
1Department of Breast Surgery, 2Department of Surgical Oncology and General Surgery, The First Affiliated Hospital of China Medical University, Shenyang, People’s Republic of China
*These authors contributed equally to this work
Abstract: Breast cancer (BC) is the most common malignant tumor among women, with high morbidity and mortality. Its onset, development, metastasis, and prognosis vary among individuals due to the interactions between tumors and host immunity. Many diverse mechanisms have been associated with BC, with immune evasion being the most widely studied to date. Tumor cells can escape from the body’s immune response, which targets abnormal components and foreign bodies, using different approaches including modification of surface antigens and modulation of the surrounding environment. In this review, we summarize the mechanisms and factors that impact the immunoediting process and analyze their functions in detail.
Keywords: immune evasion, mechanism, breast cancer, cytokines, apoptosis, PD-L1
Introduction
Breast cancer (BC) has become the leading cause of death from malignant tumors among women worldwide in the past few decades.1,2 It has an extremely high morbidity and mortality, and the molecular mechanisms of BC have attracted a large number of researchers. Cancer immunoediting is a dynamic process by which the immune system attempts to destroy tumors, and it comprises three stages: elimination, equilibrium, and escape.3,4 During the elimination stage, host immune factors respond to progressive tumors and destroy them early before clinical changes appear. In the long-lasting equilibrium stage, meager residual tumor cells survive and keep in quiescence.3 Ultimately in the immune evasion stage, tumor cells gain the capacity to evade identification and attack by host immune responses using diverse mechanisms to survive and develop in the host.5 As a significant part of immunoediting, immune evasion has been recognized as a promising hallmark of BC in recent years. Gene mutations occasionally occur in somatic cells either spontaneously or owing to the combined action of various carcinogenic factors. Some mutated cells can recover through self-repair, whereas others undergo apoptosis. A few mutated cells express neoantigens on their membrane surface. These mutated cells with neoantigens are then identified as heterologous components and killed by innate and adaptive immunity components, and then cleared before tumors develop.6 However, some primary tumors can escape host immunosurveillance and establish, develop, migrate, and relapse. It has been shown that the mechanisms adopted by tumor cells to escape immunosurveillance include the modification of surface antigens and alterations of their surrounding environment. These aspects converge at the microenvironment, which includes cancer cells and the environment by which they are surrounded. Herein, we review the current knowledge on immune evasion and the factors and pathways affecting this process during interactions between the immune system and tumors with the view of elucidating mechanisms of immune evasion and improving current immunotherapies. A summarized net of mechanisms is presented in Figure 1.
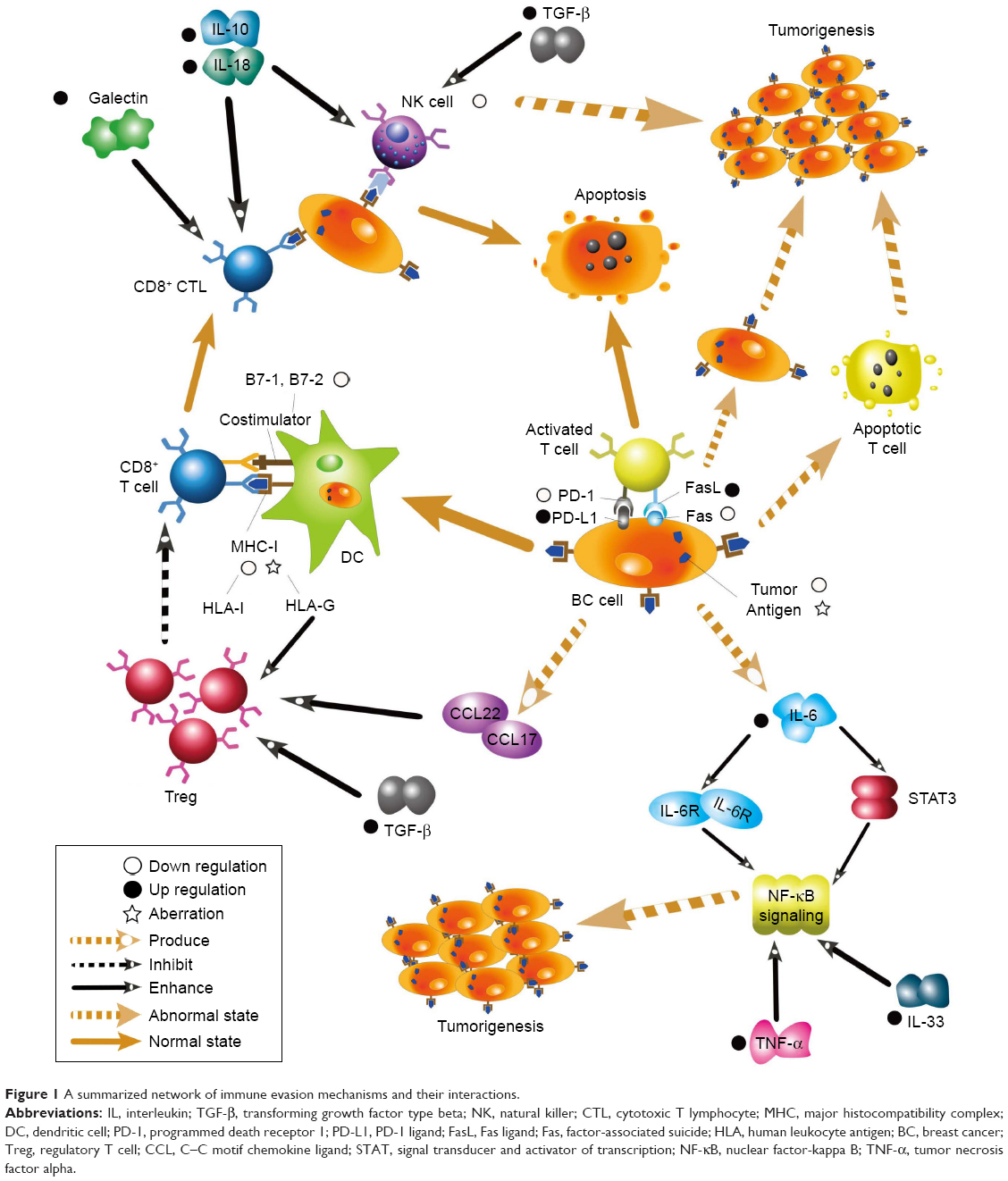 | Figure 1 A summarized network of immune evasion mechanisms and their interactions. |
Tumor-related immune evasion
Types of tumor-associated antigens
The hypothesis that tumor cells are recognized as non-autogenous cells by T cells to induce host immune responses underlies the concept of tumor immunosurveillance.5 BC cells can express specific surface antigens, distinct from those expressed by normal somatic cells, to impair the innate and acquired immune system and trigger a series of immune responses.7 There are three groups of tumor-associated antigens (TAAs): autoantigens or embryonic antigens, which are expressed excessively or abnormally; modified autoantigens, which are expressed specifically in the presence of tumors; and neoantigens, which are derived from gene mutations, chromosomal aberrations, and viral transformation.8,9 Because of these antigenic aberrations and the influence of host immunity, cells from the filial generation can become low-immunogenic tumor cells and compose the predominant cell types involved in resistance to the immune system.10 Melanoma-associated antigens (MAGEs) are a family of cancer/testis antigens which play roles as TAAs, and MAGE-A9 and MAGE-A11 are reported to be less expressed in estrogen-receptor-negative and HER2-negative BC.11 Inversely, MAGE-A9 is found to be significantly upregulated in invasive ductal BC and is positively correlated to unfavorable outcomes.12 Tchou et al discovered that mesothelin is overexpressed in triple-negative breast cancer (TNBC) via modifying the T cell receptor (TCR).13 Protein HER2/neu, known to be overexpressed in Her2-positive BC, is located primarily on the membrane of tumor cells. In addition, the expression of its extracellular domain is significantly higher in the sera of BC patients compared with tumor-free individuals, and its expression levels are positively correlated with histological tumor grade, suggesting that HER2/neu is overexpressed in and leads to the poor prognosis of Her2-positive BC.14 The low expression of HER2/neu might restrict the immune-cell-mediated destruction of tumor cells; thus, TCR gene therapy that encodes for TCR and enhances the immune response has been profoundly studied and tested in clinical trials.13,15 Additionally, a specially designed nanobody can selectively bind to TAA receptors.16 Therefore, loss or modification of surface antigens may promote immune evasion via lack of tumor cell recognition. These mechanisms may become potential targets to broaden our immunotherapeutic strategies.
Abnormal expression of major histocompatibility complex class I
Another type of antigen found on the membrane of tumors cells is presented by the major histocompatibility complex (MHC) class I, a subgroup of the MHC family. After interaction with MHC-I molecules, tumor antigens are presented on the surface of antigen-presenting cells for the tumor cells to be recognized, killed, and cleared by effector T lymphocytes.17 Therefore, the loss or modification of MHC-I on the surface of tumor cells allows these cells to escape immunosurveillance. Tumor cells silence MHC-I genes and downregulate their transcription levels to circumvent the immune system. The indispensable MHC-I antigen-processing machinery (APM) is reported to be deficient in tumors, including in BC.18 Furthermore, the beta-2 microglobulin, calnexin, and transporter-associated antigen processing 1 are downregulated in metastatic brain lesions of BC and negatively associate with cytotoxic T lymphocytes (CTLs) infiltration.19 Expression levels of human leukocyte antigen (HLA) class I molecules are significantly downregulated in BC, and this downregulation is necessary for the transformation of normal cells into abnormal cells. Gene mutation, loss of heterozygosity, and disturbance of transcriptional control can all be responsible for the low expression of HLA-I; this decreased expression of HLA-I lowers immunity and consequently is positively correlated with malignancy, metastasis, and prognosis of the tumor as a result of attenuated immunity.8
To obviate the potential for attack by CTLs and natural killer (NK) cells, HLA-G, the non-classical HLA-I antigen involved in immune mediation, is expressed on the surface of tumor cells.20 Expression levels of HLA-G increase in BC,21 which associates with poor prognosis.22 Following studies on a 14 bp InDel polymorphism in the HLA-G gene, Haghi et al performed genotyping and found that patients with higher BC stages had a higher frequency of allele deletion compared with patients with lower stages, indicating that the 14 bp InDel polymorphism in the HLA-G gene was a risk factor for the development of BC.23 In addition, the area under the curve for receiver operating characteristic values of the soluble form of HLA-G (sHLA-G) can help identify metastasis, suggesting that sHLA-G may be utilized as a biomarker for the diagnosis of BC, particularly at the metastasis stage.24 Furthermore, HLA-G may be present in exosomes to spread from tumor cells to other cells.25 König et al found that distinct sHLA-G subcomponents differentially affected the prognosis of neoadjuvant chemotherapy-treated BC.21 Due to the relationship between HLA-G expression and worsened BC prognosis, immunotherapeutic strategies to target HLA-G, and the DNA methyltransferase known to upregulate its expression are suggested.22
Antiapoptosis function
The control and reduction of apoptosis, the highly selective and programmed form of cell death, have been considered a fundamental strategy for evading the immune response in cancer development.26 Any part of the apoptosis pathway can be disturbed, causing endless proliferation of almost all types of cancers, including BC.27 All factors discussed are listed in Table 1.
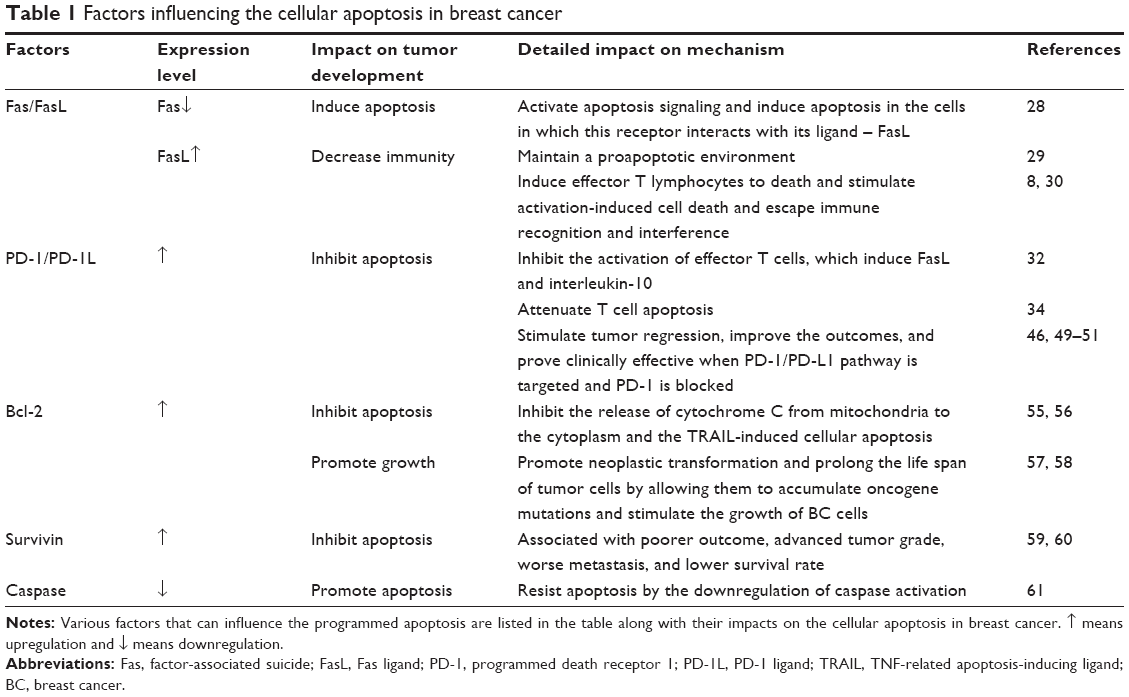 | Table 1 Factors influencing the cellular apoptosis in breast cancer |
Factor-associated suicide and its ligand
Factor-associated suicide (Fas) is a type II transmembrane protein from the tumor necrosis factor (TNF) family, also known as CD95 and Apo-1.28 Fas is an important suicide receptor because it can activate apoptosis signaling and induce apoptosis in cells in which the receptor interacts with its ligand–Fas ligand (FasL).28 FasL expresses on the activated T lymphocytes, and its presence has been implied to maintain a proapoptotic environment.29 T lymphocytes normally trigger apoptosis in Fas-expressing cells through the interaction of Fas and FasL, to limit rapid cell amplification and stimulate overactivated immune cells to downregulate the immune response (Figure 2). Increased FasL levels in BC cells induce effector T lymphocytes to die. Therefore, BC cells can stimulate activation-induced cell death and escape immune recognition and interference.8,30 Another strategy adopted by tumor cells to resist Fas-mediated apoptosis is to silence or downregulate Fas and Fas/FasL signaling pathways. The specific intracellular signaling domain of Fas, the death domain, is deficient in BC.8 Bębenek et al also demonstrated that loss of Fas and FasL was associated with worse prognosis. For this reason, these authors indicate that the Fas/FasL system can be used as a prognostic hallmark in BC.28
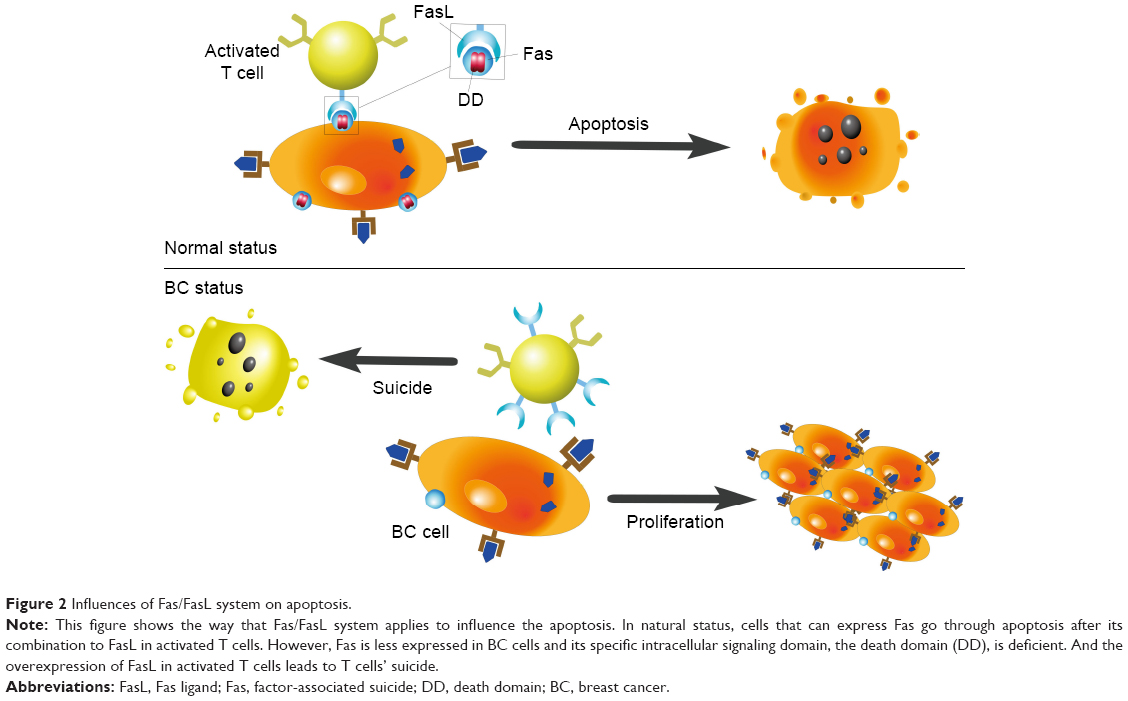 | Figure 2 Influences of Fas/FasL system on apoptosis. |
Programmed death receptor 1 and its ligand
Programmed death receptor 1 (PD-1) is a crucial immunosuppressive molecule found in CTLs, BC cells, and antigen-presenting cells.31 The interaction between PD-1 and its ligand, PD-L1, on the surface of tumor cells, inhibits the activation of effector T cells, which induce FasL and interleukin (IL) 10. PD-L1 has been found on the surface of BC cells32 and circulating tumor cells (CTCs).33 The blockade of PD-L1 can significantly attenuate T cell apoptosis in tumor models, which might be due to the important role that PD-L1 plays on regulatory T cell (Treg) induction and maintenance.33,34 While PD-L1 has been shown to directly synergize with FOXP3+ Tregs,34 it has also been revealed to be modulated by several other factors. Ubiquitination and N-glycosylation have known effects on the immunosuppressive activity of PD-L1.35 Also, increased expression of stem cell antigens Ly6K and Ly6E is reported to facilitate the activation and expression of cytokine-induced PD-L1, subsequently promoting drug resistance.36 The alimentary nutrient apigenin and its essential component luteolin have both been shown to indirectly inhibit interferon (IFN)-γ-induced PD-L1 expression via depression of signal transducer and activator of transcription (STAT) 1 phosphorylation, while not affecting constitutive PD-L1.31
The expression of PD-1/PD-L1 shows significant heterogenicity.37 It has frequently been found to be upregulated in various types of BCs, including inflammatory BCs, small-cell carcinomas of the breast, and basal tumors.37–39 Many researchers claim PD-L1 is overexpressed in TNBC,40–42 and that it is related to tumor grade,43 the local cytotoxic immune response, and prognosis.38,44 Although Mazel et al also proved that CTCs of hormone-receptor-positive and HER2-negative BCs express higher level of PD-L1,45 no specific studies on HER2-positive BC draw a similar conclusion. Moreover, PD-1/PD-L1-targeted therapies efficiently stimulate tumor regression and improve patient outcomes.46 Two agents, the PD-1 inhibitor pembrolizumab and the PD-L1 inhibitor atezolizumab, have been tested in clinical trials and are shown to increase the lasting local antitumor response to therapies40 with promising clinical benefit.47,48 As immune checkpoint inhibitors, PD-1/PD-L1 antagonists can be combined with other targeting therapies, including inhibitors of MAP2K and glucocorticoid-induced tumor necrosis factor receptor (GITR). These combinatory approaches have proven effective in clinical trials for TNBC,49–51 with the combination therapy improving outcome in vivo.52 Nevertheless, Mall et al found that the repeated use of PD-1/PD-L1 monoclonal antibodies might exacerbate an as yet uncharacterized fatal xenogeneic hypersensitivity reaction observed in a BC murine model.53 As the US Food and Drug Administration has admitted immunotherapeutic antibodies against PD-L1,54 further research and development of novel therapeutic methods are promising and necessary to elucidate the role of PD-1/PD-L1 in distinctive BC.
Bcl-2, survivin, and caspase
Other proteins are known to play significant roles in apoptosis in BC. The Bcl-2 protein encoded by the bcl-2 oncogene is involved in the endogenous apoptosis pathway by inhibiting the release of cytochrome C from mitochondria to the cytoplasm and consequently preventing tumor cell apoptosis.55 High levels of Bcl-2 in BC cells inhibit the cellular apoptosis induced by TNF-related apoptosis-inducing ligand.56 Bcl-2 promotes neoplastic transformation and prolongs the life span of tumor cells by allowing them to accumulate oncogenic mutations.57 Studies using a double-transgenic murine model reported that overexpression of both Bcl-2 and c-Myc stimulated the growth of BC cells.58 The expression of survivin, a prominent member of the family of antiapoptotic molecules, also increases in BC and is associated with poorer outcome, advanced tumor grade, worse metastasis, and lower survival rate.59,60 The decrease in caspase activation is another mechanism used by cancer cells to resist apoptosis. Accordingly, the expression of caspase-3 is downregulated in BC.61 However, the altered expression pattern of caspase-3 and caspase-7 was not strongly correlated with the clinicopathologic features of BC, indicating that the dysregulation of cellular apoptosis was much more complex than once thought.62
Tumor microenvironment
The onset and metastasis of tumors are closely correlated with the tumor microenvironment. The microenvironment is influenced by the specific structures, functions, and metabolism of neoplastic lesions, and by the inner environment of tumor cells. Tumor cells use autocrine and paracrine mechanisms to alter and maintain conditions essential for tumor survival, development, proliferation, and progression.63 Recent advancements in the areas of oncology and molecular biology have helped elucidate the association between malignant breast tumor and the immune factors around the tumors.
Tumor-infiltrating lymphocytes
Tumor-infiltrating lymphocytes (TILs) are a group of heterogenetic immune cells that primarily acquire immunosuppressive or immunomodulatory phenotypes and exert antitumor activity in the tumor environment. The synthesis and activation of TILs depend on the presence of chemokines and cytokines produced by tumor cells and immune cells that are present in tumor lesions.63
Effector CD8+ T lymphocytes
It is well supported that effector CD8+ T lymphocytes which identify TAAs may serve as biomarkers and improve the outcomes in various tumor types.64 However, these benefits depend on the interaction between the protective function of effector CD8+ T lymphocytes and the deleterious function of regulatory immune cells that are produced to help tumors escape immunity. Due to the impact of the inhibitory environment and the xenogeneic immunogenicity of antigens, T lymphocytes may ignore tumor antigens in some tumor types, including BC. Peripheral antitumor CD8+ T lymphocytes, which were different from those obtained from bone marrow, did not trigger an immune response in BC.65 Additionally, CD8+ T cells are able to restrain the anti-immune response capabilities of tumor cells. Thus, therapeutic strategies to evade or attack CD8+ T cells might provoke strong immune responses.66
Regulatory T cells
Tregs play a crucial role in immunosuppression.67 Tregs can inhibit the proliferation and activation of effector T lymphocytes and the secretion of helper T lymphocyte 1 (Th1) cytokines, resulting in the suppression of antitumor immune responses.68 Infiltration of Tregs is suggested to be significantly higher in TNBC than estrogen receptors/progesterone receptors-positive BC.69 Tregs accumulate in BC, indicating that the multiplication of these cells may be the result of both the natural circulatory system and a local trigger.8,30,70 The recruitment of Tregs to tumor lesions is dependent on C–C motif chemokine receptor (CCR) 4, modulated by several CCR family members such as CCR2,71 and induced by compounds secreted by BC cells, macrophages, and dendritic cells (DCs).70 C–C motif chemokine ligand (CCL) 22 and CCL17 are specifically attractive to Tregs, as well.72 Tregs can protect CTC from immune attack, alleviate the killing capacity of CTLs, and trigger larger quantity of myeloid-derived suppressor cells (MDSCs).33 Immune infiltration of Tregs is related to clinical prognosis,72 and can be augmented by Ly6E and Ly6K.36 The upregulation of GITR-related protein with Treg-inhibiting activity increases the proliferation, activation, and synthesis of cytokines.49 Recent studies found that the transforming growth factor type beta (TGF-β)-SMAD3/SMAD4-mediated induction of CD25/IL-2 receptor alpha on the surface of CD4+ T lymphocytes is blocked by inhibitors of MEPK/ERK signaling in BC patients. In addition, the aberrant activation of Janus kinase1/3-mediated STAT3/STAT5 decreased the expression of FOXP3.73 Due to the synergistic action of FOXP3+ Tregs and PD-L1, immune evasion is intensified when FOXP3+ Tregs and PD-L1 are both upregulated.34 Therefore, a combination therapy strategy against both FOXP3+ Tregs and PD-L1 might be promising.
Antigen presentation
T lymphocytes induce cellular immunity and target immunogenic tumor cells. The activation of non-sensitized T lymphocytes requires a secondary signal, which is triggered by the interaction of antigen-presenting cells or costimulating factors with the costimulatory molecules receptor as well as by the primary signaling.
Dendritic cells
DCs are myeloid- and plasmacytoid-derived professional antigen-presenting cells that initiate immune escape, activate naïve T cells, and induce memory T cells.74 A few mature DCs have been found in BC, revealing the complexity of antigen presentation and activation of the immune response against these tumors.75 The large number of immature DCs and the dysfunction of DCs promote immune tolerance and hinder the effective activation of cytotoxicity.76,77 The mechanisms mentioned constitute novel approaches of a clinical therapeutic method. The increased use of DC vaccine for BC patients in the past few years has increased the number of clinical adverse reactions and reminded the scientific community that important mechanisms other than dysregulation of DCs and immune escape may be occuring.74 da Cunha et al proposed that immunoinhibitory regulatory DCs are polarized by the tumor microenvironment, changing to a tolerogenic phenotype to promote tumor development and growth.78 The dysfunction of DCs is due to the abnormal differentiation of myeloid cells, and decreases the number of mature DCs and the expression of MHC-II, colony-stimulating factor (CSF), and adhesion molecules. Accordingly, tumor antigens are not adequately presented, and CSFs and adhesion molecules are downregulated on the surface of DCs.77 It has been recently shown that the differentiation of myeloid cells into DCs can be blocked by the tumor-dependent activation of STAT3 signaling in myeloid progenitor cells.79 In summary, DCs and their mechanisms of action and differentiation should be the focus of future studies.
NK group 2, member D
NK group 2, member D (NKG2D) receptors are located on the surface of NK cells and in some T cells to recognize ligands on the cellular surface.80 Tumors can exploit this by using sustained enhanced ligand production to downregulate the expression levels of NKG2D receptors and can directly blunt NKG2D via production of TGF-β as well.81,82 Crane et al also reported that tumor cells may produce soluble factors which induce expression of the NKG2D ligand on the surface of immune cells, thereby circumventing immunity.83 The results of a tumorigenicity test using an orthotopic xenotransplant BC model indicated that the self-stimulation of NKG2D could promote BC by increasing angiogenesis and promoting tumor growth, intravasation, and dissemination; however, NKG2D expression did not affect tumor proliferation or survival.84
Costimulators
T lymphocytes are activated by costimulators including the classical B7 family and the non-classical B7 homologous family.85 Tumors lack the classical costimulators CD80 and CD86 cells and, for this reason, weakly recognize HLA-II antigens.86 Therefore, T lymphocyte-activated signals would not reach the threshold for antitumor signals necessary to subvert the immune system.87 Tumor cells can thwart the expression of B7-1 and B7-2 molecules to inactivate T lymphocyte clones, and stimulate the expression of B7-H1 and B7-H4 molecules to enhance the secretion of IL-10 and trigger apoptosis in CTLs via upregulation of activated Fas/FasL on the surface of T lymphocytes. CTL-associated antigen 4 (CTLA-4) is a transmembrane receptor on T cells which shares the same B7 ligand with CD28.88 CTLA-4 transduces an intrinsic negative signal in effective T cells and directly negatively regulates the immune response.89 Accordingly, CTLA-4 has been regarded as the other essential immune checkpoint inhibitor besides PD-L1.90 Single-dose and combination therapies of the anti-CTLA-4 monoclonal antibody ipilimumab are under evaluation in clinical trials.91 Engel et al demonstrated that expression levels of B7-H1 increased in TNBC but were similar to those found in other BC subgroups.69 However, the elevated expression level of B7-H4 was correlated with the negative status of receptors and the positive state of Her2/neu.92 In addition, mechanistic studies of BC indicated that the presence of B7x correlated with a reduction in general and tumor-specific T cell cytokine responses as well as with increased infiltration of immunosuppressive cells to escape local antitumor immune responses via inhibition of the proliferation of both CD4 and CD8 T cells.93 Therefore, targeting the costimulatory pathway holds promise for improving the efficacy of immunotherapy for BC.
Inflammation and inflammatory mediators
Host immunity can affect tumors growth both in promotive and suppressive ways, and this immunoediting is a dynamic process that involves tumors and the host immune response and consists of three stages: elimination, equilibrium, and escape.3,4 During the elimination stage, inflammation is acute and tumor-inhibitory, and characterized by the infiltration of effector cells and antitumor cytokines. In contrast, during the immune escape stage, inflammation is chronic and tumor-promoting and characterized by the infiltration of inhibitory immune cells and soluble cytokines.5 Therefore, inflammation is critical for tumor onset, development, angiogenesis, and migration in BC, and cytokines play a unique role in each event.85
Transforming growth factor type beta
TGF-β is a functional bidirectional cytokine and the most studied immunosuppressive cytokine induced by BC.94 TGF-β belongs to the TGF-β superfamily and has antiproliferative activity in the early phases of cancer, acting through cell cycle arrest. TGF-β inhibits estrogen receptor alpha-mediated cancer proliferation.95,96 Mutations responsible for the antitumor effect and alterations of TGF-β signaling have been detected in BC.97 In activated T cells, TGF-β is translocated to the nucleus and induces c-Myc to promote tumor-associated inflammation in the later BC stage.98 TGF-β is a microtubule-destabilizing agent that targets p53 in human BC.99 Additionally, TGF-β was implied to interact with Wnt signaling, Her2, and focal adhesion kinase for epithelial-to-mesenchymal transition (EMT) and allows for the development of cancer stem cells.99–101 In recent years, some factors associated with TGF-β have been identified and examined. Small molecules such as galunisertib inhibit the TGF-β receptor I kinase by inhibiting the phosphorylation of SMAD2 and activation of the classical TGF-β signaling pathway.94 A subset of inducible or adaptive Tregs is enhanced by TGF-β and suppresses antitumor immunity.102
Interleukins
IL-6 can be produced by BC cells and by fibroblasts in BC. IL-6 regulates BC stem cells by activating STAT3 and the IL-6 receptor/GP130 complex.103–105 In the initiation stage, when nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling is activated by IL-6 and STAT3, NF-κB can change from a normal phenotype to an aberrant phenotype via self-regulation and inflammatory cytokines involved in NF-κB signaling.9 Increased invasion and EMT were also found to occur via IL-6 upregulation.106
IL-33 stimulates the expression and activation of MDSCs, and induces the autocrine production of granulocyte–macrophage CSF in the tumor microenvironment. Therefore, IL-33 mitigates apoptosis and promotes the survival of MDSCs. In addition, it also causes immunosuppression via activation of NF-κB and mitogen-activated protein kinase signaling.107
IL-18 is an antitumor inflammatory factor that activates CTLs and NK cells, induces antitumoral IFN-γ expression and osseous metastasis,108 and promotes tumorigenesis.109 IL-18 is essential for distal metastasis in BC.110 IL-19 and IL-20 provide a favorable microenvironment for tumor growth and foster the proliferation and progression of BC via synthesis of matrix metalloproteinases.111,112 IL-23 can modulate CD8+ T cell infiltration and promote inflammation-related development in the BC microenvironment.113 IL-10 can activate both NK cells and CTLs to inhibit development and metastasis of BC cells, and a few researchers have described its negative effect on tumorigenicity.114 The mentioned studies are summarized in Table 2.
 | Table 2 Interleukins associated with the immune evasion in breast cancer |
Tumor necrosis factor alpha
TNF-α is an inflammatory cytokine induced by toll-like receptors (TLRs) which stimulates the NF-κB signaling pathway to promote tumor development via modulation of antiapoptotic effects, tumor cell proliferation, angiogenesis, and metastasis.115 The expression level of TNF-α increases significantly in malignant breast tumors possibly due to gene polymorphisms.116 Owing to the dual effects of TNF-α, its synergic association with NF-κB is important. TNF-α stimulates cyclin D1 via activation of NF-κB to induce the proliferation of BC T47D cells;117 however, it also induces apoptosis of T47D cells in vitro.118 Activation of the TNF-α/NF-κB pathway enhances the invasion and malignancy of BC,119 whereas inhibition has the opposite effect.120 Altogether, these results indicate that TNF-α promotes apoptosis and tumorigenesis concurrently. It acts as an antitumor agent locally and induces cellular proliferation and metastasis in the tumor microenvironment.55 In addition, the increased susceptibility of TNF-α triggered by the loss of the anti-HER2 Th1 response may reflect a mechanism of immune evasion in HER2-driven tumorigenesis.121
Galectin and indoleamine-2,3-dioxygenase
Galectin is an evolutionarily conserved glycan-binding protein that contains a recognition domain identical to that of β-galactosidase and is involved in the proliferation, adhesion, migration, apoptosis, and angiogenesis of tumor cells. An analysis of BC specimens indicated that galectin-1 can be used in tumor grading.122 Stannard et al evaluated galectin inhibitor disaccharides and observed that galectin-1 could promote tumor progression by blocking the antitumor function of CD8+ CTLs.123 In addition, other studies were performed to prove that the silencing of galectin-1 could markedly reduce tumor growth and lung metastasis by decreasing the expression and immunosuppressive activity of CD4+CD25+FOXP3+ Tregs.124,125
Indoleamine-2,3-dioxygenase (IDO) is an immune inhibitor in malignant tumors.126 IDO causes tryptophan catabolism, resulting in biostatic tryptophan starvation and l-kynurenine production, the latter helping to control the interaction between BC and host immunity.127 Levina et al discovered that the immune response was enhanced when IDO1 was not expressed or expressed at low levels, demonstrating that the enhanced immune evasion associated with tumor cell proliferation and cell cycle increase as well as apoptosis resistance was due to the upregulation of IDO.128 These results indicate that the increased expression of IDO can help counterbalance host immunity and BC aggression.
Myeloid-derived suppressor cells
MDSCs are a group of heterogeneous bone marrow-derived immature cells that significantly suppress the immune response.129 MDSCs must be amplified, recruited, and activated before they successfully differentiate into DCs, macrophages, and granular leukocytes. MDSC accumulation can inhibit mature DCs and the antitumor immune response. Additionally, the enhanced stem-like qualities of BC cells and lower activation of T cells modulated by MDSCs were revealed recently.130 Mononuclear MDSCs can differentiate into macrophages and mature DCs via modulation of nitric oxide, inhibitory cytokines, and arginase-1. Granular MDSCs, the other subset of MDSCs, inhibit the immune response via direct intracellular contact and production of reactive oxygen species/intermediate nitrogen species. Cancer-associated fibroblasts, a subset of MDSCs with a similar phenotype, are an important group of interstitial tumor cells characterized by low expression of CD80 and MHC-II in BC, suggesting their involvement in immune evasion.86 In addition to these subsets, the location of MDSCs also influences their suppressive ability. Tumor-infiltrating MDSCs are more strongly inhibited by tumor-producing cytokines such as IL-33 compared to peripheral MDSCs.107 Several factors and agents targeting MDSCs have been investigated in recent years. Polyinosinic-polycytidylic acid, a ligand for TLR-3, decreases the number of MDSCs and the immunosuppressive activity in BC by augmenting the T cell response.131 In addition, previous studies have suggested that, in addition to its cytotoxicity against tumor cells, doxorubicin plays a major role in the reduction of MDSC-induced immunosuppression.132
Discussion
BC has become a major research focus because of its high susceptibility and malignancy in women. The traditional therapy of surgical excision has limited effects due to frequent distal metastasis and invasion, while chemotherapy and radiotherapy leave a great burden on the normal tissues of body.133 Thus, it is urgent to exploit promising therapeutic strategies. Since cancer immunotherapy was prized as a breakthrough,134 immune evasion has become one of the most important topics to be investigated. Herein, we reviewed the latest findings in mechanisms of immune evasion in BC. Tumor-related immune evasion includes the deficiency and modification of TAAs, the abnormal expression of MHC-I, and antiapoptosis functions. TAAs, including autoantigens, modified antigens, and neoantigens, can express in both BC cells and host immune cells when the microenvironment of tumor comes into being. Any modification or mutation of antigens might alter tumor cells’ immunogenicity, resulting in the various characteristics of different subtypes of BCs. MHC-I molecules help in antigens presentation and identification by T cells; hence, absence of MHC-I and MHC-I APM in BC might attenuate immune responses. In addition, HLA-G, normally presented on the host immune cells, is overexpressed on BC cells to escape destruction via CTLs and NK cells, and associates to poor prognosis. Inhibition of cellular programmed apoptosis might accelerate tumor cell proliferation. Targeting Fas/FasL and PD-L1/PD-1 axes can disturb the endless proliferation of tumor cells and is a potential immunotherapeutic strategy on which researchers have focused in recent years. In the tumor microenvironment, there are numerous factors such as TILs, processes related to antigen presentation, and inflammation-related cytokines. TILs play essential roles in antitumor function and the process of antigen presentation to ensure the detectability of tumor cells. Numerous stages of immunoediting show different types of inflammation, and inflammatory mediators are involved in various key pathways, while MDSCs are heterogeneous and immunosuppressive.
Limitations
It should be noted that there are still limitations existing in this review. Although these individual mechanisms are known, their joint functions remain unclear. Tumor growth and progression are associated with complex and systemic pathological processes, which must be connected in some manner. The integrity of the human body should not be disconnected from the tumor environment. This correlation will need to be determined and summarized clearly so that immunotherapeutic strategies can be designed effectively. This approach is promising to avoid clinically adverse reactions and reduce metastasis and the mortality rate.
Conclusion
Mechanisms of immune evasion in BC have been studied for decades and provide numerous promising targets for immunotherapy. The two significant immune checkpoint inhibitors, PD-L1/PD-1 ligand–receptor pairs and CTLA-4, have been studied and intervened with through immunotherapy. The improvement of basic researches and clinical trials aiming at checkpoint inhibitors might indicate applicable preclinical detection and diagnostic approaches as well as their clinically therapeutic strategies.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81372550), Program of Education Department of Liaoning Province (L2014307), and the Key Laboratory Programme of Education Department of Liaoning Province (LZ2015076, LZ2015080).
Disclosure
The authors report no conflicts of interest in this work.
References
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. | ||
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. | ||
Jiang X, Shapiro DJ. The immune system and inflammation in breast cancer. Mol Cell Endocrinol. 2014;382:673–682. | ||
Chu NJ, Armstrong TD, Jaffee EM. Nonviral oncogenic antigens and the inflammatory signals driving early cancer development as targets for cancer immunoprevention. Clin Cancer Res. 2015;21(7):1549–1557. | ||
Boyle ST, Kochetkova M. Breast cancer stem cells and the immune system: promotion, evasion and therapy. J Mammary Gland Biol Neoplasia. 2014;19(2):203–211. | ||
Töpfer K, Kempe S, Müller N, et al. Tumor evasion from T cell surveillance. J Biomed Biotechnol. 2011;2011:918471. | ||
Ernst M, Putoczki TL. Stat3: linking inflammation to (gastrointestinal) tumourigenesis. Clin Exp Pharmacol Physiol. 2012;39(8):711–718. | ||
Whiteside TL. Immune responses to malignancies. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S272–S283. | ||
Hou SY, Sang MX, Geng CZ, et al. Expressions of MAGE-A9 and MAGE-A11 in breast cancer and their expression mechanism. Arch Med Res. 2014;45(1):44–51. | ||
Xu X, Tang X, Lu M, et al. Overexpression of MAGE-A9 predicts unfavorable outcome in breast cancer. Exp Mol Pathol. 2014;97(3):579–584. | ||
Tchou J, Wang LC, Selven B, et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat. 2012;133(2):799–804. | ||
Barić M, Kulić A, Sirotković-Skerlev M, et al. Circulating Her-2/neu extracellular domain in breast cancer patients-correlation with prognosis and clinicopathological parameters including steroid receptor, Her-2/neu receptor coexpression. Pathol Oncol Res. 2015;21(3):589–595. | ||
Linnemann C, Schumacher TN, Bendle GM. T-cell receptor gene therapy: critical parameters for clinical success. J Invest Dermatol. 2011;131(9):1806–1816. | ||
Gray MA, Tao RN, DePorter SM, Spiegel DA, McNaughton BR. A nanobody activation immunotherapeutic that selectively destroys HER2-positive breast cancer cells. Chembiochem. 2016;17(2):155–158. | ||
Fu D, Geschwind JF, Karthikeyan S, et al. Metabolic perturbation sensitizes human breast cancer to NK cell-mediated cytotoxicity by increasing the expression of MHC class I chain-related A/B. Oncoimmunology. 2015;4:e991228. | ||
Seliger B, Maeurer MJ, Ferrone S. Antigen-processing machinery breakdown and tumor growth. Immunol Today. 2000;21(9):455–464. | ||
Liu Y, Komohara Y, Domenick N, et al. Expression of antigen processing and presenting molecules in brain metastasis of breast cancer. Cancer Immunol Immunother. 2012;61(6):789–801. | ||
Campoli M, Ferrone S. Tumor escape mechanisms: potential role of soluble HLA antigens and NK cells activating ligands. Tissue Antigens. 2008;72(4):321–334. | ||
König L, Kasimir-Bauer S, Hoffmann O, et al. The prognostic impact of soluble and vesicular HLA-G and its relationship to circulating tumor cells in neoadjuvant treated breast cancer patients. Human Immunol. 2016;77(9):791–799. | ||
Ishibashi K, Kumai T, Ohkuri T, et al. Epigenetic modification augments the immunogenicity of human leukocyte antigen G serving as a tumor antigen for T cell-based immunotherapy. Oncoimmunology. 2016;5(6):e1169356. | ||
Haghi M, Hosseinpour Feizi MA, Sadeghizadeh M, Lotfi AS. 14-bp insertion/deletion polymorphism of the HLA-G gene in breast cancer among women from North Western Iran. Asian Pac J Cancer Prev. 2015;16(14):6155–6158. | ||
Jeong S, Park S, Park BW, Park Y, Kwon OJ, Kim HS. Human leukocyte antigen-G (HLA-G) polymorphism and expression in breast cancer patients. PLoS One. 2014;9(5):e98284. | ||
Urosevic M, Dummer R. Human leukocyte antigen-G and cancer immunoediting. Cancer Res. 2008;68(3):627–630. | ||
Guo Y, Chang H, Li J, et al. Thymosin alpha 1 suppresses proliferation and induces apoptosis in breast cancer cells through PTEN-mediated inhibition of PI3K/Akt/mTOR signaling pathway. Apoptosis. 2015;20(8):1109–1121. | ||
Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87. | ||
Bębenek M, Duś D, Koźlak J. Prognostic value of the Fas/Fas ligand system in breast cancer. Contemp Oncol (Pozn). 2013;17(2):120–122. | ||
Olimón-Andalón V, Aguilar-Lemarroy A, Ratkovich-González S, et al. Proapoptotic CD95L levels in normal human serum and sera of breast cancer patients. Tumour Biol. 2015;36(5):3669–3678. | ||
Kim R, Emi M, Tanabe K, Uchida Y, Toge T. The role of Fas ligand and transforming growth factor beta in tumor progression: molecular mechanisms of immune privilege via Fas-mediated apoptosis and potential targets for cancer therapy. Cancer. 2004;100(11):2281–2291. | ||
Coombs MR, Harrison ME, Hoskin DW. Apigenin inhibits the inducible expression of programmed death ligand 1 by human and mouse mammary carcinoma cells. Cancer Lett. 2016;380(2):424–433. | ||
Jadus MR, Natividad J, Mai A, et al. Lung cancer: a classic example of tumor escape and progression while providing opportunities for immunological intervention. Clin Dev Immunol. 2012;2012:160724. | ||
Wang X, Sun Q, Liu Q, Wang C, Yao R, Wang Y. CTC immune escape mediated by PD-L1. Med Hypotheses. 2016;93:138–139. | ||
Li Z, Dong P, Ren M, et al. PD-L1 expression is associated with tumor FOXP3(+) regulatory T-cell infiltration of breast cancer and poor prognosis of patient. J Cancer. 2016;7(7):784–793. | ||
Li CW, Lim SO, Xia W, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. | ||
AlHossiny M, Luo L, Frazier WR, et al. Ly6E/K signaling to TGFβ promotes breast cancer progression, immune escape, and drug resistance. Cancer Res. 2016;76(11):3376–3386. | ||
Bertucci F, Finetti P, Colpaert C, et al. PDL1 expression in inflammatory breast cancer is frequent and predicts for the pathological response to chemotherapy. Oncotarget. 2015;6(15):13506–13519. | ||
Bertucci F, Finetti P, Birnbaum D, Mamessier E. The PD1/PDL1 axis, a promising therapeutic target in aggressive breast cancers. Oncoimmunology. 2015;5(3):e1085148. | ||
McCullar B, Pandey M, Yaghmour G, et al. Genomic landscape of small cell carcinoma of the breast contrasted to small cell carcinoma of the lung. Breast Cancer Res Treat. 2016;158(1):195–202. | ||
Gröschel S, Bommer M, Hutter B, et al. Integration of genomics and histology revises diagnosis and enables effective therapy of refractory cancer of unknown primary with PDL1 amplification. Cold Spring Harb Mol Case Stud. 2016;2(6):a001180. | ||
Soliman H, Khalil F, Antonia S. PD-L1 expression is increased in a subset of basal type breast cancer cells. PLoS One. 2014;9(2):e88557. | ||
Beckers RK, Selinger CI, Vilain R, et al. Programmed death ligand 1 expression in triple-negative breast cancer is associated with tumour-infiltrating lymphocytes and improved outcome. Histopathology. 2016;69(1):25–34. | ||
Guo L, Li W, Zhu X, et al. PD-L1 expression and CD274 gene alteration in triple-negative breast cancer: implication for prognostic biomarker. Springerplus. 2016;5(1):805. | ||
Sabatier R, Finetti P, Mamessier E, et al. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget. 2015;6(7):5449–5464. | ||
Mazel M, Jacot W, Pantel K, et al. Frequent expression of PD-L1 on circulating breast cancer cells. Mol Oncol. 2015;9(9):1773–1782. | ||
Chatterjee S, Lesniak WG, Gabrielson M, et al. A humanized antibody for imaging immune checkpoint ligand PD-L1 expression in tumors. Oncotarget. 2016;7(9):10215–10227. | ||
Pusztai L, Ladányi A, Székely B, Dank M. [Immunotherapy opportunities in breast cancer]. Magy Onkol. 2016;60(1):34–40. Hungarian [with English abstract]. | ||
García-Teijido P, Cabal ML, Fernández IP, Pérez YF. Tumor-infiltrating lymphocytes in triple negative breast cancer: the future of immune targeting. Clin Med Insights Oncol. 2016;10 (Suppl 1):31–39. | ||
Lu L, Xu X, Zhang B, Zhang R, Ji H, Wang X. Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J Transl Med. 2014;12:36. | ||
Loi S, Dushyanthen S, Beavis PA, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22(6):1499–1509. | ||
Black M, Barsoum IB, Truesdell P, et al. Activation of the PD-1/PD-L1 immune checkpoint confers tumor cell chemoresistance associated with increased metastasis. Oncotarget. 2016;7(9):10557–10567. | ||
Kulkarni A, Natarajan SK, Chandrasekar V, Pandey PR, Sengupta S. Combining immune checkpoint inhibitors and kinase-inhibiting supramolecular therapeutics for enhanced anticancer efficacy. ACS Nano. Epub 2016 Sep 29. | ||
Mall C, Sckisel GD, Proia DA, et al. Repeated PD-1/PD-L1 monoclonal antibody administration induces fatal xenogeneic hypersensitivity reactions in a murine model of breast cancer. Oncoimmunology. 2015;5(2):e1075114. | ||
Xu Z, Shen J, Wang MH, et al. Comprehensive molecular profiling of the B7 family of immune-regulatory ligands in breast cancer. Oncoimmunology. 2016;5(8):e1207841. | ||
Hamed EA, Zakhary MM, Maximous DW. Apoptosis, angiogenesis, inflammation, and oxidative stress: basic interactions in patients with early and metastatic breast cancer. J Cancer Res Clin Oncol. 2012;138(6):999–1009. | ||
Fulda S, Meyer E, Debatin KM. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene. 2002;21(15):2283–2294. | ||
Zhivotovsky B, Orrenius S. Carcinogenesis and apoptosis: paradigms and paradoxes. Carcinogenesis. 2006;27(10):1939–1945. | ||
Jäger R, Herzer U, Schenkel J, Weiher H. Overexpression of Bcl-2 inhibits alveolar cell apoptosis during involution and accelerates c-myc-induced tumorigenesis of the mammary gland in transgenic mice. Oncogene. 1997;15(15):1787–1795. | ||
Ryan BM, Konecny GE, Kahlert S, et al. Survivin expression in breast cancer predicts clinical outcome and is associated with HER2, VEGF, urokinase plasminogen activator and PAI-1. Ann Oncol. 2006;17(4):597–604. | ||
Khan S, Ferguson Bennit H, Asuncion Valenzuela MM, et al. Localization and upregulation of survivin in cancer health disparities: a clinical perspective. Biologics. 2015;9:57–67. | ||
Devarajan E, Sahin AA, Chen JS, et al. Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene. 2002;21(57):8843–8851. | ||
Grigoriev MY, Pozharissky KM, Hanson KP, Imyanitov EN, Zhivotovsky B. Expression of caspase-3 and -7 does not correlate with the extent of apoptosis in primary breast carcinomas. Cell Cycle. 2002;1(5):337–342. | ||
Duechler M, Peczek L, Zuk K, Zalesna I, Jeziorski A, Czyz M. The heterogeneous immune microenvironment in breast cancer is affected by hypoxia-related genes. Immunobiology. 2014;219(2):158–165. | ||
Quigley DA, Kristensen V. Predicting prognosis and therapeutic response from interactions between lymphocytes and tumor cells. Mol Oncol. 2015;9(10):2054–2062. | ||
Nagorsen D, Scheibenbogen C, Marincola FM, Letsch A, Keilholz U. Natural T cell immunity against cancer. Clin Cancer Res. 2003;9(12):4296–4303. | ||
Pourchet A, Fuhrmann SR, Pilones KA, et al. CD8(+) T-cell immune evasion enables oncolytic virus immunotherapy. EBioMedicine. 2016;5:59–67. | ||
Colombo MP, Piconese S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer. 2007;7(11):880–887. | ||
Chaput N, Louafi S, Bardier A, et al. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut. 2009;58(4):520–529. | ||
Engel JB, Honig A, Kapp M, et al. Mechanisms of tumor immune escape in triple-negative breast cancers (TNBC) with and without mutated BRCA 1. Arch Gynecol Obstet. 2014;289(1):141–147. | ||
Janikashvili N, Bonnotte B, Katsanis E, Larmonier N. The dendritic cell-regulatory T lymphocyte crosstalk contributes to tumor-induced tolerance. Clin Dev Immunol. 2011;2011:430394. | ||
Loyher PL, Rochefort J, Baudesson de Chanville C, et al. CCR2 influences T regulatory cell migration to tumors and serves as a biomarker of cyclophosphamide sensitivity. Cancer Res. Epub 2016 Sep 28. | ||
Freier CP, Kuhn C, Endres S, et al. FOXP3+ cells recruited by CCL22 into breast cancer correlates with less tumor nodal infiltration. Anticancer Res. 2016;36(6):3139–3145. | ||
Hossain DM, Panda AK, Chakrabarty S, et al. MEK inhibition prevents tumour-shed transforming growth factor-β-induced T-regulatory cell augmentation in tumour milieu. Immunology. 2015;144(4):561–573. | ||
Gelao L, Criscitiello C, Esposito A, et al. Dendritic cell-based vaccines: clinical applications in breast cancer. Immunotherapy. 2014;6(3):349–360. | ||
Fricke I, Gabrilovich DI. Dendritic cells and tumor microenvironment: a dangerous liaison. Immunol Invest. 2006;35(3–4):459–483. | ||
Bell D, Chomarat P, Broyles D, et al. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med. 1999;190(10):1417–1426. | ||
Bai XF, Liu J, Li O, Zheng P, Liu Y. Antigenic drift as a mechanism for tumor evasion of destruction by cytolytic T lymphocytes. J Clin Invest. 2003;111(10):1487–1496. | ||
da Cunha A, Michelin MA, Murta EF. Pattern response of dendritic cells in the tumor microenvironment and breast cancer. World J Clin Oncol. 2014;5(3):495–502. | ||
Farren MR, Carlson LM, Netherby CS, et al. Tumor-induced STAT3 signaling in myeloid cells impairs dendritic cell generation by decreasing PKCβII abundance. Sci Signal. 2014;7(313):ra16. | ||
Baragaño Raneros A, Martín-Palanco V, Fernandez AF, et al. Methylation of NKG2D ligands contributes to immune system evasion in acute myeloid leukemia. Genes Immun. 2015;16(1):71–82. | ||
Coudert JD, Zimmer J, Tomasello E, et al. Altered NKG2D function in NK cells induced by chronic exposure to NKG2D ligand-expressing tumor cells. Blood. 2005;106(5):1711–1717. | ||
Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. 2004;172(12):7335–7340. | ||
Crane CA, Austgen K, Haberthur K, et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc Natl Acad Sci U S A. 2014;111(35):12823–12828. | ||
El-Gazzar A, Cai X, Reeves RS, et al. Effects on tumor development and metastatic dissemination by the NKG2D lymphocyte receptor expressed on cancer cells. Oncogene. 2014;33(41):4932–4940. | ||
Esquivel-Velázquez M, Ostoa-Saloma P, Palacios-Arreola MI, Nava-Castro KE, Castro JI, Morales-Montor J. The role of cytokines in breast cancer development and progression. J Interferon Cytokine Res. 2015;35(1):1–16. | ||
Gunaydin G, Kesikli SA, Guc D. Cancer associated fibroblasts have phenotypic and functional characteristics similar to the fibrocytes that represent a novel MDSC subset. Oncoimmunology. 2015;4(9):e1034918. | ||
Garrido F, Cabrera T, Aptsiauri N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. Int J Cancer. 2010;127(2):249–256. | ||
Shepherd FA, Douillard JY, Blumenschein GR Jr. Immunotherapy for non-small cell lung cancer: novel approaches to improve patient outcome. J Thorac Oncol. 2011;6(10):1763–1773. | ||
Migali C, Milano M, Trapani D, et al. Strategies to modulate the immune system in breast cancer: checkpoint inhibitors and beyond. Ther Adv Med Oncol. 2016;8(5):360–374. | ||
Voutsadakis IA. Immune blockade inhibition in breast cancer. Anticancer Res. 2016;36(11):5607–5622. | ||
McArthur HL, Diab A, Page DB, et al. A pilot study of preoperative single-dose ipilimumab and/or cryoablation in women with early-stage breast cancer with comprehensive immune profiling. Clin Cancer Res. 2016;22(23):5729–5737. | ||
Tringler B, Zhuo S, Pilkington G, et al. B7-h4 is highly expressed in ductal and lobular breast cancer. Clin Cancer Res. 2005;11(5):1842–1848. | ||
Abadi YM, Jeon H, Ohaegbulam KC, et al. Host b7x promotes pulmonary metastasis of breast cancer. J Immunol. 2013;190(7):3806–3814. | ||
Herbertz S, Sawyer JS, Stauber AJ, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. 2015;9:4479–4499. | ||
Ewan KB, Oketch-Rabah HA, Ravani SA, Shyamala G, Moses HL, Barcellos-Hoff MH. Proliferation of estrogen receptor-alpha-positive mammary epithelial cells is restrained by transforming growth factor-beta1 in adult mice. Am J Pathol. 2005;167(2):409–417. | ||
Band AM, Laiho M. Crosstalk of TGF-β and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2011;16(2):109–115. | ||
Imamura T, Hikita A, Inoue Y. The roles of TGF-β signaling in carcinogenesis and breast cancer metastasis. Breast Cancer. 2012;19(2):118–124. | ||
Singh G, Singh SK, König A, et al. Sequential activation of NFAT and c-Myc transcription factors mediates the TGF-beta switch from a suppressor to a promoter of cancer cell proliferation. J Biol Chem. 2010;285(35):27241–27250. | ||
Chang NS. Transforming growth factor-beta protection of cancer cells against tumor necrosis factor cytotoxicity is counteracted by hyaluronidase (review). Int J Mol Med. 1998;2(6):653–659. | ||
Taube JH, Herschkowitz JI, Komurov K, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107(35):15449–15454. | ||
Jain P, Alahari SK. Breast cancer stem cells: a new challenge for breast cancer treatment. Front Biosci (Landmark Ed). 2011;16:1824–1832. | ||
Whiteside TL. Regulatory T cell subsets in human cancer: are they regulating for or against tumor progression? Cancer Immunol Immunother. 2014;63(1):67–72. | ||
Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139(4):693–706. | ||
Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010;39(4):493–506. | ||
Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121(10):3804–3809. | ||
Sullivan NJ, Sasser AK, Axel AE, et al. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28(33):2940–2947. | ||
Xiao P, Wan X, Cui B, et al. Interleukin 33 in tumor microenvironment is crucial for the accumulation and function of myeloid-derived suppressor cells. Oncoimmunology. 2015;5(1):e1063772. | ||
Kanzaki H, Shinohara F, Suzuki M, et al. A-disintegrin and metalloproteinase (ADAM) 17 enzymatically degrades interferon-gamma. Sci Rep. 2016;6:32259. | ||
Park S, Cheon S, Cho D. The dual effects of interleukin-18 in tumor progression. Cell Mol Immunol. 2007;4(5):329–335. | ||
Eissa SA, Zaki SA, El-Maghraby SM, Kadry DY. Importance of serum IL-18 and RANTES as markers for breast carcinoma progression. J Egypt Natl Canc Inst. 2005;17(1):51–55. | ||
Hsing CH, Cheng HC, Hsu YH, et al. Upregulated IL-19 in breast cancer promotes tumor progression and affects clinical outcome. Clin Cancer Res. 2012;18(3):713–725. | ||
Hsu YH, Hsing CH, Li CF, et al. Anti-IL-20 monoclonal antibody suppresses breast cancer progression and bone osteolysis in murine models. J Immunol. 2012;188(4):1981–1991. | ||
Langowski JL, Zhang X, Wu L, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442(7101):461–465. | ||
Hamidullah, Changkija B, Konwar R. Role of interleukin-10 in breast cancer. Breast Cancer Res Treat. 2012;133(1):11–21. | ||
Zhou BP, Hu MC, Miller SA, et al. HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-kappaB pathway. J Biol Chem. 2000;275(11):8027–8031. | ||
Mocellin S, Rossi CR, Pilati P, Nitti D. Tumor necrosis factor, cancer and anticancer therapy. Cytokine Growth Factor Rev. 2005;16(1):35–53. | ||
Baumgarten SC, Frasor J. Minireview: inflammation: an instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol Endocrinol. 2012;26(3):360–371. | ||
Donato NJ, Klostergaard J. Distinct stress and cell destruction pathways are engaged by TNF and ceramide during apoptosis of MCF-7 cells. Exp Cell Res. 2004;294(2):523–533. | ||
Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9(5):361–371. | ||
Connelly L, Barham W, Onishko HM, et al. Inhibition of NF-kappa B activity in mammary epithelium increases tumor latency and decreases tumor burden. Oncogene. 2011;30(12):1402–1412. | ||
Datta J, Rosemblit C, Berk E, et al. Progressive loss of anti-HER2 CD4+ T-helper type 1 response in breast tumorigenesis and the potential for immune restoration. Oncoimmunology. 2015;4(10):e1022301. | ||
Watson AP, Evans RL, Egland KA. Multiple functions of sushi domain containing 2 (SUSD2) in breast tumorigenesis. Mol Cancer Res. 2013;11(1):74–85. | ||
Stannard KA, Collins PM, Ito K, et al. Galectin inhibitory disaccharides promote tumour immunity in a breast cancer model. Cancer Lett. 2010;299(2):95–110. | ||
Salatino M, Dalotto-Moreno T, Rabinovich GA. Thwarting galectin-induced immunosuppression in breast cancer. Oncoimmunology. 2013;2(5):e24077. | ||
Dalotto-Moreno T, Croci DO, Cerliani JP, et al. Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Res. 2013;73(3):1107–1117. | ||
Ricciardi M, Zanotto M, Malpeli G, et al. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br J Cancer. 2015;112(6):1067–1075. | ||
Puccetti P, Fallarino F, Italiano A, et al. Accumulation of an endogenous tryptophan-derived metabolite in colorectal and breast cancers. PLoS One. 2015;10(4):e0122046. | ||
Levina V, Su Y, Gorelik E. Immunological and nonimmunological effects of indoleamine 2,3-dioxygenase on breast tumor growth and spontaneous metastasis formation. Clin Dev Immunol. 2012;2012:173029. | ||
Welte T, Rosen JM, Zhang XH. Fatal attraction: TICs and MDSCs. Cell Cycle. 2016;15(19):2545–2546. | ||
Peng D, Tanikawa T, Li W, et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signaling. Cancer Res. 2016;76(11):3156–3165. | ||
Forghani P, Waller EK. Poly (I: C) modulates the immunosuppressive activity of myeloid-derived suppressor cells in a murine model of breast cancer. Breast Cancer Res Treat. 2015;153(1):21–30. | ||
Alizadeh D, Trad M, Hanke NT, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014;74(1):104–118. | ||
Vinay DS, Ryan EP, Pawelec G, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35 Suppl:S185–S198. | ||
Breakthrough of the year 2013. Notable developments. Science. 2013;342(6165):1435–1441. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.