")
Back to Journals » International Journal of Nanomedicine » Volume 12
Mechanism of hepatic targeting via oral administration of DSPE–PEG–cholic acid-modified nanoliposomes
Received 18 October 2016
Accepted for publication 4 January 2017
Published 28 February 2017 Volume 2017:12 Pages 1673—1684
DOI https://doi.org/10.2147/IJN.S125047
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Linlin Sun
Ying Li, Chunyan Zhu
Department of Drug Delivery Research Center, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing, People’s Republic of China
Abstract: In oral administration, gastrointestinal physiological environment, gastrointestinal epithelial cell membranes, and blood circulation are typical biological barriers to hepatic delivery of ligand-modified nanoparticle drug delivery systems. To elucidate the mechanism of oral hepatic targeting of cholic acid receptor-mediated nanoliposomes (LPs) (distearoyl phosphatidylethanolamine–polyethylene glycol–cholic acid-modified LPs, CA-LPs), evaluations were performed on colon cancer Caco-2 cell monolayers, liver cancer HepG2 cells, and a rat intestinal perfusion model. CA-LPs, ~100 nm in diameter, exhibited sustained-release behavior and had the greatest stability in rat gastrointestinal fluid and serum for both size and entrapment efficiency. CA-LPs demonstrated highest transport across Caco-2 cells and highest cellular uptake by HepG2 cells. The enhanced endocytosis of CA-LPs was found to be mediated by Na+/taurocholate cotransporting polypeptide and involved the caveolin-mediated endocytosis pathway. Further, we used fluorescence resonance energy transfer (FRET) technology to show that the CA-LPs maintained their structural integrity in part during the transport across the Caco-2 cell monolayer and uptake by HepG2 cells.
Keywords: DSPE–PEG–cholic acid, nanoliposomes, hepatic targeting via oral administration, mechanism, FRET
Introduction
Therapeutic nanoliposomes (LPs) are lipid bilayer structures with a hydrophilic core and a lipophilic bilayer space for drugs; this space is segregated from the environment by a hydrophilic corona, usually containing polyethylene glycol (PEG). This hydrophilic PEG corona prevents recognition by macrophages and enables long-term circulation in the bloodstream.1–3 The size of LPs (10–100 nm) permits their extravasation and accumulation in tumor sites – known as the enhanced permeability and retention effect.4–7 Passive targeting is based on pathophysiological characteristics unique to solid tumors, such as hypervascularity, irregular vascular architecture, potential for secretion of vascular permeability factors, and the absence of effective lymphatic drainage that prevents efficient clearance of macromolecules. Active targeting is mainly based on the specific binding of receptors to ligands.8
Physiological obstacles have precluded oral administration of LPs for hepatic targeting. To increase intestinal uptake, LPs can be conjugated with various ligands, including bioadhesives (eg, poly [lactic acid]),9 P-glycoprotein (P-gp) pump inhibitors (eg, D-α-tocopheryl PEG succinate),10 vitamins11–14 (eg, biotin, folic acid, and vitamin B12), and transferrin.
Cholic acid receptor-mediated nanoparticle drug delivery systems have frequently been reported as oral hepatic drug delivery systems because of oral hepatic targeting properties of cholic acid15–19 and because of their ability to maintain the structural integrity in the process of physiological disposition. Therefore, it is vital to understand the related mechanisms. Currently, studies on the transport mechanism have mostly focused on the uptake pathway and partially on the intracellular trafficking of nanoparticles in different cell types. For hepatic targeting, nanoparticles need to pass from the apical (AP) membrane to the basolateral (BL) side of the gastrointestinal epithelial cells, enter the blood circulation, and diffuse into liver cells. Consequently, to elucidate the molecular mechanisms underlying nanoparticle oral hepatic targeting, it is crucial to comprehensively understand the entire gastrointestinal epithelial cell transport, blood circulation, and liver cell uptake process, especially because the roles and pathways in different steps of the process may vary.
We had previously constructed a modified distearoyl phosphatidylethanolamine (DSPE)–PEG–cholic acid LP system loaded with doxorubicin (DOX)·hydrochloric acid (HCl) (CA-LPs–DOX·HCl),20 which had the advantages of ease of synthesis, low cytotoxicity, and good safety for biomedical and pharmaceutical applications. To investigate its effectiveness in oral hepatic targeting and explain the possible underlying mechanisms thereof, we studied the release behavior and stability in vitro using gastrointestinal fluid and a CA-LP system loaded with DOX·HCl. We used the human colon carcinoma cell line, Caco-2, as an epithelial cell model to investigate the transport of LPs across the epithelial cell monolayer, because the Caco-2 system is one of the most extensively utilized assays for the assessment of permeability and it possesses many enterocytes that can express various efflux transporters, microvillar transporters, hydrolases, and conjugation enzymes, and can also display brush border region and cell tight junctions. A permanent hepatoma carcinoma cell line, HepG2, was used to investigate the uptake of LPs by hepatocytes, because it is liable to culture and can express the Na+/taurocholate cotransporting polypeptide (NTCP) receptor. Fluorescence resonance energy transfer (FRET) and fluorescence co-localization were used to monitor and study the transport across the epithelial cell monolayer and hepatic uptake of the CA-LP system.
Methods
Materials
DSPE–PEG–cholic acid was synthesized in our laboratory (batch number: 150830). Soybean phospholipids (SPC) were purchased from Lipoid (Ludwigshafen, Germany). Cholesterol, Hoechst 33258, sodium azide, genistein, methyl-β-cyclodextrin (MβCD), nystatin, and chlorpromazine were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). 3,3′-dioctadecyloxacarbocyanine perchlorate (DIO) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DII) were purchased from Beijing Fanbo Biochemicals Co. Ltd. (Beijing, China). Fetal bovine serum (FBS) was purchased from Gibco (Grand Island, NY, USA). Minimum Essential Medium/Hanks’ Balanced Salt Solution and Roswell Park Memorial Institute 1640/Hanks’ Balanced Salt Solution were purchased from HyClone (Logan, UT, USA). Penicillin–streptomycin was purchased from Invitrogen (Grand Island, NY, USA). Parenzyme was purchased from Gibco (Grand Island, NY, USA). Caco-2 and HepG2 cell lines were purchased from Cell Resource Centre, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (CAMS, Beijing, China).
Sprague Dawley rats (male, 250±20 g) were obtained from the CAMS and Peking Union Medical College (PUMC) Health Science Center (Beijing, China) and kept under specific pathogen-free (SPF) condition for 1 week before the study, with free access to standard food and water. All studies in mice were performed according to guidelines approved by the ethics committee of the CAMS and PUMC. The ethics committee of the CAMS and PUMC approved this study.
Preparation and characterization of LPs
DSPE–PEG–cholic acid, phospholipid, and cholesterol were combined to form LPs. DOX·HCl was actively loaded into the preformed LPs using the ammonium sulfate gradient method. Subsequently, DSPE–PEG–cholic acid, phospholipid, and cholesterol were dissolved at high concentrations in ethanol, and the solutions were mixed with 250 mM aqueous ammonium sulfate at 50°C. The resulting multilamellar vesicles were stepwise extruded using a Lipex extruder (Northern Lipids, Inc., Burnaby, BC, Canada). The transmembrane ammonium sulfate gradient was generated by the removal of external ammonium sulfate and subsequent replacement in three dialysis steps against 0.9% NaCl solution. Empty LPs and DOX·HCl solution were mixed and incubated for 30 min at 60°C. DII and DIO were included as fluorescent probes and were prepared using an ethanol injection method. The DIO/DII-loaded LPs modified with cholic acid or unmodified (LPs-DIO/DII or CA-LPs-DIO/DII) were prepared using the ethanol injection method. The particle sizes, polydispersity indexes (PDIs), and zeta potential values of all LPs were measured using a Nano Series ZS Zeta sizer (Malvern Instruments Ltd., Worcestershire, UK).
In vitro release test
To investigate the release of DOX·HCl from LPs, pure DOX·HCl, LPs-DOX·HCl, and CA-LPs-DOX·HCl were suspended in 1 mL aliquots of distilled water and placed in dialysis tubing (8,000–12,000 molecular weight cutoff; Greenbird Inc., Shanghai, China). Whole dialysis tubes containing CA-LPs-DOX·HCl, LPs-DOX·HCl, or DOX·HCl were then submerged in 50 mL of phosphate-buffered saline (PBS; pH 7.4) release media at 37°C and immediately shaken at a speed of 100 rpm. Subsequently, 1 mL aliquots of the release medium were sampled at 0.5 h, 1 h, 2 h, 4 h, 6 h, and 8 h and immediately replaced with equal volumes of fresh release medium. The DOX·HCl contents of the samples were determined by high-performance liquid chromatography (HPLC). Release rates (RRs) (%) were calculated using the following formula: RR = (Wi/Wtotal) × 100%, where Wi is the quantity of DOX·HCl in the release medium at the ith time point and Wtotal is the total quantity of DOX·HCl in equal volumes of corresponding LP suspensions. Experiments were performed in triplicate.
Stability studies in rat gastric fluid, intestinal fluid, or serum
The formulations were incubated in gastric fluid, intestinal fluid, or serum of mice for 2 h, 6 h, or 12 h, respectively. The encapsulation potencies before and after incubation in gastric fluid, intestinal fluid, or serum of mice were measured to investigate the stability of different formulations.
Transport of LPs across the Caco-2 cell monolayer
To investigate the influence of different DOX·HCl formulations on the adsorption properties of DOX·HCl, Caco-2 cells were used as an in vitro model of gastrointestinal epithelium. To evaluate the transport, various formulations of DOX·HCl were diluted with D-Hank’s solution to a final concentration of 5 μg/mL DOX·HCl as the test solutions, including free DOX·HCl, LPs-DOX·HCl, and CA-LPs-DOX·HCl. A 1.5 mL volume of sample was taken from the BL side at 2 h. Sample aliquots of 500 μL were mixed with 500 μL of methanol, shaken for 1 min using a vortex mixer, and centrifuged at 8,000 rpm for 10 min. The supernatant was injected into the HPLC system for measuring DOX·HCl content. The apparent permeability coefficient (Papp) was calculated according to the following equation: Papp = dQ/dt ×1/(AC0), where dQ/dt is the permeability rate, C0 is the initial concentration at the AP side, and A is the surface area of a monolayer.
The transepithelial electrical resistance (TEER) was determined using a Millicell-ER system (Millipore Corporation, Bedford, MA, USA) before and after membrane absorption. After membrane absorption, the cells were washed three times with D-Hank’s solution, complete media was added for 2 h or 24 h, and the TEER was then determined; the cell toxicity of test drugs and regeneration of cell membrane were also investigated.
After transport across the Caco-2 cell monolayer, the cells were washed three times with cold PBS, trypsinized, washed three times with PBS again, and then suspended in 0.4 mL of PBS. The mean concentrations of DOX·HCl in the cells were measured by using flow cytometry (BD, Franklin Lakes, NJ, USA).
In vitro cellular uptake assays
To investigate the in vitro cellular uptake of various formulations, HepG2 cells were seeded in six-well cell culture plates (Corning Incorporated, Corning, NY, USA), and on the following day, confluent cells were incubated with pure DOX·HCl, LPs-DOX·HCl, or CA-LPs-DOX·HCl (containing DOX·HCl at 5 μg/mL) in serum-free medium at 37°C for 2 h. After incubation, the cells were washed three times with cold PBS, trypsinized, washed three times with PBS again, and then suspended in 0.4 mL of PBS. The mean concentrations of DOX·HCl in the cells were measured by flow cytometry.
Prior to confocal microscope studies, HepG2 cells were seeded in petri dishes for 24 h and then washed with PBS. After incubation with pure DOX·HCl and CA-LPs-DOX·HCl at 37°C for 2 h, the cells were washed three times with PBS and fixed with fresh 4% paraformaldehyde for 10 min at room temperature. The cells were then counterstained with the nuclear stain Hoechst 33258 for 10 min. Blue fluorescence of Hoechst 33258 and red fluorescence of DOX·HCl were observed using a Zeiss LSM780 CLSM instrument (Zeiss Co., Oberkochen, Germany).
Endocytosis pathway activities at various temperatures
HepG2 cells were pretreated at various temperatures for 30 min and then incubated with CA-LPs for 2 h at the same temperatures. Cellular uptake of LPs was then measured as described earlier.
Endocytosis pathway activity using uptake inhibitors
HepG2 cells were pretreated with the inhibitors NaN3 (1 mg/mL), genistein (50 μg/mL), MβCD (10 mM), nystatin (50 μg/mL), chlorpromazine (10 μg/mL), and cholic acid (1 mg/mL) for 30 min. After removing the inhibitors, the cells were incubated with CA-LPs for 2 h, and the cellular uptake of LPs was determined as described in the “In vitro cellular uptake assays” section.
HepG2 cell uptake after transport across the Caco-2 cell monolayer
Caco-2 cells were seeded in the AP chamber of Transwell plates, and HepG2 cells were seeded in a glass-bottom culture dish. The AP chambers of the Transwell plates were placed on the glass-bottom culture dish. The cells in the AP chamber were washed three times with serum-free medium, and the Caco-2 cells were incubated with pure DOX·HCl or CA-LPs-DOX·HCl (containing DOX·HCl at 5 μg/mL) in serum-free medium at 37°C for 2 h. After incubation, the HepG2 cells in the glass-bottom culture dish were washed three times with PBS at 4°C, trypsinized, washed three times with PBS again, and then suspended in 0.4 mL of PBS. The mean concentrations of DOX·HCl in the cells were measured by flow cytometry.
FRET analysis
FRET analysis was performed as previously reported.21–23 A FRET pair of hydrophobic dyes, DII as acceptor and DIO as donor, was physically loaded into the LPs. To verify the occurrence of FRET, fluorescence spectra of DIO and DII double-loaded LPs diluted in deionized water or acetone were measured using a fluorospectrophotometer (Techcomp Ltd., Hong Kong, China) with excitation at 488 nm and the emission scan from 500 nm to 600 nm. The FRET efficiency of the CA-LPs was calculated according to the following formula: FRET ratio = IR/(IG + IR), where IR and IG are the fluorescence intensities at 576 nm and 508 nm, respectively.
Caco-2 cells were seeded in the AP chamber of Transwell plates and in a glass-bottom culture dish, and the AP chamber of the Transwell plates were placed on the glass-bottom culture dish. The cells in the AP chamber were washed three times with serum-free medium, and the Caco-2 cells were incubated with the CA-LPs-DIO/DII and a physical mixture of CA-LPs-DIO and CA-LPs-DII in serum-free medium at 37°C for 2 h.
HepG2 cells were seeded in a glass-bottom culture dish, and the cells were washed three times with serum-free medium and then incubated with the CA-LPs-DIO/DII and a physical mixture of CA-LPs-DIO and CA-LPs-DII in serum-free medium at 37°C for 2 h.
After incubation, the Caco-2 cells in the glass-bottom culture dish and the AP chamber and the HepG2 cells were washed three times with PBS at 4°C, fixed with fresh 4% paraformaldehyde for 10 min at room temperature, and washed three times with PBS again, and then the FRET efficiencies were observed using a Zeiss LSM780 CLSM instrument.
Intestinal localization in an in situ perfused rat intestinal model
Male Sprague Dawley rats were fasted for 12 h before the experiments and then anesthetized by intraperitoneal injection of urethane (2 g/kg). Midline incisions were then performed in the abdomen, and pylori were ligated using cotton threads. Subsequently, 1 mL aliquots of LPs-DIO/DII were injected into the jejunum and incubated for 1 h. Subsequently, 1 cm segments of the duodenum, jejunum, and ileum were excised from the intestines, washed several times with NaCl solution (0.9% w/v), and then frozen and sectioned for analysis by a DeltaVision microscope (DV Elite, Pittsburgh, PA, USA).
Statistical analysis
Statistical analysis was performed using SPSS 17.0 statistical software. The data are presented as mean ± standard error (SE). The statistical significance of differences between experimental and control groups was determined using one-way analysis of variance (ANOVA). The level of statistical significance for all tests was set to P<0.05.
Results
Characterization of LPs
The CA-LPs-DIO/DII were spherical in shape and had particle sizes of 92.5±1.93 nm, PDI of 0.15±0.027, and zeta potentials of −7.44±1.33 mV. The CA-LPs-DOX·HCl were spherical in shape and had particle sizes of 95.94±3.09 nm, PDI of 0.254±0.0015, and zeta potentials of −9.40±1.20 mV. The transmission electron microscope images and dynamic light scattering (DLS) size distribution of LPs are shown in Figure S1.
In vitro release test
A sequence of decreasing RR is shown in Figure 1: DOX·HCl solution > LPs-DOX·HCl > CA-LPs-DOX·HCl. Moreover, CA-LPs-DOX·HCl exhibited the slowest release under the same conditions, followed by LPs-DOX·HCl, which suggested that the current CA-LPs have sustained-release properties.
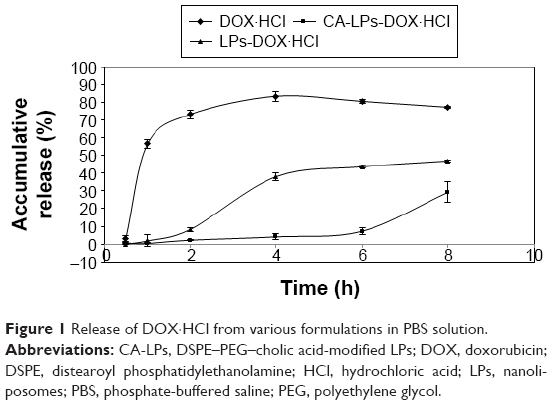 | Figure 1 Release of DOX·HCl from various formulations in PBS solution. |
Stability study in rat gastric fluid, intestinal fluid, or serum
Figure 2 shows that the particle size of CA-LPs remained <200 nm before and after incubation of LPs in rat gastric fluid, rat intestinal fluid, or rat serum for 2 h, 6 h, or 12 h, respectively, and that the encapsulation potency still exceeded 80% after their incubations; this demonstrated that the presence of DSPE–PEG–cholic acid prevented gastrointestinal enzyme destruction and protein adsorption in blood and enhanced the stability of the LPs. The longest incubation time in the following cell experiments was 2 h, and the low drug leakage during the tested period ensured that DOX·HCl was still mostly encapsulated in the LPs during the cell experiments.
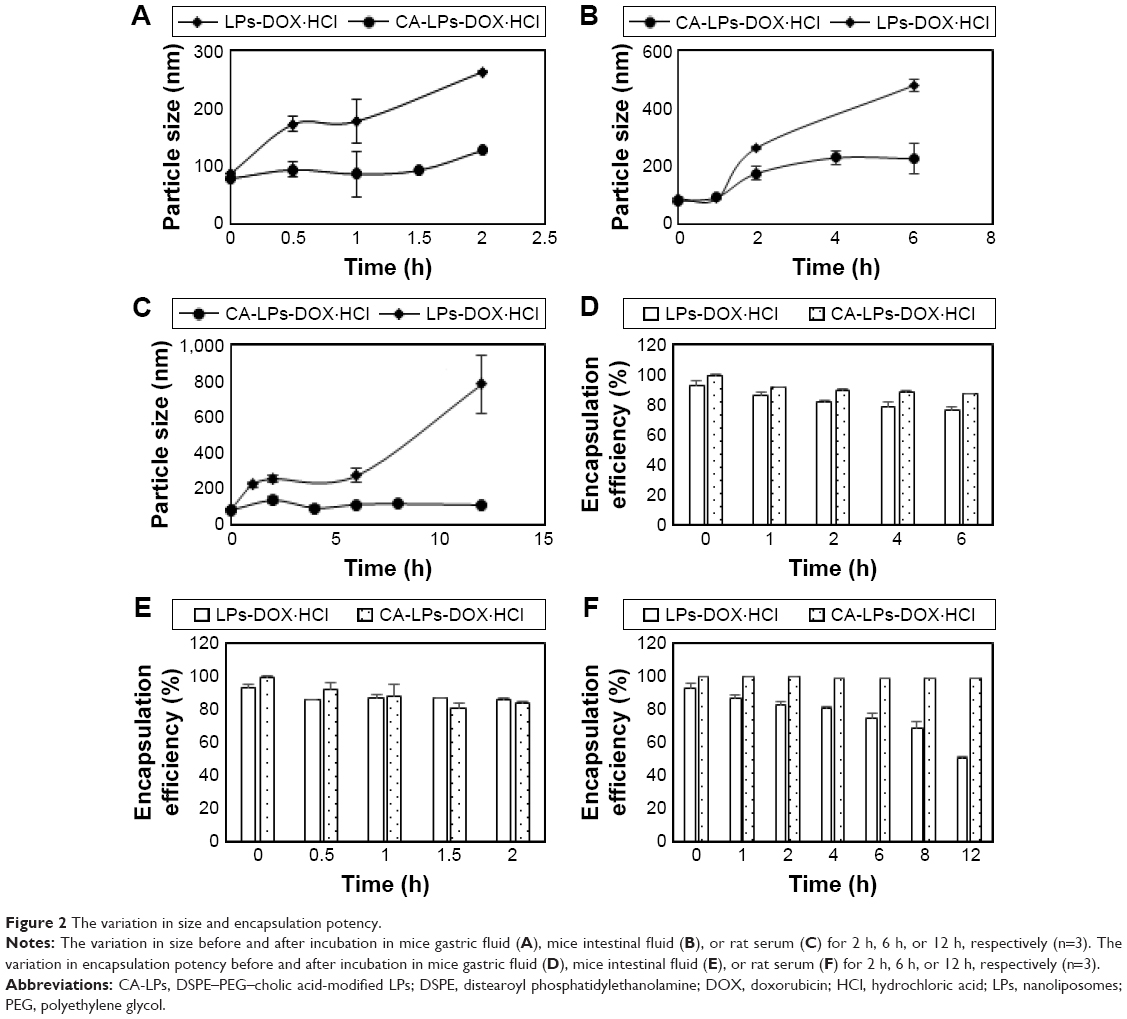 | Figure 2 The variation in size and encapsulation potency. |
Transport of LPs across Caco-2 cell monolayer
As shown in Figure 3A, the TEER showed no obvious change after transport across the Caco-2 cell monolayer of different DOX·HCl preparations, which suggested that the transport process does not involve the paracellular route. As shown in Figure 3B, the order of DOX·HCl transport rates was CA-LPs-DOX·HCl > LPs-DOX·HCl > DOX·HCl solution. Compared with the Papp of the DOX·HCl solution, the Papp of LPs-DOX·HCl improved by 3.1-fold and that of CA-LPs-DOX·HCl improved by 9.5-fold. Compared with the retention of DOX·HCl solution, the retention in Caco-2 cells of LPs-DOX·HCl improved by 4.85-fold and CA-LPs-DOX·HCl improved by 6.65-fold (Figure 3C and D).
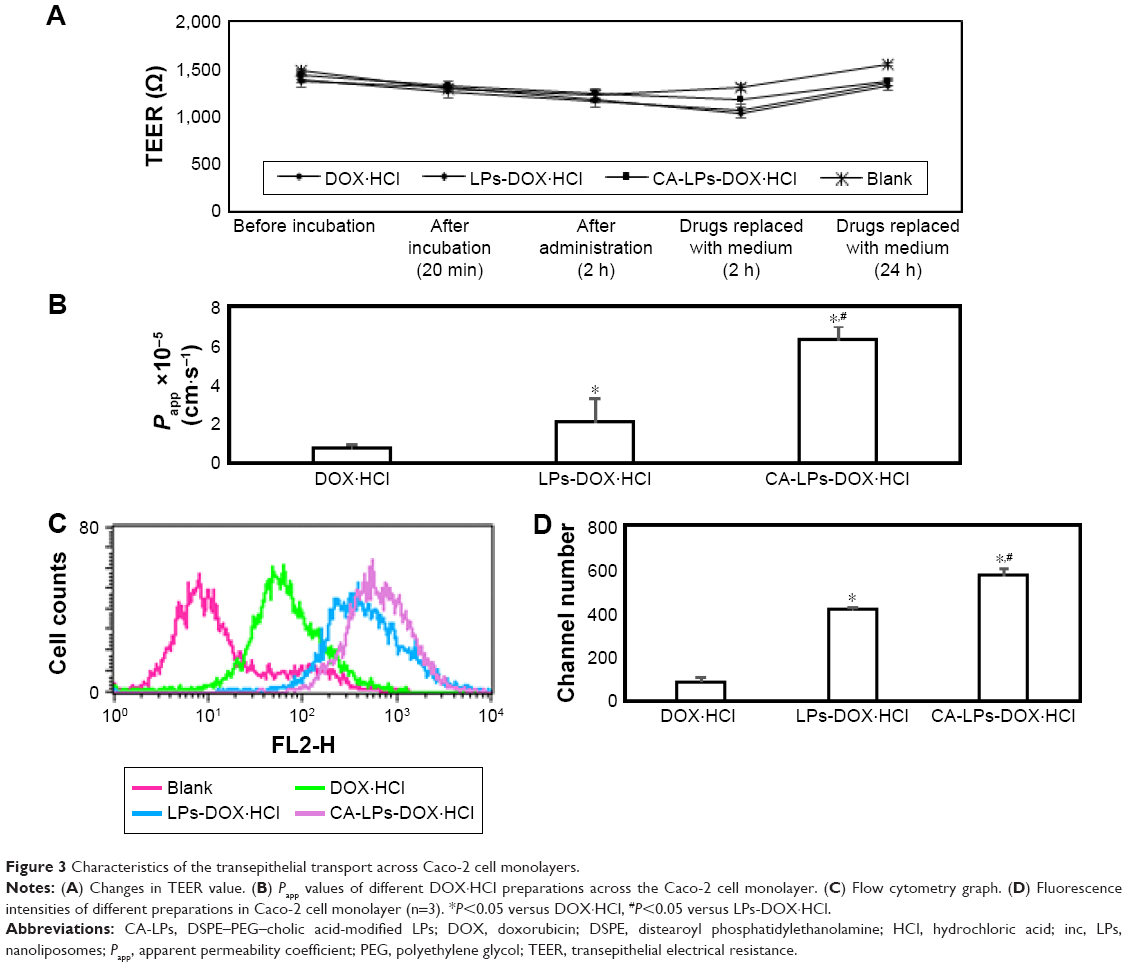 | Figure 3 Characteristics of the transepithelial transport across Caco-2 cell monolayers. |
In vitro cellular uptake of formulations
Confocal microscopy images (Figure 4A) and intracellular uptake assays (Figure 4B–E) of HepG2 cells at 2 h after applying Hoechst 33258 to one of the three drug formulations demonstrated that CA-LPs-DOX·HCl provided superior drug delivery. Specifically, nuclear fluorescence was greater in the cells treated with CA-LPs-DOX·HCl than the other formulations. Similarly, cellular uptake was correspondingly improved in the flow cytometry experiments, with 1.80- and 6.80-fold higher uptake in the presence of LPs-DOX·HCl and CA-LPs-DOX·HCl, respectively, relative to that in the presence of pure DOX·HCl.
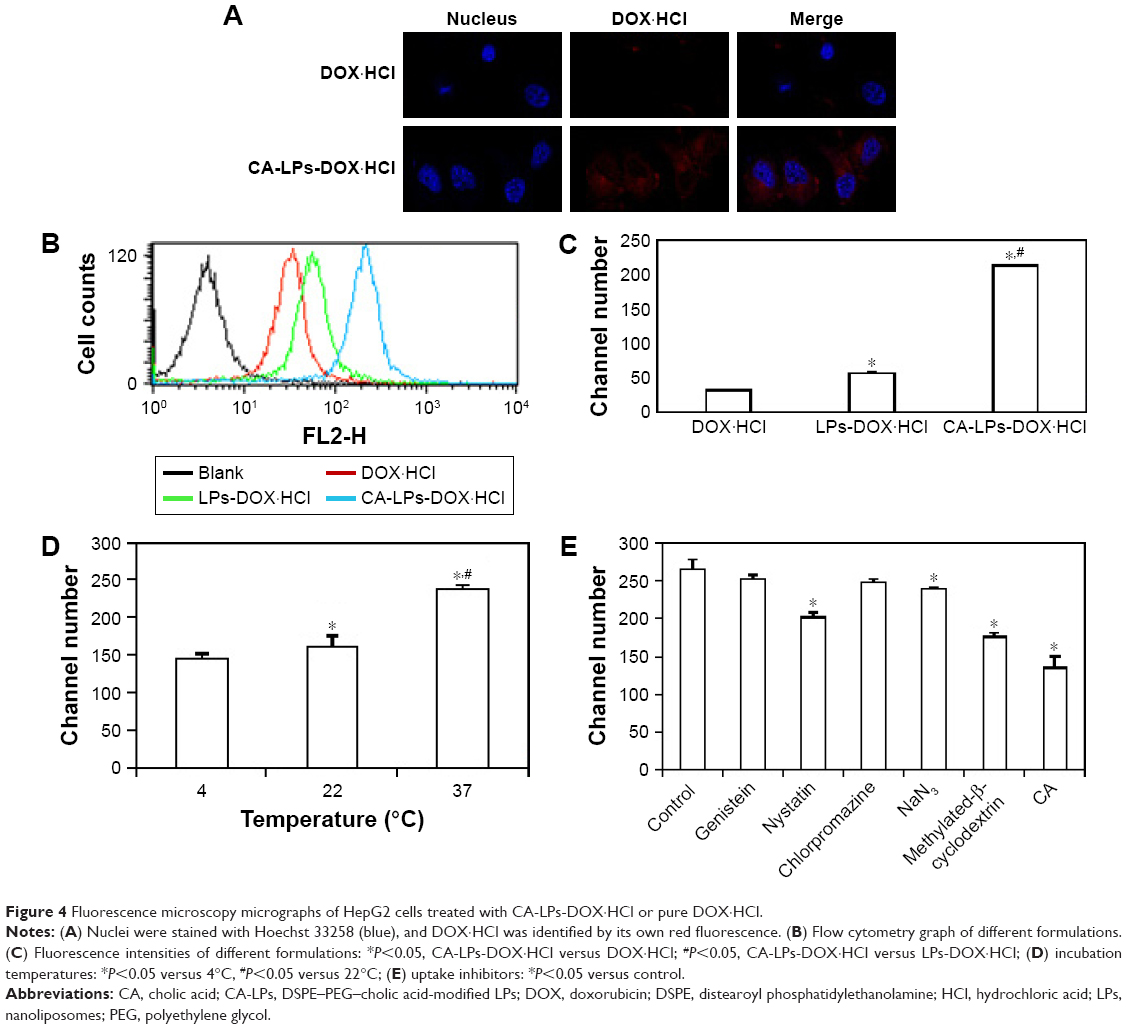 | Figure 4 Fluorescence microscopy micrographs of HepG2 cells treated with CA-LPs-DOX·HCl or pure DOX·HCl. |
Endocytosis pathway activities at various temperatures or using uptake inhibitors
Uptake of CA-LPs-DOX·HCl at 4°C and 22°C was significantly lower than at 37°C (P<0.05). In the current experiments with uptake inhibitors, NaN3, MβCD, nystatin, and cholic acid all significantly inhibited the uptake of CA-LPs-DOX·HCl (P<0.05); chlorpromazine had no obvious effect on cellular uptake of CA-LPs-DOX·HCl (P>0.05; Figure 4B–E).
HepG2 cell uptake after transport across a Caco-2 cell monolayer
DOX·HCl was taken up by HepG2 cells after transport across a Caco-2 cell monolayer to simulate the process of drug transport through the gastrointestinal tract and liver uptake (Figure 5A). The results indicated that HepG2 cell uptake of CA-LPs-DOX·HCl was 12.08 times that of free DOX·HCl. The HepG2 cell uptake after transport across a Caco-2 cell monolayer was only 6.8 times that of free DOX·HCl (Figure 5B and C), which indicated that the amount of CA-LPs-DOX·HCl uptake by HepG2 cells increased greatly after transport across a Caco-2 cell monolayer.
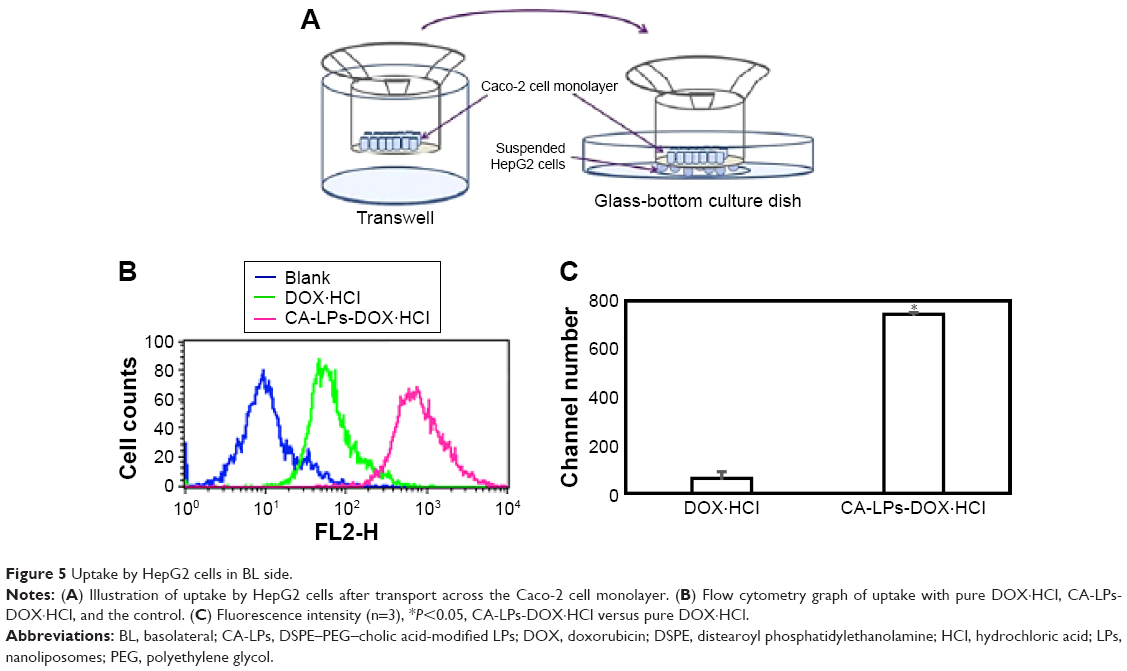 | Figure 5 Uptake by HepG2 cells in BL side. |
FRET analysis
The FRET phenomenon of LPs is shown in Figure 6A. The LPs had an average diameter of ~95 nm as measured by DLS. When the LPs were dispersed in deionized water, a strong DII signal was observed (red curve in Figure 6B) due to the close proximity of DII and DIO in the LPs core, with a FRET DII/(DIO + DII) ratio of 0.64, where DII and DIO are the fluorescence intensities at 578.1 nm and 502.7 nm, respectively. After treatment of the LPs with acetone, the FRET phenomenon disappeared (green curve in Figure 6B) because DII and DIO were no longer closely retained, resulting in a FRET ratio of 0.078. The schematic diagram of device used for uptake by Caco-2 cells after transport across the Caco-2 monolayer is shown in Figure 6C.
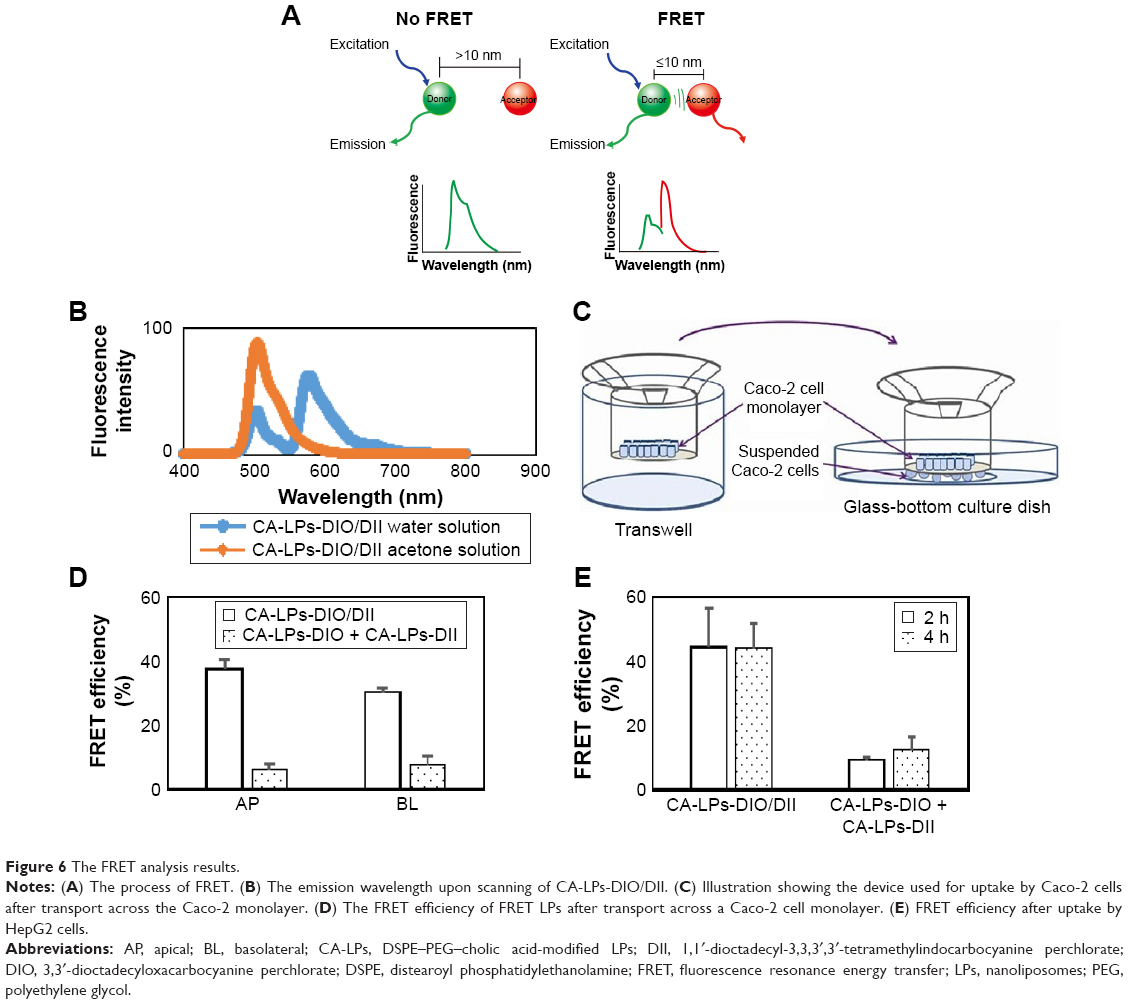 | Figure 6 The FRET analysis results. |
After a 2 h transport of the FRET LPs at 37°C, the intracellular FRET ratio in the AP and BL sides still exceeded 0.3.24 After transport across Caco-2 cell monolayer with non-FRET LPs for 2h at 37°C, the intracellular FRET ratio in the AP and BL side remained <0.1. The FRET ratio shows that the intracellular FRET phenomenon remained after the LPs were internalized and transported by cells for 2 h (Figure 6D), which confirmed the encapsulation of some lipophilic agents in the LPs after cellular uptake and transport.
After 2 h or 4 h uptake at 37°C with the FRET LPs, the intracellular FRET efficiencies of HepG2 cells were 44.52%±12.11% and 44.37%±7.29%, respectively. After 2 h or 4 h transport at 37°C of the non-FRET LPs, the intracellular FRET efficiencies of HepG2 cells were 9.37%±0.66% and 12.66%±3.60%, respectively. The FRET efficiencies show that the intracellular FRET phenomenon remained after CA-LPs were internalized by HepG2 cells for 2 h or 4 h (Figure 6E) confirming the encapsulation of some lipophilic agents in the CA-LPs after HepG2 cell uptake for 4 h.
Intestinal co-localization in an in situ perfused rat intestinal model
Distributions of DIO/DII-loaded LPs were qualitatively observed in the duodenal, jejunal, and ileal villi of the small intestines of rats. The green and red fluorescence represent DIO (DIO/DII, LPs-DIO/DII, and CA-LPs-DIO/DII) and DII (DIO/DII, LPs-DIO/DII, and CA-LPs-DIO/DII), respectively. Figure 7 shows the distributions of the preparations in the villi of the small intestine at 1 h after treatment. In these experiments, co-localization of DIO and DII is indicated by yellow staining of the villi. Following the administration of free DIO and DII, red fluorescence was distinctly separated from green fluorescence, which indicated that the free DIO and DII were not co-localized in the villi of the small intestines. In contrast, the administration of CA-LPs led to co-localization of the red and green fluorescence signals, which indicated efficient encapsulation of DIO and DII in the LPs with no leaks. Hence, CA-LPs-DIO/DII retained their structural integrity across the small intestine.
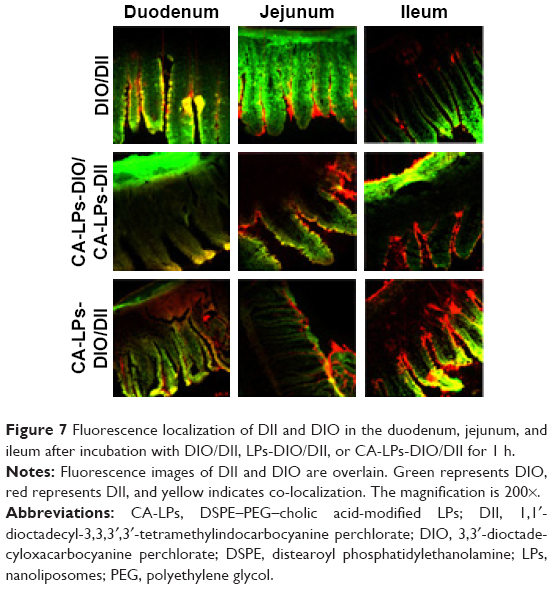 | Figure 7 Fluorescence localization of DII and DIO in the duodenum, jejunum, and ileum after incubation with DIO/DII, LPs-DIO/DII, or CA-LPs-DIO/DII for 1 h. |
Discussion
The CA-LPs were surface coated with the hydrophilic polymer PEG, which formed a hydrophilic barrier to prevent the accumulation of the LPs and to improve their physical stability; hence, the CA-LPs were characterized by slower release behavior.
The transit time of food in the body is as follows: 1–3 h in the stomach, 1.5–4.5 h in the intestine, or 12 h in blood circulation. Therefore, the incubation times in the rat gastric fluid, intestinal fluid, and serum were 2 h, 6 h, and 12 h, respectively.
The transport of DOX·HCl increased greatly after incorporation into LPs and CA-LPs, which demonstrated the positive transport enhancement effect of the LPs and cholic acid on hydrophobic molecules.
Nanoparticle drug carriers with particle sizes of <200 nm are subject to clathrin-mediated internalization, whereas larger nanoparticle drug carriers are internalized by microcapsule invagination of cytoplasmic membranes.25 Caveolae and vesicle membranes are coated with clathrin, form complexes with corresponding ligands, invaginate, separate from plasma membranes, and enter the cytoplasm as endosomes. Early endosomes then mature into late endosomes and subsequently merge with other endosomes to form lysosomes. Endocytosed substances are digested and degraded by lysosomal enzymes under conditions of low pH. In contrast, caveolae-mediated endocytosis can deliver carriers to caveoli under mild pH and enzyme conditions, which leads to subsequent transport in the endoplasmic reticulum, golgi apparatus, and cytoplasm and protection from the endosomal/lysosomal transport system.26,27
Because particle endocytosis depends on temperature and energy,28,29 NaN3 can effectively block endocytosis by undermining cellular energy sources. In the current experiments with uptake inhibitors, the uptakes of CA-LPs-DOX·HCl at 4°C and 37°C in the presence of NaN3 were significantly lower than those in control cells (P<0.05), which indicated that CA-LPs-DOX·HCl entered the cytoplasm via endocytosis.
In contrast with NaN3, MβCD is water soluble and forms complexes with cholesterol, which leads to cholesterol deficiencies in cytoplasmic membranes and structural impairments that abolish caveolae- and clathrin-mediated endocytosis. In the current experiments, MβCD significantly inhibited the uptake of CA-LPs-DOX·HCl, which indicated partial cell entry via caveolae- and clathrin-mediated endocytosis.
Nystatin also acts as a chelator of cholesterol from intact membrane rafts30–32 and inhibits caveolae-mediated endocytosis but not clathrin-mediated endocytosis. In the current experiments, nystatin significantly inhibited the uptake of CA-LPs-DOX·HCl, which indicated partial cytoplasmic entry via caveolae.
Cholic acid competitively bound NTCP on HepG2 cells and significantly inhibited the uptake of CA-LPs-DOX·HCl, which indicated that CA-LPs-DOX·HCl are also uptaken via NTCP-mediated endocytosis pathway.
Chlorpromazine prevents clathrin-mediated endocytosis by disrupting the assembly of the clathrin adaptor proteins at the cell surface. Moreover, genistein reportedly inhibits the activity of protein complexes of protein kinase (PTK) and prevents r-phosphorylation of tyrosine residues. In this study, chlorpromazine had no obvious effects on cellular uptake of CA-LPs-DOX·HCl. Thus, the current uptake data show that caveolae- and clathrin-mediated endocytosis is the main cellular uptake pathway for CA-LPs-DOX·HCl. Accordingly, protection from endosomal and lysosomal transport systems by caveolae- and clathrin-mediated endocytosis enabled improved entry of DOX·HCl into nuclei, and thus, promised improved antitumor effects.33,34
To ensure receptor and ligand binding and achieve an active targeting effect at the target site in orally administered receptor-mediated active targeting systems, the nanoparticles must maintain their integrity during absorption by the gastrointestinal tract to ensure that the contained drug does not leak before reaching the target site.
Through the energy transfer from an excited fluorophore (donor) to a nearby light-absorbing molecule (acceptor), FRET is dependent on the proximity of both fluorescent molecules, within a range of 1–10 nm of each other. The FRET technique was used in this study to monitor the state of the fluorescent probe loaded in the LPs inside of the cells.35–38 DIO and DII were used in this study, because they are both lipophilic and easy to load into LPs and the emission spectrum of DIO overlapped the excitation spectrum of DII, which is necessary for FRET.
Caco-2 cells express the human sodium salt-dependent bile acid transporter (ASBT) receptor, which can ensure the bile acid-modified formulation specificity interaction with Caco-2 cells; therefore, Caco-2 cells are used as an in vitro cell model of gastrointestinal transport, and FRET technology is used to research the structure of LPs after transport across Caco-2 cells. Preparations of DIO and DII with different fluorescence were colocated after duodenal administration to investigate the intestinal absorption mechanism of CA-LPs.
HepG2 cells express NTCP receptors to enable a specific interaction between CA-LPs and HepG2 cells; therefore, HepG2 cells are used as an in vitro cell model of hepatic uptake, and FRET technology is used to study the structure of LPs after the uptake by HepG2 cells.
Conclusion
In this study, we designed and constructed CA-LPs with good stability in rat gastrointestinal fluid and serum and sustained-release behavior. The CA-LPs demonstrated high transport efficiency across Caco-2 cells and cellular uptake by HepG2 cells. The cellular uptake process by HepG2 cells involved the caveolin-mediated endocytosis pathway and avoided lysosome internalization so that they could reach nuclei directly to kill tumor cells. FRET and confocal technologies were used to investigate the transport pathways and molecular mechanisms of CA-LPs, which confirmed that part of CA-LPs-DOX·HCl retained their structural integrity after transport across the Caco-2 monolayer and uptake by HepG2 cells. Therefore, we anticipate that the design of targeted LPs against the cholic acid receptor will provide new insights into anti-hepatoma therapy for oral administration of DOX·HCl.
Acknowledgment
This work was supported by the Beijing Natural Science Foundation (7162131).
Disclosure
The authors report no competing financial interest or conflicts of interest in this work.
References
Liu YP, Gao DW, Zhang XW, et al. Antitumor drug effect of betulinic acid mediated by polyethylene glycol modified liposomes. Mater Sci Eng C Mater Biol Appl. 2016;64:124–132. | ||
Harrison E, Nicol JR, Macias-Montero M, et al. A comparison of gold nanoparticle surface co-functionalization approaches using polyethylene glycol (PEG) and the effect on stability, non-specific protein adsorption and internalization. Mater Sci Eng C Mater Biol Appl. 2016;62:710–718. | ||
Kooijmans S, Fliervoet LAL, van der Meel R, et al. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J Control Release. 2016;224:77–85. | ||
Kibria G, Hatakeyama H, Sato Y, Harashima H. Anti-tumor effect via passive anti-angiogenesis of PEGylated liposomes encapsulating doxorubicin in drug resistant tumors. Int J Pharm. 2016;509(1–2):178–187. | ||
Szczepanowicz K, Bzowska M, Kruk T, Karabasz A, Bereta J, Warszynski P. Pegylated polyelectrolyte nanoparticles containing paclitaxel as a promising candidate for drug carriers for passive targeting. Colloids Surf B Biointerfaces. 2016;143:463–471. | ||
Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164(2):138–144. | ||
Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliver Rev. 2011;63(3):131–135. | ||
Wang JJ, Huang SW. Research progress on novel carrier-modified methods and evaluation of active targeting antitumor preparation. Chin Herb Med. 2014;6(1):22–28. | ||
Martinoa AD, Kucharczyka P, Zednikb J, Sedlarik V. Chitosan grafted low molecular weight polylactic acid for protein encapsulation and burst effect reduction. Int J Pharm. 2015;496(2):912–921. | ||
Shao Y, Yang L, Han HK. TPGS-chitosome as an effective oral delivery system for improving the bioavailability of Coenzyme Q10. Eur J Pharm Biopharm. 2015;89:339–346. | ||
Youn YS, Chae SY, Lee S, Kwon MJ, Shin HJ, Lee KC. Improved peroral delivery of glucagon-like peptide-1 by site-specific biotin modification: design, preparation, and biological evaluation. Eur J Pharm Biopharm. 2008;68(3):667–675. | ||
Loureiro A, Nogueira E, Azoia NG, et al. Size controlled protein nanoemulsions for active targeting of folate receptor positive cells. Colloids Surf B Biointerfaces. 2015;135:90–98. | ||
Netsomboon K, Feβler A, Erletz L, et al. Vitamin B12 and derivatives-In vitro permeation studies across Caco-2 cell monolayers and freshly excised rat intestinal mucosa. Int J Pharm. 2016;497(1–2):129–135. | ||
Du WW, Fan YC, Zheng N, et al. Transferrin receptor specific nanocarriers conjugated with functional 7peptide for oral drug delivery. Biomaterials. 2013;34(3):794–806. | ||
Zhang D, Li DP, Shang L, He ZG, Sun J. Transporter-targeted cholic acid-cytarabine conjugates for improved oral absorption. Int J Pharm. 2016;511(1):161–169. | ||
Vivian D, Polli JE. Synthesis and in vitro evaluation of bile acid prodrugs of floxuridine to target the liver. Int J Pharm. 2014;475(1–2):597–604. | ||
Chatterjee S, Bijsmans IT, van Mil SW, Augustijins P, Annaert P. Toxicity and intracellular accumulation of bile acids in sandwich-cultured rat hepatocytes: role of glycine conjugates. Toxicol In Vitro. 2014;28(2):218–230. | ||
Sepe V, Renga B, Festa C, et al. Investigation on bile acid receptor regulators. Discovery of cholanoic acid derivatives with dual G-protein coupled bile acid receptor 1 (GPBAR1) antagonistic and farnesoid X receptor (FXR) modulatory activity. Steroids. 2016;105:59–67. | ||
Ferrebee CB, Dawson PA. Metabolic effects of intestinal absorption and enterohepatic cycling of bile acids. Acta Pharm Sin B. 2015;5(2):129–134. | ||
Li Y, Zhu CY. Enhanced hepatic targeted delivery via oral administration using nanoliposomes functionalized with novel DSPE-PEG-cholic acid conjugate. RSC Adv. 2016;6:28110–28120. | ||
Zhao SS, Dai WB, He B, et al. Monitoring the transport of polymeric micelles across MDCK cell monolayer and exploring related mechanisms. J Control Release. 2012;158(3):413–423. | ||
Vannoy CH, Tavares AJ, Noor MO, Uddayasankar U, Krull UJ. Biosensing with quantum dots: a microfluidic approach. Sensors (Basel). 2011;11(10):9732–9763. | ||
Periasamy A, Mazumder N, Sun YS, Christopher KG, Day RN. FRET microscopy: basics, issues and advantages of FLIM-FRET imaging. Adv Time Correl Single Photon Counting Appl. 2015;111:249–276. | ||
Chen H, Kim S, Li L, Wang SY, Park K, Cheng JX. Release of hydrophobic molecules from polymer micelles into cell membranes revealed by Förster resonance energy transfer imaging. Proc Natl Acad Sci U S A. 2008;105:6596–6601. | ||
Fahmy TM, Fong PM, Goyal A, Saltzman WM. Targeted for drug delivery. Mat Today. 2005;8:18–26. | ||
Gregoridadis G, editor. Liposomes Technology. 3rd ed. New York, NY: Informa Healthcare; 2007:341–372. | ||
Cohen AW, Robert H, William S, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84(4):1341–1379. | ||
Managit C, Kawakami S, Yamashita F, Hashida M. Uptake characteristics of galactosylated emulsion by HepG2 hepatoma cells. Int J Pharm. 2005;301(1–2):255–261. | ||
Ines S, Dunay IR, Kar W, Michael B, Margitta D. An apolipoprotein E-derived peptide mediates uptake of sterically stabilized liposomes into brain capillary endothelial cells. Biochemistry. 2005;44(6):2021–2029. | ||
Charlton SA, Coym JW. The use of methyl-beta-cyclodextrin to solubilize cholesterol prior to coating onto a C18 stationary phase. J Chromatogr A. 2012;1266:69–75. | ||
Lad PJ. Activation of rat lung particulate guanylate cyclase due to filipin induced fluidity change. BiochemBiophys Res Commun. 1980;96:203–210. | ||
Sengupta D. Cholesterol modulates the structure, binding modes, and energetics of caveolin-membrane interactions. J Phys Chem B. 2012; 116:14556–14564. | ||
Kastl L, Sasse D, Wulf V, et al. Multiple internalization pathways of polyelectrolyte multilayer capsules into mammalian cells. ACS Nano. 2013;7(8):6605–6618. | ||
Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. | ||
Zhang J, Zhang L, Chai L, Yang F, Du M, Chen T. Reliable measurement of the FRET sensitized-quenching transition factor for FRET quantification in living cells. Micron. 2016;88:7–15. | ||
Morton SW, Zhao X, Quadir MA, Hammond PT. FRET-enabled biological characterization of polymeric micelles. Biomaterials. 2014; 35(11):3489–3496. | ||
Laine AL, Gravier J, Henry M, et al. Conventional versus stealth lipid nanoparticles: formulation and in vivo fate prediction through FRET monitoring. J Control Release. 2014;188:1–8. | ||
Hak S, Reitan NK, Haraldseth O, de Lange Davies C. Intravital microscopy in window chambers: a unique tool to study tumor angiogenesis and delivery of nanoparticles. Angiogenesis. 2010;13(2):113–130. |
Supplementary material
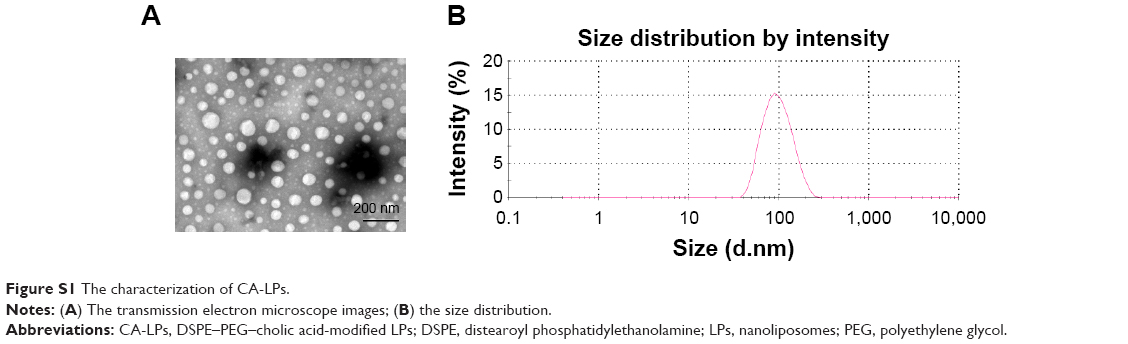 | Figure S1 The characterization of CA-LPs. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.