")
Back to Journals » OncoTargets and Therapy » Volume 7
Mechanism of action and clinical activity of tasquinimod in castrate-resistant prostate cancer
Authors Gupta N, Al Ustwani O, Shen L, Pili R
Received 25 August 2013
Accepted for publication 20 November 2013
Published 12 February 2014 Volume 2014:7 Pages 223—234
DOI https://doi.org/10.2147/OTT.S53524
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Neha Gupta, Omar Al Ustwani, Li Shen, Roberto Pili
Department of Medicine, Roswell Park Cancer Institute, Buffalo, NY, USA
Abstract: Castrate-resistant prostate cancer (CRPC) is a disease where survival is poor and treatment is challenging. Over the past 3 years, significant advances in the field have been made with US Food and Drug Administration approval of new drugs for patients with CRPC. However, despite the presence of new approved drugs such as enzalutamide, abiraterone, sipuleucel-T, cabazitaxel, and alpharadin, there is still an unmet need for novel agents with different mechanisms of action to target CRPC. Based on earlier studies demonstrating therapeutic potential of a quinoline-3-carboxamide agent roquinimex as an anticancer drug, efforts were directed to identify other useful members in this class. Tasquinimod is a second-generation quinoline-3-carboxamide agent that is currently in final stages of clinical development as a treatment for CRPC. The preclinical studies of tasquinimod have formed the basis for its success as an antiangiogenic and immunomodulatory agent in this disease. Tasquinimod is an orally available agent that has shown efficacy and favorable safety profile as deduced by the results of Phase I and II clinical trials of this drug in prostate cancer. The place of tasquinimod in the treatment of CRPC patients is currently under examination in an ongoing Phase III clinical trial. In this review, we will discuss tasquinimod, starting from its discovery and current knowledge on potential mechanisms of action to its clinical potential in CRPC.
Keywords: ABR-215050, quinoline-3-carboxamide, prostate adenocarcinoma, castration resistant
Introduction
Prostate cancer is a significant medical burden and is statistically the most common cancer in men in the Western world. It has the probability of progressing to invasive cancer in 15%–20% of cases and accounts for the second leading cause of cancer death in males.1 It has been estimated that the year 2013 will witness 238,590 new prostate cancer cases and 29,720 prostate cancer deaths.1 Castrate-resistant prostate cancer (CRPC) presents a spectrum of diseases that progress despite androgen deprivation therapy and ranges from rising prostate-specific antigen (PSA) levels without metastases or symptoms to widespread metastases and debilitating symptoms. Prognosis is associated with several factors, including performance status, presence of bone pain, extent of disease on bone scan, and serum levels of alkaline phosphatase. Bone metastases and paraneoplastic effects occur in a high proportion of patients with CRPC and can produce significant morbidity.2 Patients bearing metastatic CRPC represent a heterogeneous population that can be further classified into prognostic subgroups depending on the location of metastases (visceral, bone, or lymph nodes), as defined by Prostate Cancer Working Group 2 (PCWG2).3 CRPC is defined by a sequence of increasing PSA levels at a minimum of 1-week intervals, with a 2.0 ng/mL minimum starting value and serum testosterone less than 50 ng/dL.3,4 The recent advancement in the field of prostate cancer biology and treatment has led to the understanding that low (castrate) serum levels of testosterone do not imply hormone refractoriness. It is now known that further hormone manipulation can lead to biochemical, clinical, and radiologic response in prostate cancer with castrate levels of testosterone (previously called hormone refractory prostate cancer). Hence, there has occurred a change in terminology from “hormone refractory” prostate cancer to “castration resistant” prostate cancer. The median survival of patients with metastatic CRPC is 12–16 months from the time of diagnosis to death.5 No curative treatments are available at this stage of the disease. To date, docetaxel-based regimens (approved in 2004) have shown a modest survival advantage but are associated with the problem of resistance development in many patients that leads to decreased responsiveness.6–8 Furthermore, since cytotoxic chemotherapy lacks cancer cell specificity, there are significant limitations on dose and length of treatment with chemotherapy. This creates a need to explore other novel targeted agents in this disease process. Recent years have seen the approval of several drugs in the treatment of CRPC including chemotherapy, immunotherapy, and hormonal agents. In the year 2010, sipuleucel-T and cabazitaxel gained US Food and Drug Administration (FDA) approval for use in metastatic CRPC (mCRPC) and denosumab for bone-metastatic CRPC to prevent skeletal related events. This was shortly followed by approval of abiraterone in 2011, followed by enzalutamide approval in 2012. FDA also expanded the approved use of abiraterone to first-line chemotherapy in mCRPC in 2012.9,10 The most recent development has been FDA announcement of approval of radium Ra 223 chloride (alpharadin) in bone-metastatic mCRPC.11 These newly available agents in CRPC in chemo-naïve and chemo-refractory settings are shown in Figure 1 and briefly described in Table 1. There still exists an unmet need to explore additional agents in treatment of CRPC. Tasquinimod (ABR-215050) has been tested in preclinical studies in animal models of CRPC and has shown successful results through its unique mechanism of action. It has completed Phase I and II testing and has recently entered a Phase III clinical trial.
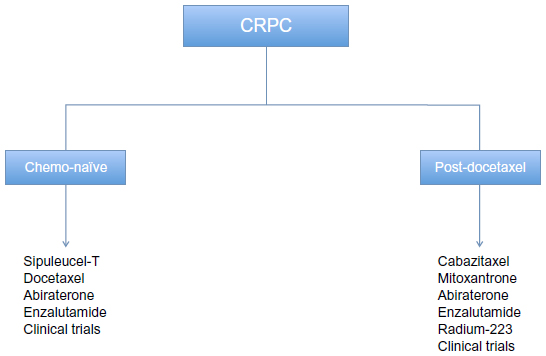 | Figure 1 Current therapeutic options for patients with metastatic CRPC. |
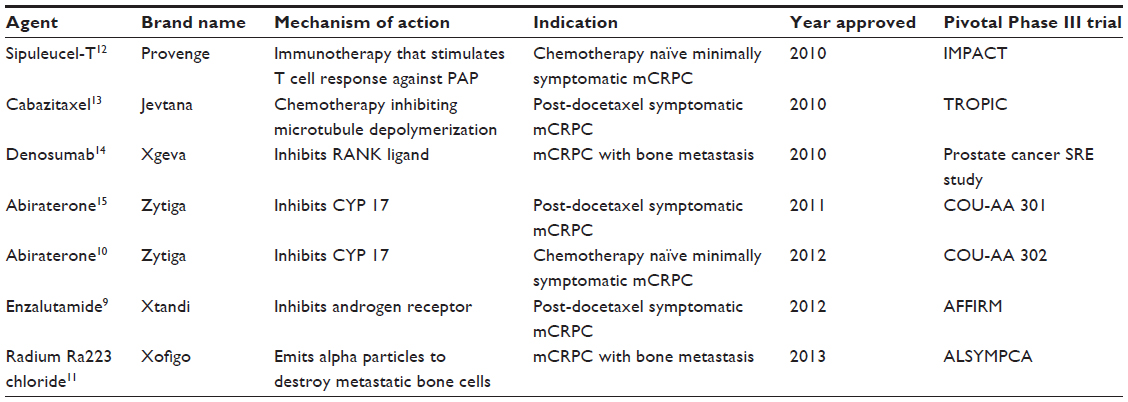 | Table 1 Treatment agents approved in CRPC |
Chemical structure of tasquinimod
Tasquinimod is the lead second-generation orally active quinoline-3-carboxamide, which is being tested for clinical development for prostate cancer. Chemically, tasquinimod is 4-hydroxy-5-methoxy-N,1-dimethyl-2-oxo-N-[4-(trifluoromethyl)phenyl]-1,2-dihydroquinoline-3-carboxamide.12,13 Figure 2 depicts the chemical structure of tasquinimod. Tasquinimod contains a substitution in the 5-position of the quinoline moiety and a substitution in the phenyl ring of the 3-carboxamide moiety. These structural modifications are possibly responsible for tasquinimod antiangiogenic activity and its lack of pro-inflammatory effects.
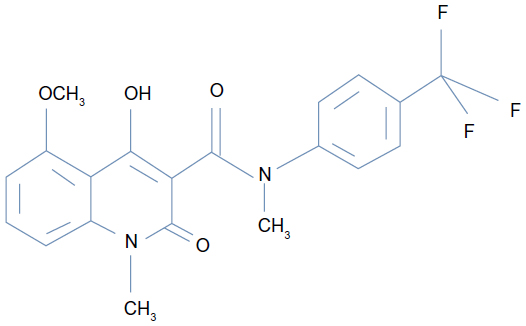 | Figure 2 Chemical structure of tasquinimod. |
History of discovery and development of tasquinimod
Development of tasquinimod has been a complex process, starting from drug discovery, small molecule recognition, and success in preclinical studies, which finally validated its clinical drug development. The first development of this agent was in 1980, when AB (Active Biotech) Leo Research Laboratories in Sweden identified a series of quinoline-3-carboxamide analogues which were found to stimulate carrageenan-induced inflammatory responses.14 Roquinimex was the first quinoline-3-carboxamide analogue identified by this drug development company, and later became famous by the trade name linomide.12 In the 1990s, roquinimex was tested in multiple animal models, and some reports of daily oral roquinimex treatment leading to anticancer effects due to stimulation of the immune system were published.15,16 Also, multiple laboratories documented the therapeutic potential of roquinimex as an anticancer drug by studies showing that roquinimex inhibited in-vivo development of prostate cancer and breast cancer in rodents mediated by inhibition of vascular endothelial growth factor (VEGF)-induced angiogenesis.17,18 Meanwhile, roquinimex was also shown to have therapeutic abilities but unanticipated serious cardiopulmonary toxicities (including myocardial infarction, pericarditis, pleural effusion, and death) in a Phase III trial in patients with multiple sclerosis (MS).19 Based on these results, AB screened the chemical library of quinoline-3-carboxamide analogues to identify second-generation compounds lacking the pro-inflammatory side effects of roquinimex for the treatment of MS and prostate cancer. Pro-inflammatory activity of roquinimex was attributed to demethylation at the carboxamide side chain nitrogen, producing highly planar metabolites that are pro-inflammatory.12 Therefore, a library of second-generation compounds was created to restrict planar metabolite production, thereby reducing pro-inflammatory side effects.12 Laquinimod was the lead second-generation analogue used in a variety of MS animal models, after which tasquinimod was identified for clinical development for treatment of prostate cancer based on its 30–60-fold higher potency (in comparison with roquinimex) and significant inhibition of prostate cancer growth in mice.20,21 Tasquinimod is a second-generation quinoline-3-carboxamide compound which has high efficacy against cancer and lacks the pro-inflammatory side effects, unlike its initial counterparts from the group of quinoline-3-carboxamide.
Pharmacokinetic parameters
Our current understanding of pharmacokinetics of tasquinimod is based on the results of completed Phase 1 and Phase II clinical trials.22,23 Tasquinimod has been found to have a low clearance of 0.19 L/h at 0.5 mg and 0.22 L/h at 1 mg dose level, making increase in systemic exposure lesser than the dose increase. The volume of distribution is 5.9 L, the elimination half-life is 40±16 hours, and the maximum plasma concentrations occur at 2.6 hours. Area under the curve steady state amounts to 4.8 μmol/h. Co-administration with food has not been found to affect the pharmacokinetic properties of tasquinimod. No relationship between pharmacokinetic parameters and race, ethnicity, or hepatic function has been identified.
Mechanism of action
The mode of action of quinoline-3-carboxamide compounds is not fully understood. Several studies have demonstrated that this group of agents interferes with tumor angiogenesis, macrophage infiltration, cytokine production, and autoimmune/inflammatory diseases.21,24–26 Although further studies are needed to elucidate all the pathways by which quinoline-3-carboxamide agents function, recent preclinical studies have led to significant improvement in our understanding of actions of this class. Most of these studies involve tasquinimod, which is the lead second-generation quinoline-3-carboxamide employed for clinical development. Tasquinimod has unique pharmacotherapeutic properties and acts through multi-targeted mechanisms to treat prostate cancer. Figure 3 shows a representation of potential mechanisms of activity of tasquinimod. In preclinical human prostate cancer models, it has been demonstrated that tasquinimod has both antitumor and anti-metastatic activities, which are affected mainly through its anti-angiogenic and immunomodulatory effects.27 The anti-metastatic action likely occurs through interference with the tumor establishment process. In a preclinical study by Jennbacken et al, it was observed that tasquinimod seems to mainly inhibit the initial establishment of metastatic deposits and had lesser effect on inhibition of growth of already metastasized deposits.27
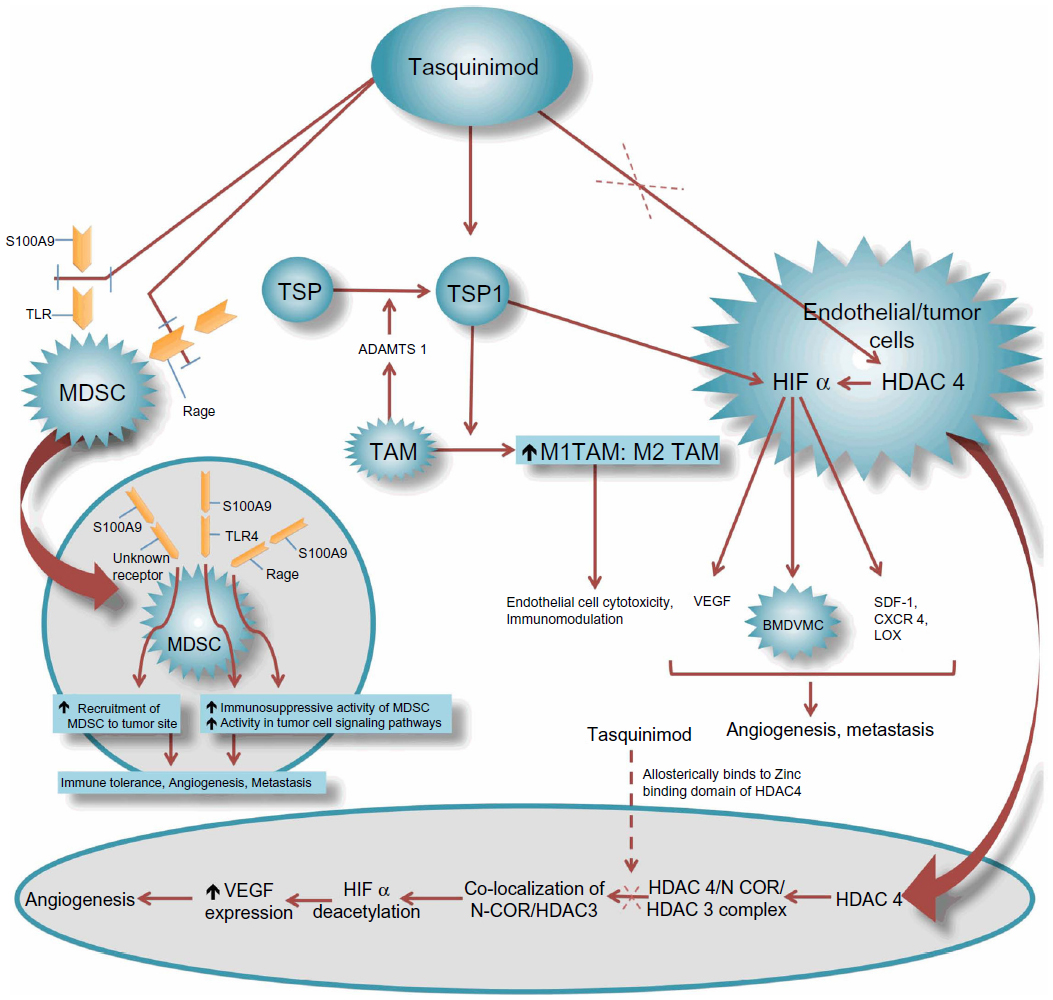 | Figure 3 Mechanism of action of tasquinimod. |
Inhibition of angiogenesis
Angiogenesis is known to play a central role in the progression of CRPC.28 Microvessel density in prostate cancer, a histological measure of tumor angiogenesis, has been shown to correlate with Gleason score and predict disease progression.28,29 This observation provided a rationale for investigating anti-angiogenic therapy as a treatment strategy for this disease. Tumor angiogenesis involves the complex interplay between pro- and anti-angiogenic factors influencing tumor cells, endothelial cells, and surrounding stroma. Key angiogenic factors implicated in prostate cancer progression and metastasis include VEGF, angiopoietins, and fibroblast growth factor (FGF).30 The anti-angiogenic activity of tasquinimod has been demonstrated in several preclinical studies of CRPC. In a study by Isaacs et al, it was shown that tasquinimod’s ability to prevent growth of tumor in human and rodent prostate cancer models correlated with inhibition of angiogenesis in several in-vitro and in-vivo assays such as endothelial capillary tube formation, aortic ring assay, chorioallantoic membrane assay, real time tumor blood flow, tumor blood pO2 (oxygen partial pressure) measurements, tumor blood vessel density, and tumor hypoxic and apoptotic fractions.21
Tasquinimod has been also reported to upregulate the expression of thrombospondin (TSP)-1, which further inhibits tumor growth and metastases by inhibition of angiogenesis and migration or by transforming growth factor (TGF)-β activation.13 The increase in TSP-1 expression by tasquinimod appears to be a direct effect, as elevated TSP mRNA (messenger ribonucleic acid) levels have been achieved in the in-vitro cell cultures where no other cell types were present.13 TSP-1 is a 450 kDA glycoprotein synthesized by several cell types and is known to suppress tumor growth, hinder matrix metalloproteinase-9 activation, and participate in mobilization of VEGF.31 TSP-1 negatively regulates the tissue levels of pro-angiogenic inducers (ie, VEGF and FGF) and blocks their receptors on endothelial cells, thereby inhibiting neovascularization and converting “angiogenic switch” into an anti-angiogenic state.30–32 Some studies have shown that prostate tumor growth inhibition is accompanied by downregulation of hypoxia inducing factor (HIF)-1α protein, androgen receptor protein, and glucose transporter-1 protein in addition to decreased tumor tissue levels of VEGF which is a downstream target of HIF-1α.13 HIF-1α is also known to act by recruiting the bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion.33 Tumor associated macrophages (TAMs) also participate in the process of attaining optimal activity of tasquinimod by secreting a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-1 leading to TSP proteolysis and fragmentation.34 This fragmentation liberates the soluble N-terminal deletion fragments of TSP-1, which are the active particles involved in preventing angiogenesis via blocking the action of VEGF and FGF on endothelial cells. Tasquinimod has also been reported to bind to histone deacetylase (HDAC)-4. This binding locks HDAC-4 in a conformational status that inhibits the formation of the HDAC-4/nuclear receptor co-repressor (NCOR)/HDAC-3 repressor complex. The HDAC-4/NCOR/HDAC-3 complex is needed for deacetylation of HIF-1α (transcription factor) and of histone proteins around the DNA (deoxyribonucleic acid). The prevention of deacetylation of HIF-1α by tasquinimod leads to inhibition of VEGF transcription.35
Immunomodulation
The immune-modulatory properties of tasquinimod may be mediated by TAMs. A study by Martin-Manso et al shed light on the involvement of M1 polarized TAMs in mediating the actions of TSP-1 to produce angiogenesis inhibition.36 This study showed that TSP-1 leads to increased recruitment of M1-polarized TAMs which potentiate reactive oxygen species-mediated cytotoxicity of endothelial cells. This causes inhibition of angiogenesis leading to tumor suppression. It is possible that tasquinimod induced levels of TSP-1 changes the balance between M1 and M2 cells by facilitating infiltration of TAMs with M1 phenotype over TAMs of pro-angiogenic M2 phenotype. Another mechanism by which tasquinimod produces immunomodulation is via its binding to a myeloid-related protein S100A9.37 S100A9 pro-inflammatory protein is secreted by myeloid-derived suppressor cells (MDSCs) and is reported to attract MDSCs into tumor site and enhance their immunosuppressive activity in addition to promoting tumor cell signaling pathways.38 MDSCs are immature myeloid cells derived from bone marrow hematopoietic precursors that failed to differentiate into granulocytes, macrophages, or dendritic cells due to partial block in differentiation caused by pathological conditions like cancer.39,40 These cells are very heterogeneous and can lead to immunosuppression by multiple mechanisms.40,41 MDSCs have also been demonstrated to promote angiogenesis, tumor growth, and metastasis.42 The correlation of tumor growth with MDSC numbers has also been demonstrated specifically in murine prostate cancer models.43 Bjork et al showed in their study that S100A9 is an efficient ligand for Toll-like receptor (TLR)-4 and a receptor for advanced glycation end products (RAGE) in the presence of zinc.37 Binding of tasquinimod to S100A9 protein impedes its interaction with TLR-4 and RAGE along with inhibiting the recruitment of MDSCs, thereby preventing tumor growth and metastasis.37,44 In murine prostate cancer models, low dose tasquinimod has been shown to affect maturation of MDSCs into other immunosuppressive cell types.43 Also, tasquinimod has been shown to cause a 60% decrease in tumor infiltrating MDSCs without affecting peripheral MDSCs or monocytes, implicating that tasquinimod targets MDSCs by interfering with their trafficking to tumor sites.45 Inhibiting granulopoiesis is another proposed mechanism of action of tasquinimod, as tasquinimod has reduced the number of peripheral and tumor-infiltrating granulocytes.45
Other mechanisms of action of tasquinimod
In a preclinical study by Jennbacken et al, treatment with tasquinimod in an animal model led to downregulation of hypoxia driven HIF-1α regulated genes like stromal derived factor-1 (SDF-1), C-X-C chemokine receptor (CXCR)-4, and lysyl oxidase (LOX).27 SDF-1, the chemo-attractant for CXCR-4-expressing tumor cells, was found to be decreased in specific organs of the treated animals.27,33 CXCR-4 is known to participate in metastatic dissemination to a few particular sites.46 CXCR-4-positive tumor cells are known to be attracted to the tissues where the chemo-attractant SDF-1 is abundantly expressed.46 LOX is known to play a part in formation of pre-metastatic niche and inducing metastatic tumor growth.47 Hence, by blockade of the SDF-1/CXCR-4 axis and negative regulation of LOX, tasquinimod likely plays an important part in inhibition of metastasis.27 Interestingly, tasquinimod also upregulates the gene expression of GDF-15, which is a divergent member of TGF-β superfamily and its role in prostate cancer, whether being that of a tumor suppressor or promoter is currently being debated.48 In TRAMP (transgenic adenocarcinoma of the mouse prostate) mouse prostate cancer models, GDF-15 has been demonstrated to slow cancer development but increase metastases.49 Nevertheless, GDF-15 has been recently shown to suppress in-vitro angiogenesis through its interaction with connective tissue growth factor.13,50 It is involved in apoptotic pathways and is thought to play a role in antitumor activity of tasquinimod.13 Other possible mechanisms of action of tasquinimod may be mediated by upregulation of CYP1A1 gene.13,51 CYP1A1 protein is currently being considered as a novel anticancer drug target based on the findings that its selective overexpression in tumor cells induces the metabolism of benzothiazole and aminoflavone compounds to their reactive species, which are responsible for DNA adduct formation and cell death.52 Table 2 shows target proteins/molecules, their respective levels in CRPC, and the effect of tasquinimod on the expression/action of these proteins/molecules.
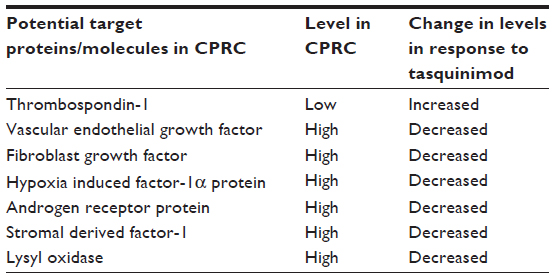 | Table 2 Potential target proteins/molecules modulated by tasquinimod |
Clinical activity of tasquinimod
Based on its effects against human prostate cancer xenografts, tasquinimod entered clinical development as a novel treatment for prostate cancer. The Phase I and II clinical trials of tasquinimod in CRPC have suggested a promising clinical potential. Table 3 shows a summary of study characteristics and results of these trials. This has led to tasquinimod development in a Phase III trial, which is currently ongoing.
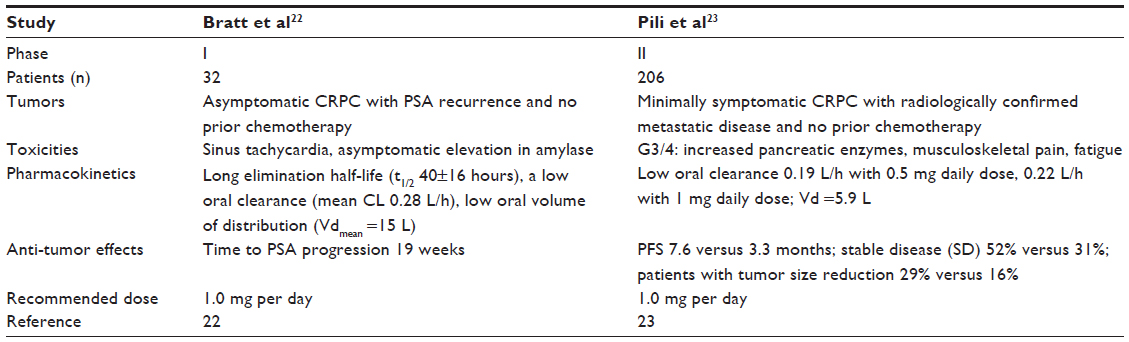 | Table 3 Summary of published early phase trials of tasquinimod in CRPC |
Phase I clinical trial
Bratt et al reported two Phase I trials of tasquinimod in chemotherapy naïve CRPC, demonstrating good clinical efficacy as reflected by development of new bone metastases in only a few patients receiving tasquinimod.22 Patients with CRPC and PSA recurrence along with not having received any previous chemotherapy were enrolled in this study. Patients received tasquinimod up to 1 year, either at fixed doses of 0.5 or 1.0 mg per day or at an initial dose of 0.25 mg per day that escalated to 1.0 mg per day. The eligibility criteria included histologically or cytologically proven CRPC with PSA progression, ECOG (Eastern Cooperative Oncology Group) score of 0–1, and normal bone marrow, renal, and hepatic functions. The patient population that was excluded from the study constituted patients with clinical symptoms associated with prostate tumor progression, patients having received previous anticancer therapy (except for hormonal treatment), patients requiring systemic corticosteroids or warfarin, and patients with previous significant cardiac events or prior uncontrolled inter-current illness. The study design was that of two open-label studies. The first study was a dose-escalation study to determine the maximum tolerated dose of tasquinimod. The second study conducted was an intra-patient stepwise dose-escalation study. A total of 32 patients were enrolled. Twenty-one patients were maintained for 4 months or more.
In this Phase I trial, the maximum tolerated dose was determined to be 0.5 mg per day, but when using stepwise intra-patient dose escalation, a dose of 1.0 mg per day was well tolerated. Dose-limiting toxicity was sinus tachycardia and asymptomatic elevation in amylase. Common adverse events included transient elevation in inflammatory laboratory markers, anemia, and symptoms such as nausea, fatigue, myalgia, and extremity pain.
Phase II clinical trial
After the favorable side-effect profile and potential efficacy suggested by the Phase I trial, a Phase II trial of tasquinimod in men with minimally symptomatic metastatic CRPC was performed recently. This was a randomized, double-blind, placebo-controlled 2:1 study, where disease progression was shown to be significantly delayed. A total of 206 men were enrolled in the study across 45 centers, out of which 201 started treatment and were included in safety and efficacy analyses. Of these, 134 patients were assigned to tasquinimod group whereas 67 assigned to placebo group.
Eligibility criteria is this study were histologically confirmed prostate adenocarcinoma, Karnofsky performance score of 70–100, adequate hematologic, renal, and liver function, castrate levels of testosterone <50 ng/dL, visual analog pain score of 3 or more (scale 0–10), and radiologically confirmed metastatic disease. Progression was defined by rising serum PSA levels, progression of bi-dimensionally measurable soft tissue metastasis, or new bone lesions detected by bone scan within 12 weeks before screening. The major exclusion criteria comprised of: opiate intake; prior cytotoxic chemotherapy within 3 years; history of pancreatitis or cardiovascular disease, including recent myocardial infarction, congestive heart failure, ventricular arrhythmias, or unstable angina; and concomitant use of warfarin.
Patients were randomly allocated in a ratio of 2:1 to receive tasquinimod or placebo in a double-blind fashion. Patients received either placebo or tasquinimod for treatment. The treatment plan was such that patients with tasquinimod received an oral daily dose of 1.0 mg after a titration phase of 0.25 mg/d for 2 weeks followed by 0.5 mg/d for 2 weeks, leading to a maximum of 6 months of double-blind treatment. Switchover to tasquinimod arm was offered to asymptomatic patients in the placebo group with disease progression during the first 6 months or without progression at 6 months, whereas patients receiving tasquinimod with no disease progression at 6 months were offered open-label treatment until progression, and patients who had disease progression while on tasquinimod were withdrawn. The primary endpoint was the progression-free proportion at 6 months and was analyzed when all patients had completed 6 months of treatment, progressed or withdrawn from the study, whichever came first. Six months progression-free proportions for tasquinimod and placebo groups were 69% and 37%, respectively (P<0.001). After 6 months of treatment, 63% of patients in the placebo had progressed, as compared with 31% in the tasquinimod group. Median progression-free survival (PFS) improved from 3.3 to 7.6 months (P=0.0042), representing a clinically meaningful halving of the ongoing risk of progression or death over time over placebo. Improvement in PFS was observed despite crossover of a small number of men (n=17) who were progression free at 6 months in the placebo arm. In PCWG2 CRPC clinical subgroups, PFS in months was as follows: nodal metastasis 6.1 versus 3.1, bone metastasis 8.8 versus 3.4, and visceral metastases 6.0 versus 3.0 for patients receiving tasquinimod versus placebo. In patients with measurable disease, 7% of tasquinimod-treated patients (4/61) versus 0% of those receiving placebo (0/39) had a best partial response, whereas 52% versus 31% had stable disease. A plot of RECIST (Response Evaluation Criteria in Solid Tumors) responses showed reduction in tumor sizes in 29% patient receiving tasquinimod versus 16% patients receiving placebo. Tasquinimod treatment also inhibited the increase in bone alkaline phosphatase levels observed during progression, which may reflect a positive therapeutic effect on delayed progression of osteoblastic bone metastasis. In the double-blind phase, 84% of patients with progressive disease had radiographic progression, whereas symptomatic or clinical progression was documented in 22% of patients. No significant differences in time to PSA progression or PSA kinetics were observed between the two treatment groups.
In this Phase II trial, tasquinimod showed an acceptable toxicity in the majority of men <75 years old, with infrequent discontinuation because of adverse effects. Discontinuation rate because of toxicity was 22% versus 1% for patients receiving placebo, and most of these were grade 1–2 toxicities (89% in tasquinimod arm versus 94% in placebo arm). In most cases, adverse effects were transient and reversible and could be managed through dose reductions or supportive measures, with continuation of drug at individual MTDs.
Tasquinimod treatment led to transient asymptomatic rise in several lab parameters such as amylase, lipase, CRP, and fibrinogen reported in 40% of patients receiving tasquinimod versus 10% receiving placebo. Grade 3–4 adverse events in the tasquinimod group were increased lipase, muscular weakness, deep vein thrombosis, anemia, asthenia, renal failure, and pneumonia. Adverse events occurring more frequently in the tasquinimod arm included gastrointestinal disorders, fatigue, musculoskeletal pains, and elevations of pancreatic and inflammatory biomarkers. The most common reason for treatment discontinuation because of toxicity was muscle and/or joint pain.
Combination strategies with tasquinimod in CRPC
In a preclinical study by Dalrymple et al, tasquinimod has been shown to exhibit enhanced antitumor properties when combined with androgen ablation in a human prostate cancer model (CWR-22R-H).25 This study showed that delayed addition of either androgen ablation followed by tasquinimod or tasquinimod followed by androgen ablation produced a greater antitumor growth response in comparison to either monotherapy. Both surgical or chemical androgen ablation were utilized as androgen ablation methods in this study. Tasquinimod was shown to have a superior anticancer efficacy and improved survival when combined with docetaxel in human CRPC xenografts.25 The results from this study have provided a rationale for clinical testing of a tasquinimod combination with chemotherapy in CRPC patients. As cabazitaxel in combination with prednisone has emerged as a standard chemotherapy in the post-docetaxel setting,53,54 the CATCH prostate cancer trial (NCT01513733)55 has been initiated where a combination of tasquinimod and cabazitaxel is being tested in metastatic CRPC after progression on docetaxel (see below).
In another preclinical study, the anticancer efficacy of fractionated radiation against prostate cancer was significantly enhanced when tasquinimod was sequentially administered after completion of fractionated radiation.56 By combining the results of these studies showing improved antitumor response in prostate cancer with combination of tasquinimod and androgen ablation or radiation therapy, it is conceivable that these combinations may be a successful therapeutic approach which warrants further testing.25,55
Interestingly, in murine prostate cancer models, combination of tasquinimod with peptide vaccine SurVaxM has been reported to improve antitumor immune responses and reduced tumor growth when compared with vehicle and single-agent tasquinimod treatment.45 These results of combination of tasquinimod with vaccine therapy establish a rationale for the clinical testing of tasquinimod in combination with sipuleucel-T in CRPC patients.45
Toxicities
Overall, the side-effect profile of tasquinimod seems to be favorable with minimal grade 3–4 toxicities. Based on the Phase I trial of tasquinimod, adverse effects of elevated amylase and sinus tachycardia were the only reported dose-limiting toxicities.22 Based on Phase II trial, tasquinimod treatment has been found to be safe, with similar but slightly greater adverse-effect profile as observed in phase I.23 This has been attributed to more elderly patients in Phase II (22% elderly patients) versus Phase I (6%), as tolerability appears to decrease with age, possibly due to slower hepatic clearance of tasquinimod.
Ongoing clinical trials and future directions
Currently, there are three ongoing clinical trials on tasquinimod in prostate cancer (NCT01234311, NCT01732549,57,58 NCT0151373355) as described below. Table 4 shows the list of ongoing clinical trials related to tasquinimod in prostate cancer. A Phase III randomized, double-blind, placebo-controlled study of tasquinimod (NCT0123431157) is being conducted currently in asymptomatic to mildly symptomatic patients with metastatic CRPC to confirm the effect of tasquinimod on delaying disease progression compared with placebo. Approximately 1,200 eligible patients with metastatic CRPC are to be randomly assigned in a 2:1 ratio to either tasquinimod or placebo. Variable doses of tasquinimod ranging from 0.25 to 1.00 mg daily are being used in the trial. Eligible patients include males with histologically confirmed diagnosis of adenocarcinoma of prostate, with evidence of bone-metastatic disease on radiographic examination, castrate levels of serum testosterone <50 ng/dL, people with Karnofsky score of >70%, and no evidence of prior malignancies. Major exclusion criteria constitute prior cytotoxic chemotherapy for prostate cancer within 2 years, previous radiotherapy, biological therapy or antiandrogens within 4 weeks, and opiate or radiotherapy requirement for prostate cancer pain. Although the study is powered for OS analysis, the primary endpoint is PFS defined as the time from the date of randomization to the date of radiological progression or death. Figure 4 shows the schema of the inclusion criteria and clinical endpoints of the Phase III study.
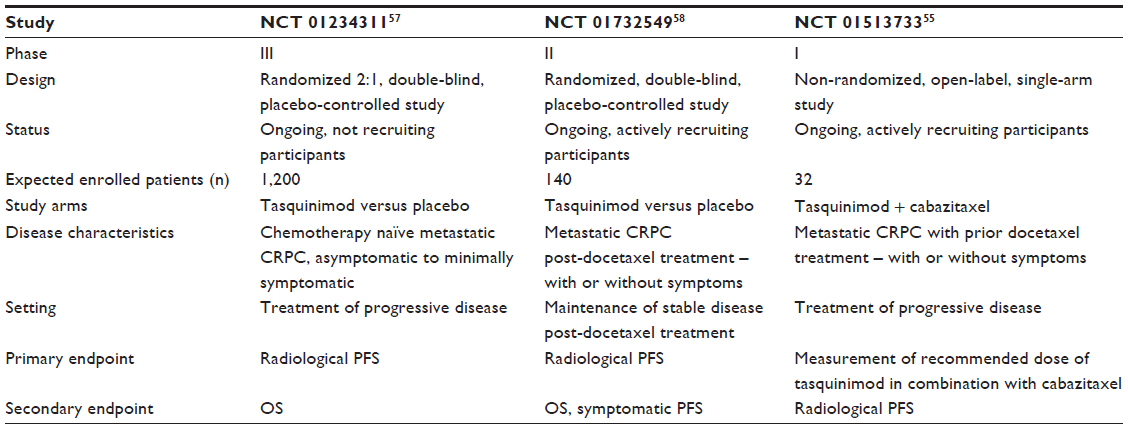 | Table 4 Ongoing trials involving tasquinimod in prostate cancer |
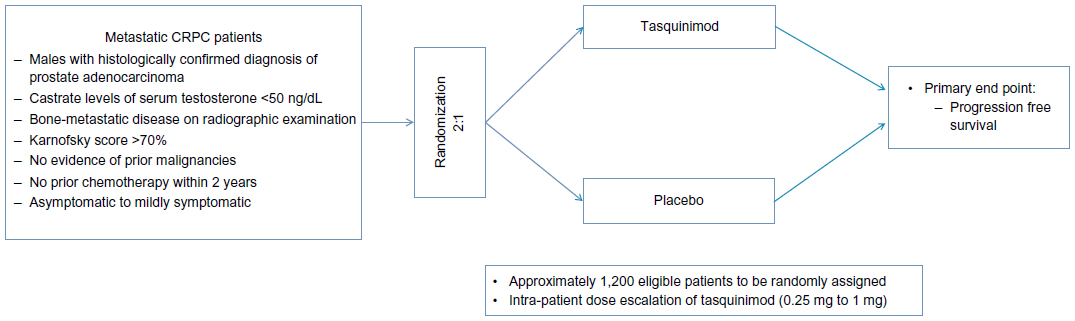 | Figure 4 Schema for current tasquinimod Phase III study. |
Recently, a clinical trial of tasquinimod maintenance therapy (NCT0173254958) has been initiated to confirm whether tasquinimod used as maintenance therapy is active and tolerable in patients with metastatic CRPC not progressing following a prior first-line docetaxel based chemotherapy. It is a Phase II randomized, double-blind, placebo-controlled trial being performed by Ipsen (Paris, France). Primary outcome measure is time to radiological PFS. Overall survival, symptomatic PFS, time to PFS on next-line therapy and safety profile of tasquinimod are the secondary outcome measures. This study is currently recruiting patients and is expected to enroll a total of approximately 140 patients. Patients are required to have histologically confirmed prostate cancer with evidence of metastatic disease on radiological evaluation, with or without symptoms along with castrate levels of testosterone. Other major requirements for inclusion are prior treatment with a first-line docetaxel-based chemotherapy of 75 mg/m2 every 3 weeks with corticosteroids for a minimum of six cycles and absence of progressive disease at the end of docetaxel treatment, which is defined according to RECIST criteria, no new lesion(s) assessed by bone scan, and no elevated PSA for the three last tests. Major exclusion criteria involves patients with history of other malignancies, concurrent use of other anticancer agents, or prior radiation therapy since starting docetaxel.
CATCH prostate cancer trial (NCT0151373355) as mentioned above is an ongoing Phase I clinical trial to test the outcome of combination therapy of tasquinimod and the recently approved cabazitaxel in prostate cancer. Approximately, 32 patients are estimated to become enrolled in this study. The primary objective is to determine the recommended dose of tasquinimod in combination with cabazitaxel and prednisone based on safety and tolerability in men with chemo-refractory metastatic CRPC. The secondary outcome measures in this study are radiologic PFS (modified PCWG defined) and evidence of efficacy (based on PSA declines, circulating tumor cell changes over time, and rates of radiographic response along with duration of responses).
Conclusion
CRPC is a disease with complex pathogenesis, poor survival, and limited treatment options. Recent drug approvals have represented a significant advance but have not totally fulfilled the need for more effective drugs for this disease. With ongoing improvement of understanding of molecular mechanisms in the pathogenesis of CRPC, targeted therapies are being tested as a new approach to fight this disease. Tasquinimod is an orally active targeted agent that has shown potential benefit in preclinical studies and is currently being tested in a Phase III clinical trial. Anti-angiogenesis effect and immunomodulation represent the major mechanisms through which tasquinimod mediates its antitumor and antimetastatic effects. The molecular basis of angiogenesis inhibition by tasquinimod in CRPC may be due to modulation of TSP-1, HIF-1α, and HDAC-4. Mounting evidence suggests that tasquinimod may have a significant immunomodulatory activity via inhibition of S100A9 on MDSCs and TAMs. Ongoing preclinical and future clinical studies will shed additional light on the molecular mechanisms responsible for the modulation of immune response by this agent that can be exploited in novel, rational combination strategies.
Disclosure
Roberto Pili has received a research grant from Active Biotech and is a paid consultant for Ipsen. The other authors have no conflicts of interest to declare.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. | |
Hotte SJ, Saad F. Current management of castrate-resistant prostate cancer. Curr Oncol. 2010;17 Suppl 2:S72–S79. | |
Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26:1148–1159. | |
Cheng HH. Advanced Clinical States in Prostate Cancer. In: Kibel AS, editor. Urologic Clinics of North America; 2012. | |
Heidenreich A, Schrader AJ. The treatment of hormone refractory prostate cancer. EAU Update Series. 2003;1:40–50. | |
Petrylak DP. The treatment of hormone-refractory prostate cancer: docetaxel and beyond. Rev Urol. 2006;8 Suppl 2:S48–S55. | |
Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–1520. | |
Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–1512. | |
Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. | |
Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–148. | |
Hafeez S, Parker C. Radium-223 for the treatment of prostate cancer. Expert Opin Investig Drugs. 2013;22:379–387. | |
Isaacs JT. The long and winding road for the development of tasquinimod as an oral second-generation quinoline-3-carboxamide antiangiogenic drug for the treatment of prostate cancer. Expert Opin Investig Drugs. 2010;19:1235–1243. | |
Olsson A, Bjork A, Vallon-Christersson J, et al. Tasquinimod (ABR-215050), a quinoline-3-carboxamide anti-angiogenic agent, modulates the expression of thrombospondin-1 in human prostate tumors. Mol Cancer. 2010;9:107. | |
Stalhandske T, Eriksoo E, Sandberg B. A novel quinoline-3-carboxamide with interesting immunomodulatory activity. Int J Immunopharmacol. 1982;4:336. Available from: http://www.sciencedirect.com/science/article/pii/0192056182903125. Accessed January 8, 2014. | |
Harning R, Szalay J. A treatment for metastasis of murine ocular melanoma. Invest Ophthalmol Vis Sci. 1988;29:1505–1510. | |
Kalland T. Effects of the immunomodulator LS 2616 on growth and metastasis of the murine B16-F10 melanoma. Cancer Res. 1986;46:3018–3022. | |
Joseph IB, Vukanovic J, Isaacs JT. Antiangiogenic treatment with linomide as chemoprevention for prostate, seminal vesicle, and breast carcinogenesis in rodents. Cancer Res. 1996;56:3404–3408. | |
Hartley-Asp B, Vukanovic J, Joseph IB, et al. Anti-angiogenic treatment with linomide as adjuvant to surgical castration in experimental prostate cancer. J Urol. 1997;158:902–907. | |
Tan IL, Lycklama a Nijeholt GJ, Polman CH, et al. Linomide in the treatment of multiple sclerosis: MRI results from prematurely terminated Phase-III trials. Mult Scler. 2000;6:99–104. | |
Brunmark C, Runstrom A, Ohlsson L, et al. The new orally active immunoregulator laquinimod (ABR-215062) effectively inhibits development and relapses of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;130:163–172. | |
Isaacs JT, Pili R, Qian DZ, et al. Identification of ABR-215050 as lead second generation quinoline-3-carboxamide anti-angiogenic agent for the treatment of prostate cancer. Prostate. 2006;66:1768–1778. | |
Bratt O, Haggman M, Ahlgren G, et al. Open-label, clinical Phase I studies of tasquinimod in patients with castration-resistant prostate cancer. Br J Cancer. 2009;101:1233–1240. | |
Pili R, Haggman M, Stadler WM, et al. Phase II randomized, double-blind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J Clin Oncol. 2011;29:4022–4028. | |
Diab A, Michael L, Wahren B, et al. Linomide suppresses acute experimental autoimmune encephalomyelitis in Lewis rats by counter-acting the imbalance of pro-inflammatory versus anti-inflammatory cytokines. J Neuroimmunol. 1998;85:146–154. | |
Dalrymple SL, Becker RE, Isaacs JT. The quinoline-3-carboxamide anti-angiogenic agent, tasquinimod, enhances the anti-prostate cancer efficacy of androgen ablation and taxotere without effecting serum PSA directly in human xenografts. Prostate. 2007;67:790–797. | |
Vukanovic J, Hartley-Asp B, Isaacs JT. Inhibition of tumor angiogenesis and the therapeutic ability of linomide against rat prostatic cancers. Prostate. 1995;26:235–246. | |
Jennbacken K, Welen K, Olsson A, et al. Inhibition of metastasis in a castration resistant prostate cancer model by the quinoline-3-carboxamide tasquinimod (ABR-215050). Prostate. 2012;72:913–924. | |
Weidner N, Carroll PR, Flax J, et al. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol. 1993;143:401–409. | |
Gettman MT, Pacelli A, Slezak J, et al. Role of microvessel density in predicting recurrence in pathologic Stage T3 prostatic adenocarcinoma. Urology. 1999;54:479–485. | |
Doll JA, Reiher FK, Crawford SE, et al. Thrombospondin-1, vascular endothelial growth factor and fibroblast growth factor-2 are key functional regulators of angiogenesis in the prostate. Prostate. 2001;49:293–305. | |
Rodriguez-Manzaneque JC, Lane TF, Ortega MA, et al. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc Natl Acad Sci U S A. 2001;98:12485–12490. | |
Ren B, Yee KO, Lawler J, et al. Regulation of tumor angiogenesis by thrombospondin-1. Biochim Biophys Acta. 2006;1765:178–188. | |
Du R, Lu KV, Petritsch C, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. | |
Lee NV, Sato M, Annis DS, et al. ADAMTS1 mediates the release of antiangiogenic polypeptides from TSP1 and 2. EMBO J. 2006;25:5270–5283. | |
Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. | |
Martin-Manso G, Galli S, Ridnour LA, et al. Thrombospondin 1 promotes tumor macrophage recruitment and enhances tumor cell cytotoxicity of differentiated U937 cells. Cancer Res. 2008;68:7090–7099. | |
Bjork P, Bjork A, Vogl T, et al. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009;7:e97. | |
Sinha P, Okoro C, Foell D, et al. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–4675. | |
Schmid MC, Varner JA. Myeloid cells in the tumor microenvironment: modulation of tumor angiogenesis and tumor inflammation. J Oncol. 2010;2010:201026. | |
Sevko A, Umansky V. Myeloid-derived suppressor cells interact with tumors in terms of myelopoiesis, tumorigenesis and immunosuppression: thick as thieves. J Cancer. 2013;4:3–11. | |
Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. | |
Ye XZ, Yu SC, Bian XW. Contribution of myeloid-derived suppressor cells to tumor-induced immune suppression, angiogenesis, invasion and metastasis. J Genet Genomics. 2010;37:423–430. | |
Shen L, Ciesielski M, Miles KM, et al. Targeting myeloid derived suppressor cells as novel strategy to enhance immunotherapy in murine prostate cancer models. [abstract]. In: Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; 2012 Mar 31-Apr 4; Chicago, IL. Philadelphia (PA): AACR; Cancer Res 2012;72(8 Suppl):Abstract nr 1551. doi:1538-7445.AM2012-1551. Available from: http://cancerres.aacrjournals.org/cgi/content/short/72/8_MeetingAbstracts/1551. | |
Cheng P, Corzo CA, Luetteke N, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–2249. | |
Shen L, Ciesielski M, Miles KM, et al. Modulation of suppressive myeloid populations by tasquinimod. [abstract]. In: Proceedings of the 104th Annual Meeting of the American Association for Cancer Research; 2013 Apr 6-10; Washington, DC. Philadelphia (PA): AACR; Cancer Res 2013;73(8 Suppl):Abstract nr 4746. doi:10.1158/1538-7445.AM2013-4746. Available from: http://cancerres.aacrjournals.org/cgi/content/meeting_abstract/73/8_MeetingAbstracts/4746. | |
Taichman RS, Cooper C, Keller ET, et al. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–1837. | |
Erler JT, Bennewith KL, Cox TR, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. | |
Vanhara P, Hampl A, Kozubik A, et al. Growth/differentiation factor-15: prostate cancer suppressor or promoter? Prostate Cancer Prostatic Dis. 2012;15:320–328. | |
Husaini Y, Qiu MR, Lockwood GP, et al. Macrophage inhibitory cytokine-1 (MIC-1/GDF15) slows cancer development but increases metastases in TRAMP prostate cancer prone mice. PLoS One. 2012;7:e43833. | |
Whitson RJ, Lucia MS, Lambert JR. Growth differentiation factor-15 (GDF-15) suppresses in vitro angiogenesis through a novel interaction with connective tissue growth factor (CCN2). J Cell Biochem. 2013;114(6):1424–1433. | |
Beresford AP. CYP1A1: friend or foe? Drug Metab Rev. 1993;25:503–517. | |
Nandekar PP, Sangamwar AT. Cytochrome P450 1A1-mediated anticancer drug discovery: in silico findings. Expert Opin Drug Discov. 2012;7:771–789. | |
Keating GM. Cabazitaxel: a guide to its use in hormone-refractory metastatic prostate cancer. Drugs Aging. 2013;30(5):359–365. | |
Heidenreich A, Scholz HJ, Rogenhofer S, et al. Cabazitaxel plus prednisone for metastatic castration-resistant prostate cancer progressing after docetaxel: results from the german compassionate-use programme. Eur Urol. 2013;63(6):977–982. | |
Armstrong AJ. The CATCH prostate cancer trial: cabazitaxel and tasquinimod in men with prostate cancer. Available from: http://clinicaltrials.gov/show/NCT01513733. NLM identifier: NCT01513733. Accessed November 22, 2013. | |
Dalrymple SL, Becker RE, Zhou H, et al. Tasquinimod prevents the angiogenic rebound induced by fractionated radiation resulting in an enhanced therapeutic response of prostate cancer xenografts. Prostate. 2012;72:638–648. | |
Active Biotech AB. A study of tasquinimod in men with metastatic castrate resistant prostate cancer. Available from: http://clinicaltrials.gov/show/NCT01234311. NLM identifier: NCT01234311. Accessed November 22, 2013. | |
Ipsen. A proof of concept study of maintenance therapy with tasquinimod in patients with metastatic castrate-resistant prostate cancer who are not progressing after a first line docetaxel based chemotherapy. Available from: http://clinicaltrials.gov/show/NCT01732549. NLM identifier: NCT01732549. Accessed November 22, 2013. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.