")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Mechanism involved in insulin resistance via accumulation of β-amyloid and neurofibrillary tangles: link between type 2 diabetes and Alzheimer’s disease
Authors Rad SK , Arya A, Karimian H, Madhavan P , Rizwan F, Koshy S , Prabhu G
Received 12 May 2018
Accepted for publication 20 September 2018
Published 22 November 2018 Volume 2018:12 Pages 3999—4021
DOI https://doi.org/10.2147/DDDT.S173970
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Tuo Deng
Sima Kianpour Rad,1 Aditya Arya,2–4 Hamed Karimian,2 Priya Madhavan,5 Farzana Rizwan,5 Shajan Koshy,5 Girish Prabhu5
1Department of Molecular Medicine, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia; 2Department of Pharmacology and Therapeutics, School of Medicine, Faculty of Health and Medical Sciences, Taylor’s University, Subang Jaya, Malaysia; 3Department of Pharmacology and Therapeutics, Faculty of Medicine, Dentistry and Health Sciences, The University of Melbourne, Parkville, VIC, Australia; 4Malaysian Institute of Pharmaceuticals and Nutraceuticals (IPharm), Bukit Gambir, Gelugor, Pulau Pinang, Malaysia; 5School of Medicine, Faculty of Health and Medical Sciences, Taylor’s University, Subang Jaya, Malaysia
Abstract: The pathophysiological link between type 2 diabetes mellitus (T2DM) and Alzheimer’s disease (AD) has been suggested in several reports. Few findings suggest that T2DM has strong link in the development process of AD, and the complete mechanism is yet to be revealed. Formation of amyloid plaques (APs) and neurofibrillary tangles (NFTs) are two central hallmarks in the AD. APs are the dense composites of β-amyloid protein (Aβ) which accumulates around the nerve cells. Moreover, NFTs are the twisted fibers containing hyperphosphorylated tau proteins present in certain residues of Aβ that build up inside the brain cells. Certain factors contribute to the aetiogenesis of AD by regulating insulin signaling pathway in the brain and accelerating the formation of neurotoxic Aβ and NFTs via various mechanisms, including GSK3β, JNK, CamKII, CDK5, CK1, MARK4, PLK2, Syk, DYRK1A, PPP, and P70S6K. Progression to AD could be influenced by insulin signaling pathway that is affected due to T2DM. Interestingly, NFTs and APs lead to the impairment of several crucial cascades, such as synaptogenesis, neurotrophy, and apoptosis, which are regulated by insulin, cholesterol, and glucose metabolism. The investigation of the molecular cascades through insulin functions in brain contributes to probe and perceive progressions of diabetes to AD. This review elaborates the molecular insights that would help to further understand the potential mechanisms linking T2DM and AD.
Keywords: Alzheimer’s disease, type 2 diabetes mellitus, insulin deficiency, insulin signaling pathway, cholesterol
Introduction
Many reports suggest a strong pathophysiological links between type 2 diabetes mellitus (T2DM) and Alzheimer’s disease (AD). Prevalence of T2DM and its associated complications leads to AD that increases with time in the aging population, with profound oxidative stress (OS) potentially relating the molecular mechanisms involved in T2DM–AD linkage.1,2 Insulin action in the brain stimulates the modulation of numerous molecular cascades, such as cholesterol metabolism, energy expenditure, glucose homeostasis, feeding behavior, synaptogenesis, neurotrophy, neurotransmitters, cognition, memory, inflammation, apoptosis, and reproduction.3 In addition, insulin regulates the metabolism of peripheral β-amyloid peptide (Aβ) and hyperphosphorylated tau protein. In AD, the extracellular accumulation of Aβ plaques, intracellular aggregation of hyperphosphorylated tau protein in neurofibrillary tangles (NFTs), and neuronal loss occur in the cortex and hippocampus.4,5 Hence, the disruption of insulin functions in diabetic conditions, like hyperinsulinemia and hyperglycemia, interrupts insulin signaling involved in the clearance of Aβ plaques and NFTs pathology. This leads to the accelerated formation of neurotoxic Aβ and NFTs via various mechanisms, including GSK3β, JNK, CamKII, CDK5, CK1, MARK4, PLK2, Syk, DYRK1A, PPP, and P70S6K contributing to the aetiogenesis of AD.1,2,6 In this review, we discuss the roles of aberrant brain insulin signaling in T2DM leading to AD and the mechanisms in the deposition of Aβ and NFTs and their therapeutic potential in restoring the brain pathways that might contribute to T2DM and AD treatment.
Insulin hormone linking type 2 diabetes and AD
The excessive insulin finds way into the brain and interrupts the biochemistry.7–9 One of the possible mechanisms could be the modification of insulin signaling involved in a variety of neuronal functions of brain, such as abnormal protein O-GlcNAcylation, alteration of mitochondria, OS, glucose metabolism, and cholesterol, as well as amyloid plaques (APs) formation, changed Aβ metabolism, and tau hyperphosphorylation protein deposition.1,8,10 Reduced insulin plasma levels in T2DM can impair this signaling pathway forming two core neuropathological hallmarks of AD, ie, NFTs and Aβ plaque, which leads to impaired memory and cognitive dysfunction.11,12 The progression of diabetes to AD and their molecular cascades involved in the function of insulin are discussed below (Table 1).
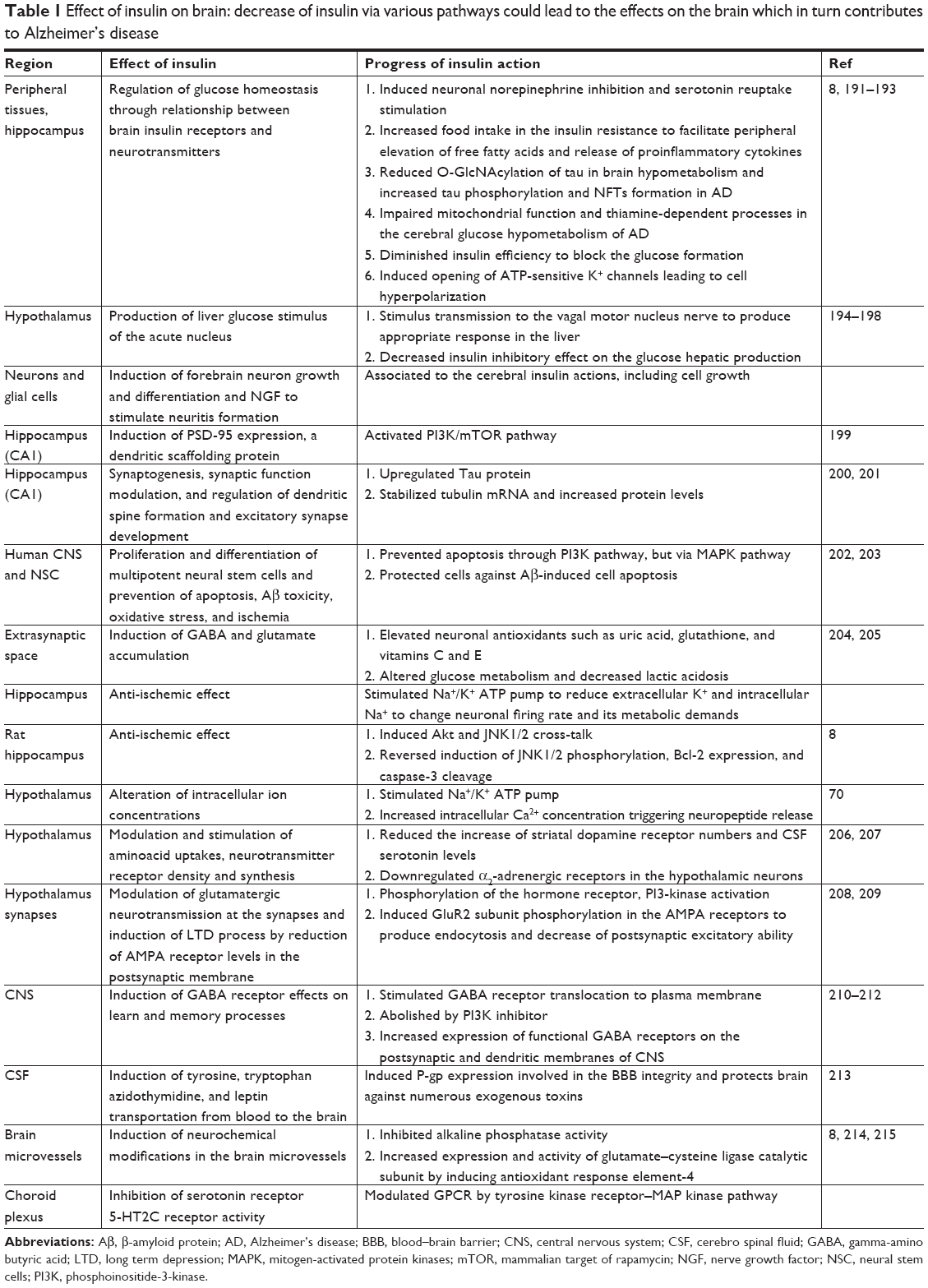 | Table 1 Effect of insulin on brain: decrease of insulin via various pathways could lead to the effects on the brain which in turn contributes to Alzheimer’s disease |
Insulin and brain
Regulation of carbohydrate and fat metabolism is mediated by insulin hormone via stimulating the absorption of glucose from the blood to fat tissue and skeletal muscles. Disturbance in insulin in the periphery system may cause diabetic mellitus (DM), but in the brain develop certain neurodegenerative states like mild cognitive impairment (MCI) and AD. However, brain itself can also synthesize some portion of insulin and crosses the blood–brain barrier (BBB) through a saturable transporter within the central nervous system (CNS) that affects feeding and cognition through CNS mechanisms that are independent of glucose utilization.3,8,13,14 Studies on the mechanisms of insulin production and secretion in the CNS show similarities between beta cells and neurons, remarkably in the context of ATP-sensitive K+ (KATP) channel depolarization.15,16 Increased number of insulin receptors (IRs) during cell differentiation in the brain recommends important role of IR signaling in neuronal proliferation during development, maturation, regeneration of axons, and neurite outgrowth in developing neurons projections as they grow (Figure 1).8,17,18
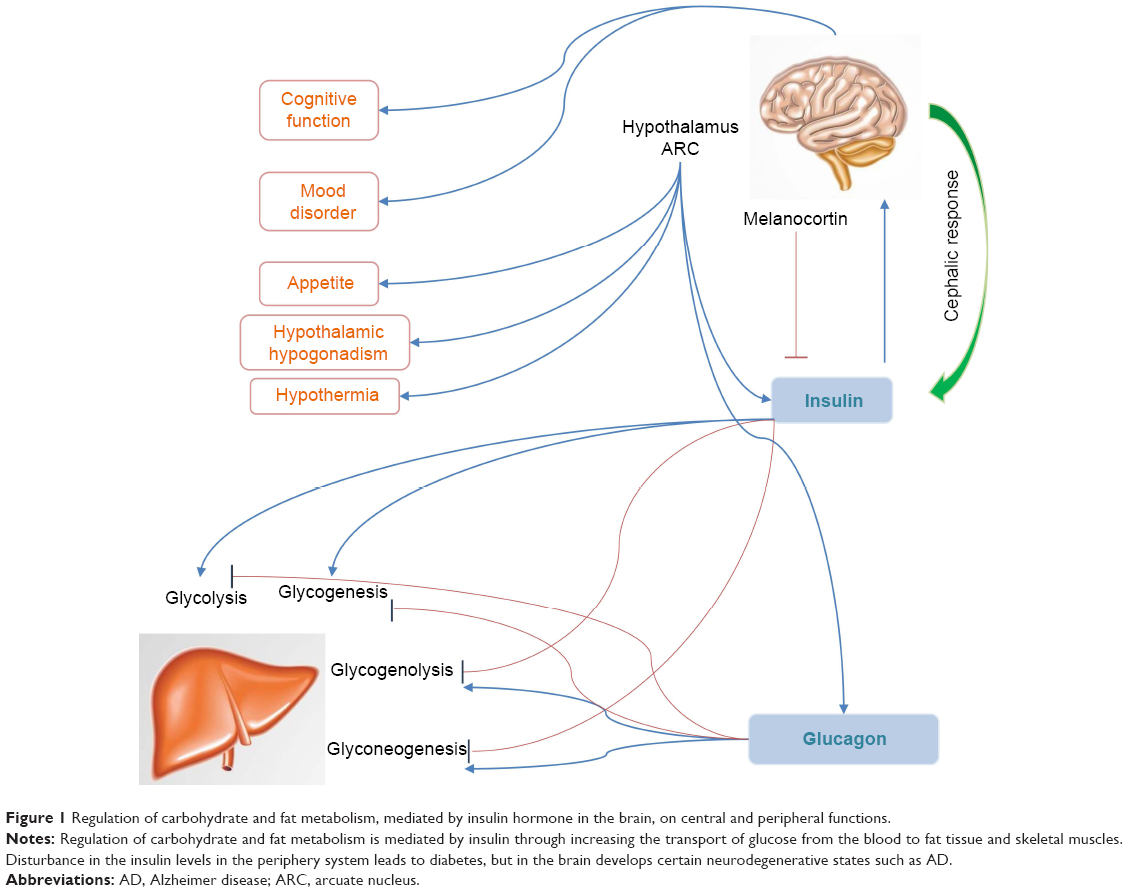 | Figure 1 Regulation of carbohydrate and fat metabolism, mediated by insulin hormone in the brain, on central and peripheral functions. |
Cholesterol metabolism
Cholesterol which is metabolized in the brain plays a crucial role in cell membrane, independent from peripheral tissues featuring BBB, where it plays important membrane function, acts as an antioxidant, and serves as the raw material to produce steroidal progesterone which modulates neuroendocrine functions that alter physiology and behavior in the CNS. Interestingly, in adipose tissue breakdown of fat is inhibited by insulin which is responsible for the intracellular lipase inhibition that demands triglycerides to hydrolyze and release fatty acids. Moreover, insulin stimulates entry of glucose into adipocytes to synthesize glycerol within the cells, thereby enhancing the rate of glucose translocation across the cell membrane, muscles, and in adipose tissue. Then, apolipoprotein E (ApoE)-cholesterol particle is processed to free the cholesterol in the lysosomes and is then transported to the membrane. ApoE isoform ε4 is the most common risk factor for AD that correlates with escalation of Aβ clearance and accumulation in the brain during AD.19–22 Thus, insulin alteration in diabetes can interrupt brain cholesterol metabolism leading to metabolic dysfunction, thereby causing neurological disorders (Figure 2).23
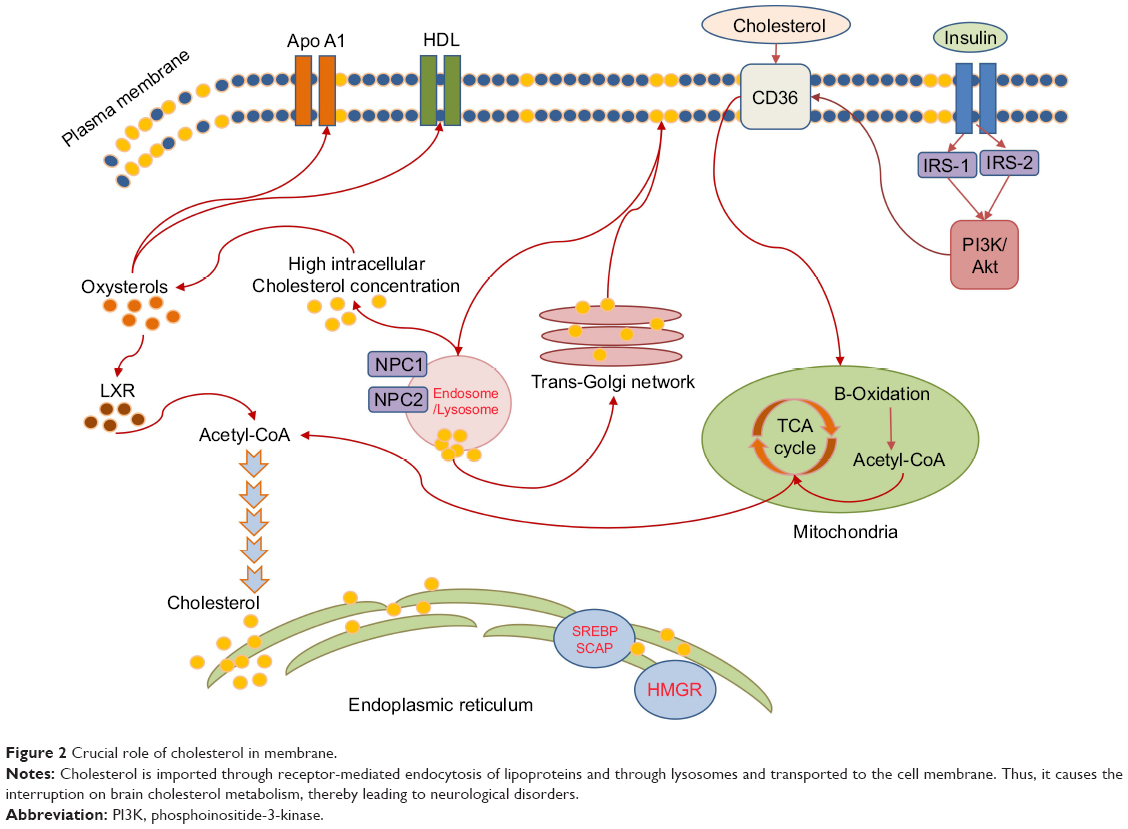 | Figure 2 Crucial role of cholesterol in membrane. |
Glucose uptake
Uptake of glucose, the main fuel in body, varies among the tissues depending on the tissue metabolic needs and glucose availability. The glucose transporter (GLUT) protein isoforms are involved in facilitating the translocation of glucose in which the prominent isoforms are GLUT1-4. Glucose uptake is stimulated by the movement of GLUT4 transporters from the intracellular membrane into the plasma membrane which demands GLUT4-containing vesicles to facilitate the process.24 In the kidney, glucose uptake is accomplished by the secondary active transport mechanism through GLUT2 transporter, linked to Na+/K+ pump reliant on the sodium gradient generated by NaKATPase. Malfunctioning of GLUT4 protein in the hippocampus affects the biochemical reactions and cognitive flexibility offered by hippocampal neurons, thus developing depression and lowering the cognitive function which in turn increases the risk of Alzheimer development (Figure 3).25–27
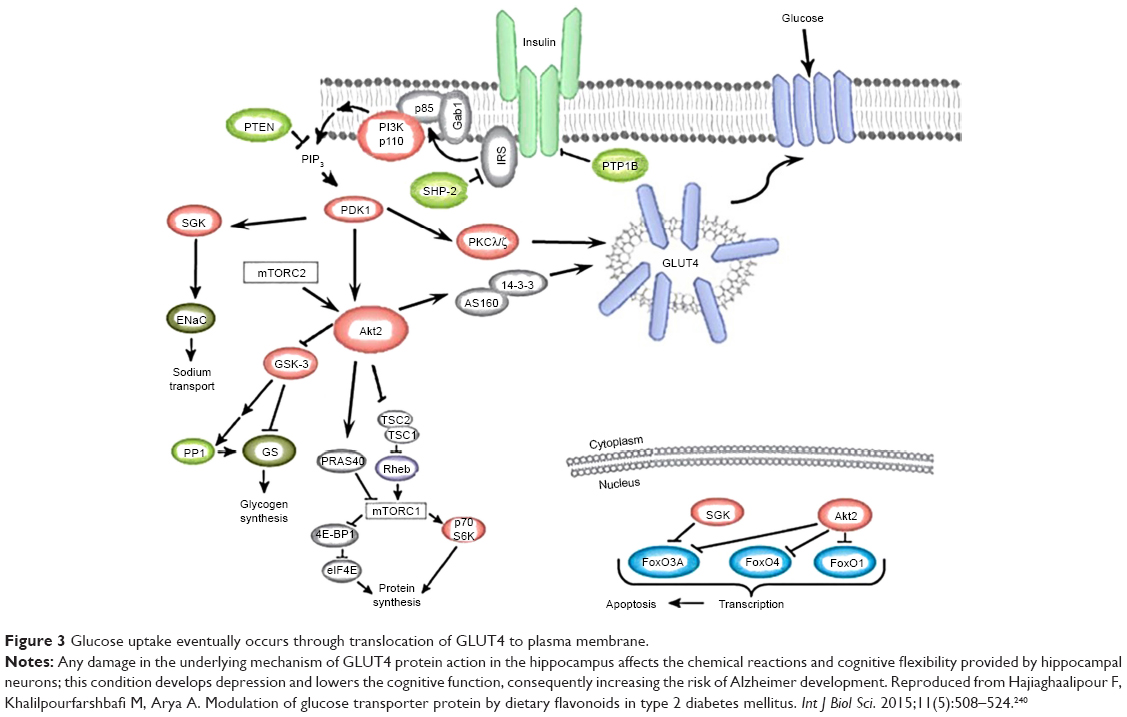 | Figure 3 Glucose uptake eventually occurs through translocation of GLUT4 to plasma membrane. |
GLUT function can be regulated by insulin-like growth factor (IGF) family. IGF is very close to the natural human growth hormone and consists of three ligands (insulin, IGF-1, and IGF-2), six IRs (IRα [fetal], IRβ [adult], IGF-1 receptor [IGF-1R], IGF-2R, hybrid IGF-1R/IRα, hybrid IGF-1R/IRβ), and up to seven IGF-binding proteins (IGFBP1-7).28,29 IGF-1 and insulin can control the neuronal excitability, metabolism, and survival through insulin/IGF-1 signaling pathway. Abnormality and disruption in the activity of these pathways trigger the continuous dwindle of neurons in AD brain.30,31 Few evidences on the brain of AD patients showed deficit ratio of insulin and resistance in IGF-1, suggesting that AD might be a brain-type diabetes or diabetes type 3.32,33 Altered neuronal IGF-1 function seems to be an important aspect of the overall synaptic and neuronal pathology induced by Aβ protein precursor (AβPP)-Aβ clearance in the apoE4 carriers. Hence, both hyperinsulinemia and hyperglycemia can increase the neuritic plaque formation and progress in AD.30,34
Energy expenditure
The amount of energy which is consumed for the performance of physical activities, such as inhalation and exhalation, blood circulation, breakdown of food particles, or physical movement, is known as energy expenditure. This energy is obtained by the electrochemical gradient generated by the electron transport chain (ETC) which drives ATP synthesis via ATP synthase. The ATP production capacity and/or efficiency is performed via mitochondrial dynamics in beta cells.35,36
Leptin as an adipocytokine which is produced in the peripheral system as well as in the brain possesses key role in phenomena such as food intake, obesity, glucose homeostasis, and energy expenditure. It is proved that both leptin expression levels and signaling pathways could be connected to the pathophysiology of many neurodegenerative diseases, such as AD. It is illuminated that leptin receptors are highly expressed in the hippocampus involved with learning and memory, and are found critically affected in AD patients. In vivo and in vitro studies suggest that leptin supplementation could decrease both Aβ production and tau phosphorylation which contribute to the development process and pathogenesis of AD.37 Insulin secretion occurs in blood stream based on the availability of free fatty acid, amino acid, and beta cell measures glucose through mitochondrial respiration and nutrient oxidation accordingly. The main stimulator of insulin secretion signal is elevation of cytosolic ATP/ADP ratio or high glucose oxidation, mitochondrial ATP synthesis, and low ATP demand in the beta cells.38,39 Permanent excess nutrient or continuous exposure to fat in T2DM damages mitochondria or decreases its function in the beta cell through reduced antioxidant activity and sustained overproduction of reactive oxygen species (ROS), independent of changes in mitochondrial ATP synthesis.40 Besides, NF-kβ, TNF-α, and IL-6 as acute inflammatory cytokines lead to negative energy balance and promote energy expenditure and increase ROS.41,42
It is shown that abnormal production of AT-derived proteins contributes to the pathogenesis of insulin resistance and metabolic syndrome such as T2DM.9,43 The inflammatory cytokines elevation, energy expenditure, and insulin deficiency result in high glucose expenditure and accumulation of β-amyloid peptide as a hallmark of AD.8,9,28
Role of leptin in glucose homeostasis
A balance between the insulin and glucagon maintains blood glucose levels or glucose homeostasis. Insulin exerts its pleiotropic effects through binding to the insulin receptor substrate (IRS) proteins which mediate regulation of glucose transport, protein metabolism, and control of cell growth and survival. IRS proteins connect insulin receptor activation to essential downstream kinase cascades, such as the phosphoinositide-3-kinase (PI3K) or mitogen-activated protein kinases (MAPK) pathways. Decreased IRS-1 contributes to reduction of glucokinase and increases blood glucose levels in diabetes.44–46
As we know, hypothalamus regulates leptin signaling that has a role in food intake and energy homeostasis in mammals which results in the downregulation of orexiogenic peptides, such as neuropeptide Y (NPY) and agouti-related peptide (AgRP), and in reverse, it can increase the expression of anorexiogenic peptides, such as α-MSH, which promotes energy expenditure in either adipose or skeletal muscle tissue. It is indicated that leptin-mediated ObRb receptors are expressed in vast density in the arcuate nucleus (ARC), dorsomedial nucleus (DMH), and the ventromedial nucleus (VMH) of the hypothalamus. In the ARC, obRb is rarely expressed in two different neuronal cell types of the hippocampus (CA1 and CA3 regions) and the dentate gyrus,47 which express both NPY and AgRP, and proopiomelanocortin (POMC) is mainly expressed.48,49 Also, leptin increases synaptogenesis and aids in memory formation in the hippocampus and is pretended to be a cognitive promoter.50 Similarly, it was shown to elevate neurogenesis in the dentate gyrus in rodents.51 Leptin also plays a vital role in hippocampal neuronal survival via activating the PI3K/Akt/mammalian target of rapamycin (mTOR), as well as the AMP-activated protein kinase (AMPK)/SIRT1, JAK2/STAT3, ERK pathway signal transduction pathways through binding to its long-form receptor obRb.52 Leptin upregulates the expression of some potent endogenous antioxidant enzymes involved in apoptosis, such as manganese superoxide dismutase and the anti-apoptotic protein Bcl-xL in the hippocampus.52
Leptin can modulate Aβ production and metabolism. Interestingly, chronic peripheral leptin administration in Tg2576 mice reduced tau phosphorylation explicitly at residues Ser202, Ser396, and Ser404 in retinoic acid. Such reduction is suggested to be mediated via AMPK, Akt, and p38 pathways.53,54 All these evidences could illuminate the role of leptin in T2DM and highlight AD linkage. Moreover, epidemiological studies have also found depleted leptin levels in the pathogenesis of AD. In a study by Narita et al, it was found that higher leptin levels positively correlate with higher hippocampal volumes.55 It should be noted that all the leptin-induced signaling pathways link to phosphorylation of glycogen synthase GSK3β and decrease in hyperphosphorylation of tau.56 It is interesting that the low circulating leptin levels, in turn, could potentially contribute to cognitive decline and worsen the pathology, leading to a downward spiral of further weight loss and progression of AD.57 The presence of amyloid plaques, NFTs, and neurodegeneration in the hypothalamus of human AD brains suggest that Aβ-mediated suppression of leptin-responsive cells in the hypothalamus is extremely possible.58,59 Hence, leptin may possess a bidirectional role in the dysfunction of leptin signaling that exacerbates AD pathology.
In addition, insulin activates PKB (or Akt) which is a serine/threonine kinase, composed of various members, including PKBα (Akt1) and PKBβ (Akt2). Only PKBα drives islet and β-cell proliferation. Interestingly, PKBα-deficient mice also revealed normal insulin-stimulated disposal of blood glucose.60,61
TNF-α is a key cytokine that influences intermediary glucose metabolism which compromises IR/IRS-1 signaling independently of transcriptional regulation. This effect is mediated by minimum of seven serine kinases, which include c-JUN-NH2-terminal kinase (JNK), Akt/PKB, and IKK. Tyrosine phosphorylation of IRS-1 and IRS-2 serine at 307 residues is an essential factor to actively downstream effector pathways impairment in the insulin homeostasis. Insulin homeostasis impairment affects glucoregulatory mechanisms characterized by altered glucose tolerance and causes insulin resistance leading to escalation of Aβ peptide, APP, NFTs neurotoxicity, and tau phosphorylation associated with AD.8,61,62
Feeding behavior
Controlling the body mass by maintaining food consumption underlies a twisted flow. Excess of food intake contributes to the onset and progression of the metabolic syndrome. Some hormones such as insulin, glucagon-like peptide 1 (GLP-1), and leptin are involved in the regulation of food uptake and energy consumption.63–66 Insulin with leptin exerts their acute effect by altering cells’ function and nutritional behavior via PI3K pathway. PI3K increases α-MSH release and decreases NPY release which induces depolarization of AgRP neurons and increases food intake. GLP-1 also regulates glucose homeostasis and reduces food intake. Food intake and the anorexic brain-gut peptide GLP-1 activate amygdala dopamine signaling through D2 receptor which is necessary and sufficient to alter the feeding behavior.67–69
Hypothalamic AMPK regulates food intake as well as body weight through altering the expression of NPY, AgRP, POMC, and CART in the ARC nucleus. Unlike feeding, fasting increases hypothalamic AMPK activity. In the hypothalamus, minimum of two mechanisms exert impact on the anorexigenic effects on AMPK inhibition which results in the activation of acetyl-CoA carboxylase (ACC) and mammalian target of rapamycin (mTOR), and the phosphorylation of p70S6 kinase (p70S6K).68–71 In T2DM, increased mTOR and p70S6 kinase expressions elevate production of leptin which has direct effects on food intake. Feeding behavior dysfunction in T2DM modulates brain functionality leading to neurodegeneration process such as AD through overproduction of APP, Aβ 1–42, and thereby accumulations of Aβ, and also contributes to NFTs production.2,72
Synaptogenesis feeding behavior
Synaptogenesis is a multi-step process of synapse formation which is promoted by IGF-1 and IGF-2 through several pathways during all the major phases of neurodevelopment. The activation of protein kinase C (PKC) also regulates synaptogenesis through phosphorylation, binding to signaling lipids, and translocation from the cytosol to the membrane.17,29,73–75 Phosphorylation of PKC at the first step is essential for its activation and formation of its catalytically active competent conformation. PI3K activation by insulin also induces synaptogenesis and controls the expression of synaptic markers in addition to their accumulation in the nerve cells. PI3K, accompanied with the existing elements of the InR signaling pathway, controls cellular magnitude, growth and multiplication, and creation of synapses in between the neurons. PI3K and B/Akt protein kinase regulate the development of synapses as well as their preservation. PI3K acts via its binding to synapsin, actin filaments, and high phosphoinositide levels that are linked to the cAMP pathway and cAMP response.60,76 The sites of expression of IRSp53 in the synapses are located on the granular layer of the cerebellum and hippocampal neurons, which suggests that these molecules are components of insulin-dependent signaling pathway at the postsynaptic apparatus. IRSp53 is a key factor in cytoskeleton which is phosphorylated upon stimulation with insulin and involved in neurite outgrowth and neurodegenerative disorders.8,77 Insulin stimulates translation, but not the transcription, of postsynaptic density PSD-95 in the hippocampal CA1 neurons, through the PI3K–Akt–mTOR pathway, an important intracellular signaling pathway in regulating the cell cycle.74,78 There is a linkage between the phosphorylation of IGF1-induced Akt and release as well as the translocation of GLUT4 from intracellular pools to nerve process membranes in the normal developing brain. High glutamate levels phosphorylate the Ser (307) residue in the IRS-1 protein which develops less reactivity and induces IGF-I via activation of pathway associated with protein kinase A (PKA) and PKC. This action arises due to a reciprocal activity between IGF-1 and nerve growth factor (NGF) in the peripheral nerves, where the PI3K/Akt/GSK3 pathway underlies the impact raised from the cooperation of both agents on axonal growth. Insulin signaling pathways activate PKC and its substrates, many of which are vital components of synaptogenesis, cognition and neuronal repair, differentiation, growth, and apoptosis.79
Insulin also activates MAPK pathway through tyrosine phosphorylation of certain prototypical signaling adaptors such as Shc/Grb2, SOS/Grb2, and Gab-1/Shp2. Diabetes declines the activity of PI3K/AKT/mTOR in the enteric neurons, which impairs retrograde NGF transport in the vagus nerve. Activation of PI3-phosphatase decreases cellular contents of lipid products by PI3K. Any defects in the intracellular PI3K translocation or phosphatase activation may modify Akt/PKB activity.74,78,80 Synaptogenesis and synaptic remodeling increase Aβ oligomers which can directly produce neuronal insulin resistance and directly bind to PKC and inactivates it. GSK-3β phosphorylates multiple sites of tau protein in the intact cells. Aberrant hyperphosphorylated tau protein is a critical feature in AD pathogenesis that signifies a close molecular relationship between diabetes and AD.74,80,81
Neurotrophy
Neurotrophy or nerve damage is strongly regulated by insulin which is essential for neuronal development and survival via IGF-1 and ROS signaling pathways. IGF-1 pathway coordinates growth, proliferation, differentiation, development, metabolism, and glucose homeostasis. The graft of IGF-1 and the related receptor trigger the phosphorylation of essential adaptor proteins together with Shc and IRSs, which leads to the activation of two prosurvival signaling pathways.75,82,83 Phosphorylation of IRS-1 or IRS-2 stimulates PI3K–PDK1–AKT signaling pathway, whereas phosphorylation of Shc induces RAS, RAF, and ERK/MAPK signaling pathway which leads to regulation of neurotrophy. Moreover, phosphorylation of threonine 308 via PDK1 or phosphorylation of serine 473 via mTORC2 results in the activity of AKT; this activation increases the life span of cells through abundant mechanisms, like deterrence of apoptosis and giving rise to prosurvival gene expression. Decreased levels of serum IGF-I in DM patients with sensory and autonomic neuropathy compared with nonneuropathic DM or nondiabetic controls and IGF-I and IGF-II lead to sympathetic neuroaxonal dystrophy. IGF-1 blocks amyloid toxicity by increasing survival signaling through PI3–AKT and ERK which accumulate high levels of Aβ from overexpressing APP. The Aβ oligomers elevate pro-inflammatory cytokines in the brain that mimic the trophic factor/insulin resistance as observed in AD brain.18,30,71,75,84
Apoptosis
Several studies suggest the protective role of insulin against apoptosis through various signaling pathways that suppress the excessive accumulation of ROS within the cells.85 The insulin/IGF/Akt is one of these pathways in promoting β-cell survival. However, ER stress–induced apoptosis is mediated at least in part by signaling through the phosphatidylinositol 3-kinase/Akt/GSK3β pathway. Moreover, presence of advanced glycation end products (AGEs) and advanced lipoxidation end products (ALE) that merely resulted from a long-term accumulation of modified protein can be considered as a stress. Chronic hyperglycemia-induced OS such as nitric oxide (NO) plays a central role in the formation of AGEs in DM.86,87 G-protein-adenylyl cyclase signaling, diacylglycerol (DAG)/PKC pathway, and calcium movement play a vital role in diabetes-induced galactooligosaccharide (GOS). Normalization of mitochondrial superoxide production blocked AGEs overproduction, PKC activation, and increased glucose flux through the aldose reductase pathway and NF-κB activation.88,89
Impaired immune system in diabetes is almost affiliated with the reduction of antioxidant activity and antioxidant enzymes manifestation as well as eccentric performance or abnormal enzyme activities. Among the diversity of the existing antioxidant enzymes, SOD, GPx, and CAT reflect greater impact on the regulation of ROS formation and entire antioxidant content available in a certain tissue as well as in DM.90–92 NADPH oxidase and dysfunction of mitochondrial respiratory chain (MRC) are also a major source of ROS in diabetes. Thus, blocking the overexpression and activation of this enzyme and subsequent ROS production together with its ROS scavenging property reduce OS in diabetes. Mitochondrial-derived superoxide anion is common in complications with diabetes. Overexpression of manganese-dependent superoxide dismutase (Mn-SOD), which is the major scavenger of mitochondrial superoxide anion and mitochondrial DNA damage in T2DM, prevents high glucose-induced OS and cell apoptosis. OS-related pathways interconnect AD and T2DM. It is a well-known connection of Aβ protein and hyperphosphorylated tau with glucose metabolic intermediates and IRs. Insulin transporters cause this interconnection in the AD brain, Aβ accumulates in the plaques, and receptor for AGE mediates Aβ 1–42-induced perturbations of APP and NFTs neurotoxicity (Figure 4).18,87,89,93
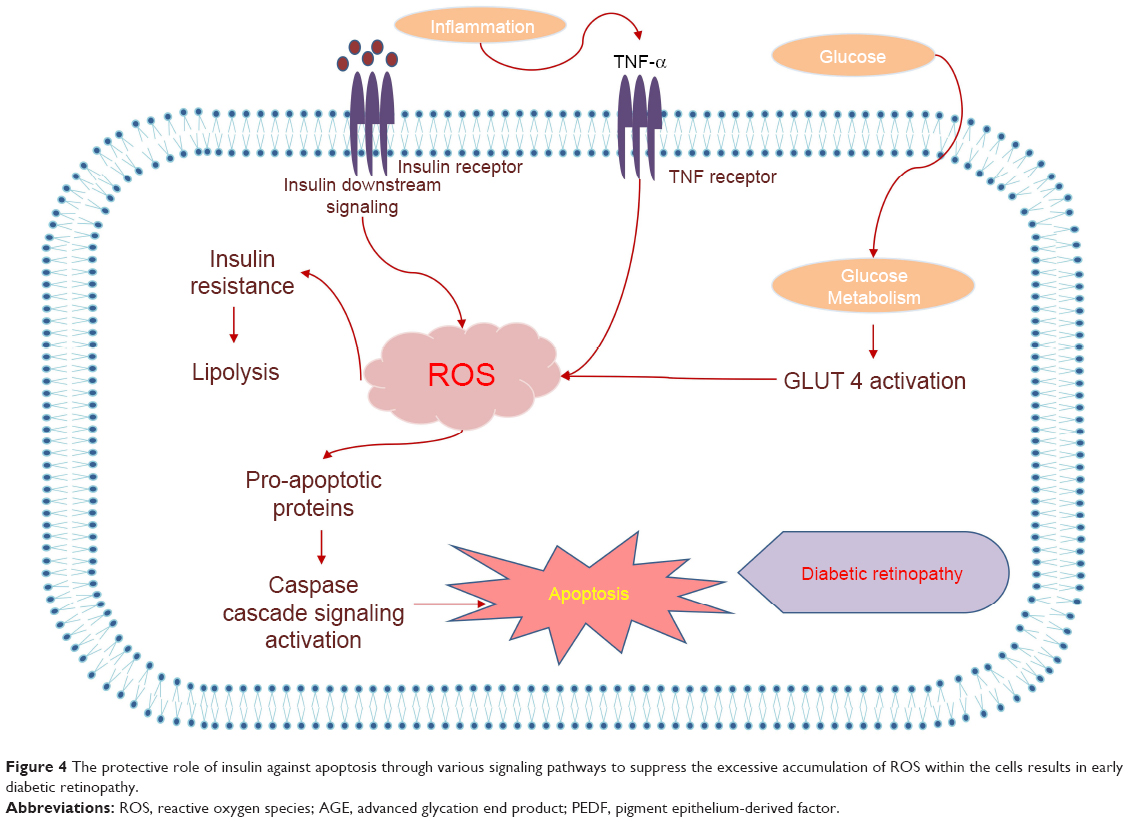 | Figure 4 The protective role of insulin against apoptosis through various signaling pathways to suppress the excessive accumulation of ROS within the cells results in early diabetic retinopathy. |
Neurotransmitters
Endogenous chemical messengers that enable transmission of signals from one neuron to the target neuron, muscle cell, or gland cell stimulating insulin and glucagon are demarcated as neurotransmitters. Glutamate is one of the most abundant neurotransmitters in the brain; an excess of glutamate overstimulates brain cells, which results in neurological inflammation and cell death. A high glutamate concentration triggers insulin release to lower glucose levels which in turn increases glutamate. Another neurotransmitter, gamma-amino butyric acid (GABA) in the CNS, prevents nerve transmission in the brain and has a calming nervous activity. Heavy secretion of insulin results in a protracted secretion of GABA, glutamate, aspartate, and taurine through the number of GABAA receptors. Moreover, insulin therapy could be considered as an efficient therapy to bridle the toxic activity of neurotransmitters to preserve neurons. Therefore, there is a close linkage between GABAergic signaling system and various aspects of AD pathology, including tau hyperphosphorylation, Aβ toxicity, and apoE4 effect. Low levels of GABA and glutamate cause significant limitation in the activities of synapses and synaptic transmission of neurons in the temporal cortex of AD patients, and activation of GABAA receptors induces tau hyperphosphorylation.18,94–96
Promoting glycogen synthesis
Synthesis of glycogen from glucose in skeletal muscle is regulated via the activity of certain hormones such as insulin. Muscle and liver uptake the available glucose upon stimulation by insulin hormone and leads to the activation of glycogen synthase (GS) through dephosphorylation of three specific serine residues, collectively termed sites 3.97–99
GS kinase (GSK)-3 is principally responsible for phosphorylation of sites 3, whereas phosphatase (PP)-1, a glycogen-bound form of protein, dephosphorylates these sites.100 However, defeat in tracing the fall in quantity of cAMP localization in muscles is associated with a secondary glycogen synthase kinase which is not influenced by cyclic nucleotides. Immediate effect of insulin is to redirect synthesized glucose-6-phosphate to glycogen without affecting the rate of gluconeogenesis which requires hepatic Akt2-dependent redirection of glucose-6-phosphate to glycogen independently of GSK3α and GSK3β phosphorylation. Downstream defects at the level of glycogen synthase kinase (GSK)-3 or impaired regulation of the GSK3 target site of GS (site 3a/b/c and 4) which leads to abnormal phosphorylation in activation of GS and dysregulation of CaMKII seems as a major cause of insulin resistance phenomenon. In AD brain, increased activities of Akt, PKA, and GSK3α, and β and Aβ-induced GSK3β phosphorylation increase tau phosphorylation which leads to mitochondrial dysfunction.99–102
Cognition and memory
Controlling the transmission of ions through neuroreceptors located on the membrane is activated by neurotransmitters and synaptic transmission which influence the cognitive function in the presence of insulin.77 Suppression of Wnt or PI3-kinase signaling ruin the synaptic connections between neurons which is known as long-term potentiation (LTP) and results in the less synaptic strength which affects the process of learning and memory function. GSK3β at high expression level suppresses LTP impact, thus providing lesser spatial learning.103,104 GSK3 also phosphorylates and inhibits cAMP-responsive element-binding protein, a universal modulator of memory.105 Moreover, GSK3 promotes actin and tubulin assembly, processes required for synaptic reorganization during memory formation. PI3K/Akt/GSK-3β is another pathway to impair the ability of insulin in activation of glucose disposal and glycogen synthase in T2DM. Overexpression of GSK3 induces a series of pathological changes, most of which are hallmarks of AD and T2DM incurring severe pathology, such as cognitive decline. Adiponectin is an important target for AD by induction of Aβ and Tau phosphorylation in hippocampus and extrahypothalamic region. GSK3β together with GSK3α causes AD by inducing tau hyperphosphorylation to form NFTs through PI3K/Akt/GSK-3β signaling pathway.103,106–108
Inflammation
Insulin suppresses the pro-inflammatory proteins such as JNK, IKKβ/NF-κB, AP1, CAM1, PSD95, and MCP1 which downregulate the inflammatory response. Inflammation together with insulin resistance is increased by expression of several pro-inflammatory cytokines such as interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF)-α. In contrast, IKKβ acts selectively against the physiological substrates and the IκB protein inhibitors of NF-κB. Phosphorylation by IKKβ targets IκBα to degrade proteasome that liberates NF-κB for translocation from cytoplasm into nucleus to promote expression of numerous target genes and consequently induce insulin resistance.42,62,109–111
PI3K–Akt–mTOR signaling pathway facilitates the synthesis of PSD-95 protein via insulin induction in Dendron’s and hippocampal area. PSD-95, a 95-kDa scaffolding protein of PSD, is degraded by IL-1β. T2DM enhances pro-inflammatory factors in the brain cells such as microglia and astrocytes that contribute and provoke AD. Inflammatory agents like toxicants and pollutants when accumulated in higher proportion lead to cellular stress, amyloid precursor protein (APP), and rise in genesis state, thereby stimulating amyloid-β–42 (Aβ–42) peptide production.78,112
Role of insulin resistance and tau protein in AD
Insulin resistance, impaired glucose tolerance, and formation of insoluble protein aggregates, as well as the loss of neurons and synapses, extend risk factors in the development process of AD, but evidence for this assertion is not consistent. Impaired insulin signaling certainly does not preclude evidence from having a deficit effects on cognition independent of its role in AD pathology, such as diminished learning, memory, problem solving, and mental flexibility. Mechanisms of T2DM progression to AD by insulin are classified into two main categories: NFTs and Aβ formation. Various experimental paradigms suggest that Aβ and tau have been found to exert synergistic modes of toxicity, while the effect of insulin on the brain is complex and not confined to Aβ production (Figure 5).2,9,18,113,114 Tau protein plays a wider role in cellular shape, motility, and signal transduction in AD. The C-terminal of this protein is probably responsible for tubulin-binding and the acidic N-terminal region interacts with other cytoskeletal elements. The proline-rich middle region contains the target sites of many kinases. Moreover, R1–R4 are four repeat domains called microtubule-binding domains (MBDs) and each of them repeats and conserves consensus motif KXGS, which can be phosphorylated at serine.115,116
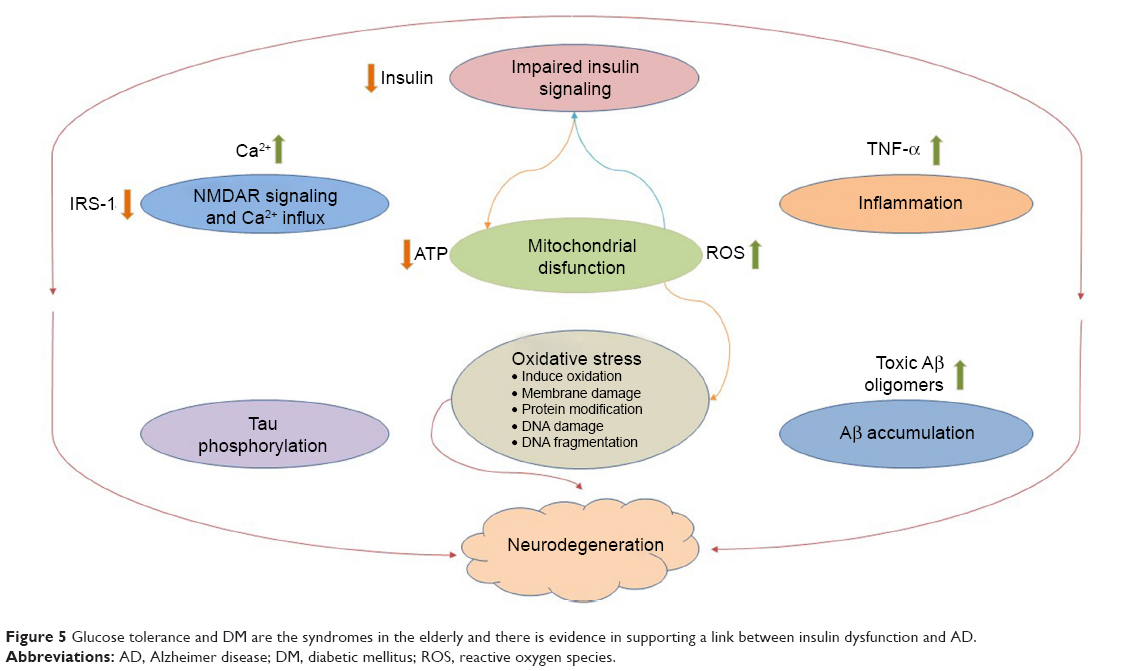 | Figure 5 Glucose tolerance and DM are the syndromes in the elderly and there is evidence in supporting a link between insulin dysfunction and AD. |
Insulin can modulate phosphorylation of tau protein which are MBD molecules involved in microtubule assembly and stabilization. Dysfunction of insulin can cause tau hyperphosphorylation through two different mechanisms at specific amino acids including Ser and Thr: glucose/energy metabolism and temperature independent.117,118 Out of 85 phosphorylatable residues in tau protein, 28 sites are exclusively phosphorylated in AD brains (Table 2). Reduced insulin plasma levels in T2DM can impair this signaling pathway resulting in tau hyperphosphorylation and disintegration of microtubules and thereby formation of NFTs.115 Most promising candidate kinases for tau phosphorylation which are responsible to provoke AD and T2DM are listed in Table 2. The details and the functions of each kinase are as follows:
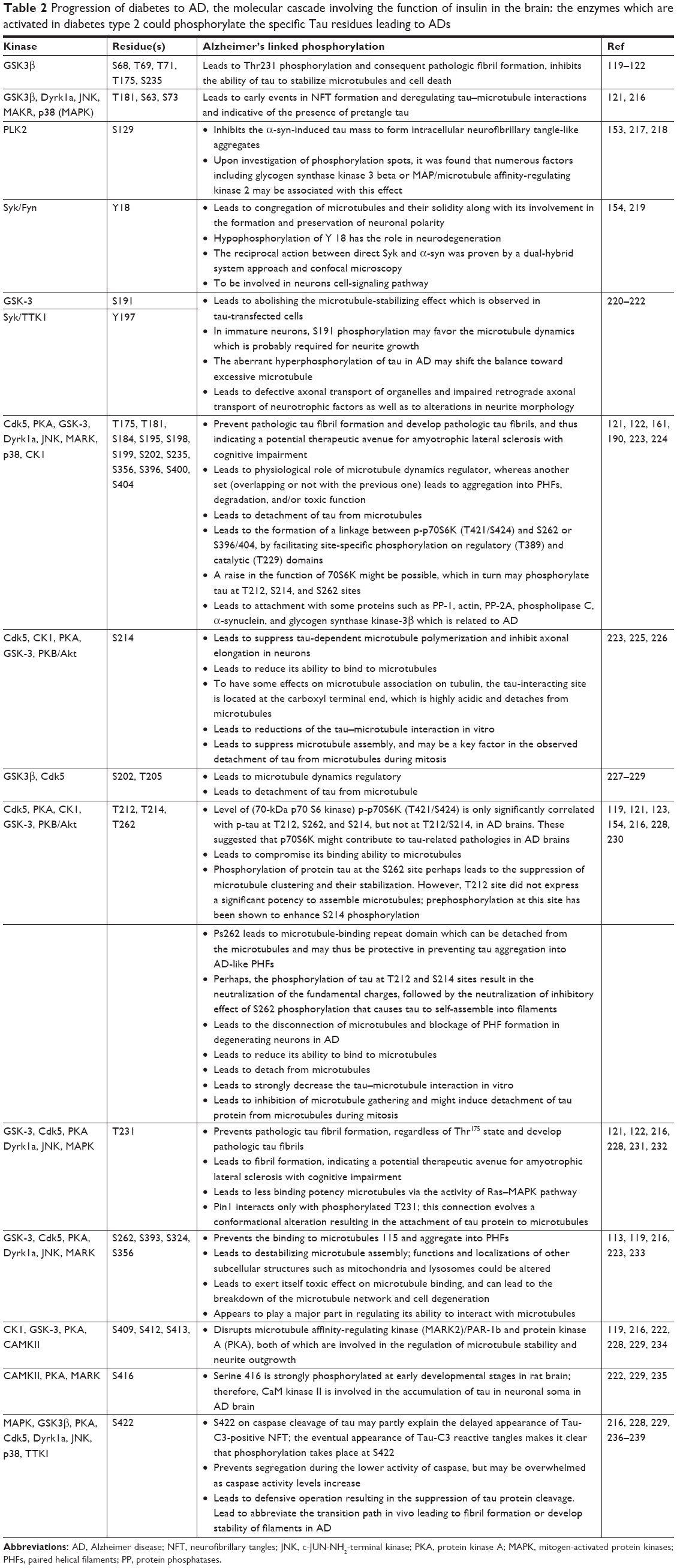 | Table 2 Progression of diabetes to AD, the molecular cascade involving the function of insulin in the brain: the enzymes which are activated in diabetes type 2 could phosphorylate the specific Tau residues leading to ADs |
GSK3β
It was shown that H2O2 increases GSK-3β activity in human embryonic kidney 293/Tau cells which leads to tau hyperphosphorylation at Ser396, Ser404, and Thr231. GSK3β is involved in the formation of both Aβ deposits and NFTs, two pathological features of AD. The Aβ promotes GSK-3β activity in the neuronal cells which is at Thr231 residue, but it enhances phosphorylation at the S9, S68, T69, T71, T175, and Ser396/404 sites which decreases tau–microtubule interactions and the pathologic fibril formation, thereby reducing tau binding to microtubules.119–122
c-JUN-NH2-terminal kinase
The JNK is a subfamily of the MAPK that binds and phosphorylate c-Jun on Ser-63 and Ser-73 within its transcriptional activation domain. The β2-adrenergic receptor (β2AR)–PKA–JNK pathway phosphorylates tau protein at Ser-214, Ser-262, Thr231, and Thr-181, which are utilized by Aβ signals in the primary neurons of prefrontal cortex (PFC) in mouse brain.123 Knocked-out β2AR mice showed an extreme decrease of phosphorylation of presenilin 1 (PS1) and APP associated to the Aβ-induced tau pathology in AD. Aβ also induces the clusterin/p53/Dkk1/wnt–PCP–JNK pathway, which drives the upregulation of several genes that mediate development of AD-like neuropathologies; these studies are in agreement with the animal experiments.124
JNK plays a critical role in regulation of insulin signaling, inflammatory response, apoptosis, and caspase-3 activity in diabetes and increases the expression of IL-6, IL-8, monocyte chemotactic protein-1, and tumor necrosis factor-α 35 in AD pathology.8,125,126
CamKII
Calcium-/calmodulin-dependent protein kinase type II is one of the abundant Ca2+-regulated protein kinases in the brain; these kinases are expressed primarily in neurons. CaMKII is regulated by Ca2+-/CaM-induced autophosphorylation at multiple sites such as Thr 286/287.127 The tau phosphorylation is upregulated at Ser214, Ser262, Ser131, Thr212, and Thr135 by CaMKII kinase in frontal cortex and hippocampus, and is found in PHF-tau of AD brains and T2DM which leads to the inactivation of protein phosphatases (PP).118,128 Obviation of synapses results in depleting the memory function in preliminary stage of AD which is expected to be induced via Aβ oligomers by CaMKII. O-GlcNAc modification by CaMKII at Ser 279 activates CaMKII autonomously, creating molecular memory even after Ca2+ concentration declines in T2DM.129 Furthermore, it was shown that the activation of CaMKII directly inhibits AGEs formation significantly and reverses D-ribose-induced tau hyperphosphorylation which links T2DM to AD.115,128
CDK5
Cdk5 belongs to the Cdk family which is expressed in the CNS and other tissues. It regulates several cell processes such as neuronal migration, actin, and microtubule dynamics. Cdk5 acts through PP1 and regulates several proteins such as inhibitor-1 (I-1) and I-2. P25 is a neurotoxic activator of cdk5 which triggers tau phosphorylation and NFTs formation in AD pathophysiology.130 Moreover, p35 is a neuronal-specific protein that is nonhomologous to cyclins, and is responsible for the identification and activation of Cdk5 in association with Cdk5R1 or Cdk5R2. Interestingly, the study on gene-targeted therapy opens up the clue with potential role of p35/Cdk5 kinase in the migration process of neurons as well as in the development stage of mammalian cortex.131 Expression of Cdk5 and its co-activator p35 is upregulated in the presence of high glucose concentration and strong bounding with Cdk5 kinase is maintained. In contrast, transforming growth factor beta receptor I (Tgfbr1) inhibitors downregulate the expression of Cdk5 and p35 kinase activity. Similarly, early growth response protein 1 (Egr-1) has a capacity to highly express in the presence of glucose by mediating TGF-β1-ERK1/2 pathway and its inhibition by siRNA downregulates p35 and Cdk5 scenario.132 Moreover, protein–protein interactions regulate the activity of Cdk5 with the intervention of regulatory and target molecules having substantial association with nestin in approaching p35, demonstrating streamlined flow and continuation of Cdk5/p35 activity. The truncated form of p35 molecule, p25, acquires and gathers in higher amount in the brain of AD patients’ neurons. This increases Cdk5 kinase activity by repelling the degradation of p35 to p25 and binding of p25 to Cdk5 reflecting its cellular location and alters its substrate specificity. Hyperphosphorylation of tau molecules by p25/Cdk5 complex reduces its ability to associate with microtubules, thereby inducing cytoskeletal disruption, morphological degeneration, and apoptosis. Therefore, various findings support the idea of indicating p35 cleavage and accumulation of p25 involvement in the pathogenesis of cytoskeletal abnormalities and neuronal death.133 Cdk5 is also expressed in adipocytes to phosphorylate proliferator-activated receptor gamma (PPARγ) which leads to metabolic syndromes such as T2DM.134,135 High glucose levels induce the expression of p35 and Cdk5 through TGF–β1–ERK1/2–Egr-1 pathway leading to create high ROS.132 ROS also induces tau hyperphosphorylation and neuroinflammation in AD and T2DM via increasing proinflammatory mediators and the expression of TNF-α, IL-1β, and IL-6, and apoptosis.136,137
CK1
CK1, a ubiquitous serine/threonine-selective protein kinase, is mainly expressed in the neurons. CK1 is involved in the tau hyperphosphorylation and Aβ production which has been evidenced by the increased levels of CK1ε protein or mRNA leading to elevated phosphorylation of many sites of tau protein such as S262, S356, and S214, involved in AD and T2DM.121,138
PKA and PKB
PKA and PKB, the two members of phosphoinositide-dependent PK, play a central role in cellular signaling by the process of phosphorylation. PKA phosphorylates many sites of tau such as Ser262 and Ser409 to increase cAMP levels as a prime for CK1 and GSK-3, whereas PKB phosphorylates this protein at Thr212 and Ser214 which promotes tau attachment to the 14-3-3 as studied in the in vitro model. Phosphorylation of tau at S241 by both PKA and PKB is associated with organization of microtubule cytoskeleton and formation of NFTs in AD.139–141 These two kinases increase glucose uptake and inotropic effects in adipocytes and pancreatic cells, and glucotoxicity, as well as promote proliferation in the beta cells which are involved in progression of T2DM.142 Blocking IP modulation of hepatic gluconeogenesis through PKA/CREB and PI3K-γ/PKC-ζ/TRB3/AKT pathway can also contribute to the T2DM progression.143
P38
P38 enzymatic activity in the MAPK reacts to the stress induction, in addition to apoptosis, leading to hyperglycemia that induces OS. This phenomenon, p38 MAPK pathway activation via tau protein phosphorylation, initiates development of AD and T2DM.144,145 Many studies have shown that activated p38 is exclusively localized to the NFTs and coimmunoprecipitated with PHF-tau in the hippocampal and cortical brain regions of AD brain.146,147
MARK4
MARK4, also known as Par-1d/MarkL1, is a member of the AMPK, which is implicated in the regulation of glucose and energy homeostasis. Phosphorylation of the microtubule by this kinase causes its detachment from the microtubules. MARK selectively phosphorylates existing S262 and S356 emerged in every MBD and other proteins that influence microtubules to facilitate the formation of cell processes.148,149 It was reported that MARK4 deficiency mitigated insulin resistance enhancing insulin-stimulated AKT phosphorylation in major metabolic tissues.150
PLK2
Upregulation of PLK2 (SNK) is mediated by the increased α-syn phosphorylation at S129 site which elevates pre-form of α-syn fibrils and with Aβ leading to tau hyperphosphorylation and reduction of tau binding to microtubules to promote the formation of NFTs-like aggregates in AD.151,152 Tau phosphorylation leads to aggregation of this protein by co-expressing glycogen synthase and kinase 3 beta or MAP/microtubule affinity-regulating kinase 2 involved in the progression of T2DM.153
Syk
Syk, a tyrosine kinase of tau protein at tyrosine 18 and α-syn, probably could influence the function and physiology of neurons in the brain.154 The tau in the detergent-resistant membranes is a tyrosine phosphorylated form which harbors lipid rafts. This form of tau protein is expected to facilitate a neurotoxic reactance towards Aβ. Syk can phosphorylate tau protein at Y18, Y197, and Y394 sites, respectively. Although other src family kinases may phosphorylate tau in the brain, PHF-tau is phosphorylated at tyrosine 394 and Fyn is the strongest candidate for tyrosine phosphorylation.117,155,156
DYRK1A
This kinase plays an important role in the signaling pathways which regulate cell proliferation and probably brain development. Dyrk1A mediates phosphorylation at the Thr356 and T181 residues of GSK3β that can inhibit its activity. Since DYRK1A pathway involves in the regulation of β cell mass and carbohydrate metabolism, defect in this protein could lead to T2DM.157,158
PPP
PPP group includes serine/threonine PP 1, 2A, 2B, and 5. Activity of PP2A in the normal brain is more than in AD brain (71% vs 50%), but activity of PP1 and PP5 in normal is less than in AD brain (11% and 10% vs 20% and 20%, respectively). PP1, PP2A, PP2B, and PP5 dephosphorylate tau protein at various sites, implicated in the stability and function regulation of microtubule. PP1 and PP2A are associated in a state where tau protein is hyperphosphorylated significantly than the tau protein in normal brain. PP5 at a higher expression level affects phosphorylation spot by removing the phosphate groups. Thus, it promotes neurons preservation vs apoptosis induced by Aβ.159,160
P70S6K
Protein, p70S6K, accompanied with Ser-Thr kinase, phosphorylates the ribosomal S6 subunit, the fundamental sequel in cell cycle control, growth, and differentiation leading to tau accumulation by translation and upregulating the expression. At protein level, the epitope T421/S424 of p-p70S6K is associated with tau phosphorylation. These epitopes phosphorylate tau at S214, S262, and T212 sites in AD brain, and inhibit recombinant tau assembly in vitro. Activated p70S6K in NFT-bearing neurons might be caused by the aberrant regulation of P13K and MAPK pathways, as well as the reduced activity of PP2A in AD brain. Deposition of Aβ in the AD brain also contributes to activation of p70S6K and consequential formation of tau-associated pathologies in AD brain, P70s6k plays a critical role in the early development process of T2DM as well. IRs mediate PI3K and p70S6K activation during insulin stimulation.161–163
Aggregation and degradation of hyperphosphorylated Tau
Approximately 90% of APP can be processed by nonamyloidogenic pathway and the remaining is processed by amyloidogenic pathway.
Nonamyloidogenic pathway
In nonamyloidogenic or nonplaque-forming pathway, a transmembrane protein known as APP is segregated via α-secretase enzyme leading to the formation of carboxy terminus fragment α (CTFα) and soluble APP fragment α (sAPPα). Later, γ-secretase segregates CTFα, which ends up with the induction of APP intracellular cytoplasmic domain (AICD) and p3 peptide. Probably, the sAPPα, which is considered as neuroprotective factor, is associated with the establishment of synapses within the neurons, neurite outgrowth, and neuronal survival. AICD may be involved in nuclear signaling via transcriptional regulation as well as axonal transport through its ability to associate with various proteins.164,165
Amyloidogenic pathway
In the amyloidogenic or plaque-forming pathway, APP and β-secretase are interposed within the endosome with an acidic environment, inducing β-secretase to segregate APP protein, following the formation of CTFβ and soluble APP fragment β (sAPPβ). Consequently, CTFβ is cleaved by γ-secretase enzyme to form AICD and Aβ fragments. Later, sAPPβ together with Aβ liberates into the extracellular environment where Aβ fragments accumulate to form plaques.
Aβ aggregation and plaque formation
Aβ peptide chain contains 38 (Aβ38), 40 (Aβ40), or 42 (Aβ42) amino acids. Aβ42 is chemically stickier compared with the other peptides. All three genetic mutations that cause early-onset AD change the role of gamma secretase, leading to an increased production of Aβ42.166,167 Aβ peptides aggregate into oligomers to organize fibrils with the formation of AP. Aβ plaques block signaling pathways and cells connection, which can be lethal to cells. Further, it can cause NTFs formation and Aβ is thought to cause oxidative damage to the cells. Along with the development of NFTs, low levels of insulin can increase the Aβ levels and forms AP in the brain. The Aβ peptide acts as monomers, dimers, or multimers on cell membranes and binds to its receptors on neuronal and glial cells at the nanomolar concentration to interfere with neurotransmission and memory before the AP builds up.5,168,169
Insulin–amyloid plaque–neurofibrillary tangles
Insulin regulates peripheral Aβ and tau metabolism which influences the Aβ release in the brain through regulating APP metabolism to modulate the balance between Aβ anabolism and catabolism.170 Lack of insulin or its action may link T2DM to AD by modification of Aβ production and degradation. Defect in the insulin-dependent pathways may increase the activation of GSK3 associated with the risk of AD. T2DM also modifies mitochondrial antioxidant defense system and assists brain weakness in the presence of Aβ toxicity.
A link between the involvement of insulin-degrading enzyme (IDE) in hyperinsulinemia and AD is closely related to dysfunction in the metabolic and neurological pathways.171,172 IDE is a thiol zinc-metallo-endopeptidase that cleaves small proteins such as insulin, Aβ, glucagon, calcitonin, and amylin which leads to the formation of β-pleated sheet-rich amyloid fibrils under certain conditions; levels of insulin together with Aβ in the brain are regulated by IDE. Interestingly, the hypofunction of this enzyme triggers the formation of AD and T2DM.
Role of antidiabetic drugs on Alzheimer’s disease
The incidence of MCI more often seen in T2DM patients may develop to AD. Therefore, improvement in cognition with antidiabetic drugs could be a strategy rather than mere glycemic control. Interestingly, these drugs could benefit AD patients associated with T2DM and it remains to be determined whether the potential is due to glucose lowering or the neuroprotective effects. However, further research is warranted to investigate their links between cognitive impairment and AD, and their safety measure is important too when considered in the management setting.
We highlight the potential of antidiabetic drugs with experimental and clinical observation through numerous studies that would be of interest to the researchers in developing strategies and linking in-depth mechanisms.
Biguanides
Metformin is an oral hypoglycemic drug under biguanide class used in the treatment of diabetes. In experimental studies, metformin showed neuroprotective role by preventing etoposide-induced apoptotic cell death in primary neurons and improved oxygen-glucose in neuronal injury. McNeilly, in 2013, demonstrated that in high fat-diet-induced animals, metformin attenuated the insulin resistance and weight gain, but had no effect on performance in either massive transfusion protocol (MTP) or no MTP (nMTP) tasks. In addition, metformin has shown to prevent the appearance of molecular and pathological characteristics of AD in neuroblastoma cell line model of insulin resistance. Interestingly, in diabetic rat model, metformin has revealed the reduction of cell proliferation and neuroblast differentiation in hippocampal dentate gyrus.173–176
Ng et al investigated the effect of metformin on the risk of cognitive impairment and its possible modulation by apolipoprotein E (ApoE) ε4 gene polymorphism. Metformin did not show any significant interactive role with ApoE ε4 and depression. Interestingly, among individuals with diabetes, long-term treatment (>6 years) reduced the risk of cognitive decline.
On other hand, the clinical studies on metformin show that the subjects aged 50 years and older significantly decreased the risk of dementia when compared with non-medication group after adjustment for cerebrovascular disease.177 In contrast, a case–control study displayed that long-term users of metformin were at greater risk of developing AD.178 Similarly, a study which included AD and cognitively intact patients showed worse cognitive performance in metformin users compared with those who were taking metformin and calcium together.179 Altogether, these studies raise the possible confounding effects of metformin in the management process of AD/neurological function, and therefore needs further understanding through molecular biomarkers approaches in clinical studies.
Sulfonylurea
Sulfonylureas such as glyburide and glipizide inhibit mTOR activation in the experimental model, as we know aberrant PI3K/mTOR activation is commonly experienced in diabetes and AD.180 Glyburide has been shown to inhibit inflammasomes responsible for the elevation of proinflammatory cytokines resulting in neuroinflammation associated with AD.181
In clinical studies, sulfonylureas do not alter the risk of developing AD in a long-term population-based case–control study.178 However, combination of metformin and sulfonylureas in a prospective cohort study over the period of 8 years reduced the risk of dementia by 35%, but their efficacy in preventing or improving memory and cognition needs to be determined.177
Thiazolidinediones
Thiazolidinediones such as rosiglitazone and pioglitazone might have role in reducing the risk of neurodegeneration.182 Rosiglitazone has shown protective effects in experimental models against neuronal insulin resistance induced by beta amyloid oligomers.183 On the other hand, pioglitazone showed improved cognitive performance in a rat model of memory impairment.184
In randomized controlled trial (RCT), rosiglitazone preserved memory function in patients with early AD and amnestic MCI but beta amyloid continued to be stable in plasma.172 Another small randomized double-blind trial on rosiglitazone demonstrated improvement in cognitive function in mild-to-moderate AD patients who were not carriers of the ApoE ε4 allele.185 In multicenter randomized concept clinical trial, rosiglitazone ameliorated impairment of brain glucose metabolism in mild-to-moderate AD subjects, but did not show evidence of slowing clinical progression.186 Another RCT on pioglitazone significantly decreased AD assessment scale (ADAS) score in AD/MCI subjects.187 In contrast, Phase III trial on rosiglitazone monotherapy failed to show a benefit on cognitive outcomes in mild-to-moderate AD.188 Similarly, another population–based case–control study did not change the risk of developing AD.178
Glucagon-like peptide 1
Another study on GLP-1 receptor agonists, liraglutide and lixisenatide, reduced the hippocampal burden and improved spatial memory in AD transgenic mice.189 Liraglutide ameliorated tau hyperphosphorylation and restored brain insulin sensitivity in type 2 diabetic rats.190 Thus, liraglutide diminishes neurodegenerative developments in AD.
Overall, preclinical and clinical studies support the efficacy of antidiabetic drugs in cognitive enhancement; some studies have failed to confirm reports of improved cognition in patients with T2DM even after good glycemic control. However, more clinical studies on antidiabetic drugs in agreement with preclinical approaches would enhance the chance of correlating MCI/AD for better therapeutic strategy and thereby increase the quality of life in AD patients.
Conclusion
This review extracted valuable outcomes from the studies that described the underlying common mechanisms between T2DM and AD, and the molecular determinants which could have significant therapeutic potential in treatment of T2DM- and/or AD-related damages. It was concluded that those patients who develop T2DM often suffer from dementia which might be AD. These patients could also suffer from hyperglycemia, hypercholesterolemia, and insulin signaling dysfunctions which are common features to T2DM. In addition, some antidiabetic drugs could have beneficial effects against some AD hallmarks, such as tau hyperphosphorylation, Aβ plaque formation, and apolipoprotein particularly ApoE4. Therefore, cardiometabolic signaling needs appropriate crosstalk to understand the mechanism and linkage with neuroinflammation process in the neurodegenerative disorders.
Acknowledgment
This review was supported by the Taylor’s University Flagship Research (Project No: TUFR/2017/002/01) under the theme “Ageing and Quality of Life.”
Author contributions
SKR was involved in the writing and original draft preparation, AA was involved in the supervision, writing, and editing of the manuscript. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Li L, Hölscher C. Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev. 2007;56(2):384–402. | ||
Yang Y, Song W. Molecular links between Alzheimer’s disease and diabetes mellitus. Neuroscience. 2013;250:140–150. | ||
Kleinridders A, Ferris HA, Cai W, Kahn CR. Insulin action in brain regulates systemic metabolism and brain function. Diabetes. 2014;63(7):2232–2243. | ||
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. | ||
Danysz W, Parsons CG. Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine – searching for the connections. Br J Pharmacol. 2012;167(2):324–352. | ||
Moreira PI, Santos MS, Seiça R, Oliveira CR. Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J Neurol Sci. 2007;257(1–2):206–214. | ||
Derakhshan F, Toth C. Insulin and the brain. Curr Diabetes Rev. 2013;9(2):102–116. | ||
Blázquez E, Velázquez E, Hurtado-Carneiro V, Ruiz-Albusac JM. Insulin in the brain: its pathophysiological implications for states related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front Endocrinol. 2014;5(2):161. | ||
Kim B, Feldman EL. Insulin resistance in the nervous system. Trends Endocrinol Metab. 2012;23(3):133–141. | ||
Bond MR, Hanover JA. O-GlcNAc cycling: a link between metabolism and chronic disease. Annu Rev Nutr. 2013;33(1):205–229. | ||
Sims-Robinson C, Kim B, Rosko A, Feldman EL. How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol. 2010;6(10):551–559. | ||
Correia SC, Santos RX, Carvalho C, et al. Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012;1441:64–78. | ||
Banks WA, Owen JB, Erickson MA. Insulin in the brain: there and back again. Pharmacol Ther. 2012;136(1):82–93. | ||
Hanson LR, Frey WH. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008;9(Suppl 3):1. | ||
Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev. 1999;20(2):101–135. | ||
Plum L, Belgardt BF, Brüning JC. Central insulin action in energy and glucose homeostasis. J Clin Invest. 2006;116(7):1761–1766. | ||
Russo VC, Gluckman PD, Feldman EL, Werther GA. The insulin-like growth factor system and its pleiotropic functions in brain. Endocr Rev. 2005;26(7):916–943. | ||
Vignini A, Giulietti A, Nanetti L, et al. Alzheimer’s disease and diabetes: new insights and unifying therapies. Curr Diabetes Rev. 2013;9(3):218–227. | ||
Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43(8):1467–1472. | ||
Chauhan NB. Membrane dynamics, cholesterol homeostasis, and Alzheimer’s disease. J Lipid Res. 2003;44(11):2019–2029. | ||
Hung YH, Bush AI, La Fontaine S. Links between copper and cholesterol in Alzheimer’s disease. Front Physiol. 2013;4:111. | ||
Lane RM, Farlow MR. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease. J Lipid Res. 2005;46(5):949–968. | ||
Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22(1):129–157. | ||
Sayem ASM, Arya A, Karimian H, Krishnasamy N, Ashok Hasamnis A, Hossain CF. Action of phytochemicals on insulin signaling pathways accelerating glucose transporter (GLUT4) protein translocation. Molecules. 2018;23(2):258. | ||
Scheepers A, Joost HG, Schürmann A. The glucose transporter families SGLT and GLUT: molecular basis of normal and aberrant function. JPEN J Parenter Enteral Nutr. 2004;28(5):364–371. | ||
Dehghan F, Hajiaghaalipour F, Yusof A, et al. Saffron with resistance exercise improves diabetic parameters through the GLUT4/AMPK pathway in-vitro and in-vivo. Sci Rep. 2016;6(1):25139. | ||
Arya A, Looi CY, Cheah SC, et al. Anti-diabetic effects of Centratherum anthelminticum seeds methanolic fraction on pancreatic cells, β-TC6 and its alleviating role in type 2 diabetic rats. J Ethnopharmacol. 2012;144(1):22–32. | ||
Talbot K, Wang HY, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122(4):1316–1338. | ||
Chiu S-L, Cline HT. Insulin receptor signaling in the development of neuronal structure and function. Neural Dev. 2010;5(1):1. | ||
Carro E, Torres-Aleman I. The role of insulin and insulin-like growth factor I in the molecular and cellular mechanisms underlying the pathology of Alzheimer’s disease. Eur J Pharmacol. 2004;490(1–3):127–133. | ||
Giuffrida ML, Tomasello F, Caraci F, Chiechio S, Nicoletti F, Copani A. Beta-amyloid monomer and insulin/IGF-1 signaling in Alzheimer’s disease. Mol Neurobiol. 2012;46(3):605–613. | ||
Sowers JR. Effects of insulin and IGF-I on vascular smooth muscle glucose and cation metabolism. Diabetes. 1996;45(Suppl 3):S47–S51. | ||
Leroith D, Werner H, Faria TN, Kato H, Adamo M, Roberts CT. Insulin-like growth factor receptors. Implications for nervous system function. Ann N Y Acad Sci. 1993;692(1 The Role of I):22–32. | ||
de La Monte SM. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res. 2012;9(1):35–66. | ||
Gropper SS, Smith JL. Advanced Nutrition and Human Metabolism. Belmont: Cengage Learning; 2012. | ||
Ahima RS. Principles of energy homeostasis. In: Ahima RS, editor. Metabolic Syndrome: A Comprehensive Textbook. Cham: Springer International Publishing; 2016:311–326. | ||
Bonda DJ, Stone JG, Torres SL, et al. Dysregulation of leptin signaling in Alzheimer disease: evidence for neuronal leptin resistance. J Neurochem. 2014;128(1):162–172. | ||
Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17(4):491–506. | ||
Varshosaz J. Insulin delivery systems for controlling diabetes. Recent Patents on Endocrine, Metabolic & Immune Drug Discovery. 2007;1(1):25–40. | ||
Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. 2010;12(4):537–577. | ||
Apostolova N, Victor VM. Molecular strategies for targeting antioxidants to mitochondria: therapeutic implications. Antioxid Redox Signal. 2015;22(8):686–729. | ||
de Lemos ET, Oliveira J, Pinheiro JP, Reis F, Teixeira de Lemos E, Páscoa Pinheiro J. Regular physical exercise as a strategy to improve antioxidant and anti-inflammatory status: benefits in type 2 diabetes mellitus. Oxid Med Cell Longev. 2012;2012(3):1–15. | ||
Lihn AS, Richelsen B, Pedersen SB, et al. Increased expression of TNF-alpha, IL-6, and IL-8 in HALS: implications for reduced adiponectin expression and plasma levels. Am J Physiol Endocrinol Metab. 2003;285(5):E1072–E1080. | ||
Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365(9467):1333–1346. | ||
Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11(3):272–280. | ||
Luttrell LM, Daaka Y, Lefkowitz RJ. Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr Opin Cell Biol. 1999;11(2):177–183. | ||
Elmquist JK, Bjørbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395(4):535–547. | ||
Elmquist JK, Elias CF, Saper CB. Hypothalamic control of body weight. Neuron. 1999;22:221–232. | ||
Schwartz MW, Woods SC, Porte D, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404(6778):661–671. | ||
Harvey J, Shanley LJ, O’Malley D, Irving AJ. Leptin: A Potential Cognitive Enhancer? London: Portland Press Limited; 2005. | ||
Garza JC, Guo M, Zhang W, Lu XY. Leptin increases adult hippocampal neurogenesis in vivo and in vitro. J Biol Chem. 2008;283(26):18238–18247. | ||
Guo Z, Jiang H, Xu X, Duan W, Mattson MP. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J Biol Chem. 2008;283(3):1754–1763. | ||
Greco SJ, Sarkar S, Johnston JM, et al. Leptin reduces Alzheimer’s disease-related tau phosphorylation in neuronal cells. Biochem Biophys Res Commun. 2008;376(3):536–541. | ||
Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun. 2009;380(1):98–104. | ||
Narita K, Kosaka H, Okazawa H, Murata T, Wada Y. Relationship between plasma leptin level and brain structure in elderly: a voxel-based morphometric study. Biol Psychiatry. 2009;65(11):992–994. | ||
Greco SJ, Sarkar S, Casadesus G, et al. Leptin inhibits glycogen synthase kinase-3beta to prevent tau phosphorylation in neuronal cells. Neurosci Lett. 2009;455(3):191–194. | ||
Mcguire MJ, Ishii M, Dysfunction L. Leptin dysfunction and Alzheimer’s disease: evidence from cellular, animal, and human studies. Cell Mol Neurobiol. 2016;36(2):203–217. | ||
Mcduff T, Sumi SM. Subcortical degeneration in Alzheimer’s disease. Neurology. 1985;35(1):123–126. | ||
Ishii M, Wang G, Racchumi G, Dyke JP, Iadecola C. Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide Y neurons. J Neurosci. 2014;34(27):9096–9106. | ||
Fayard E, Tintignac LA, Baudry A, Hemmings BA. Protein kinase B/Akt at a glance. J Cell Sci. 2005;118(Pt 24):5675–5678. | ||
Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9(1):59–71. | ||
Popa C, Netea MG, van Riel PL, van der Meer JW, Stalenhoef AF. The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res. 2007;48(4):751–762. | ||
Breier BH, Vickers MH, Ikenasio BA, Chan KY, Wong WP. Fetal programming of appetite and obesity. Mol Cell Endocrinol. 2001;185(1–2):73–79. | ||
Shah M, Simha V, Garg A. Review: long-term impact of bariatric surgery on body weight, comorbidities, and nutritional status. J Clin Endocrinol Metab. 2006;91(11):4223–4231. | ||
Arshadi S, Azarbayjani MA, Hajaghaalipor F, et al. Evaluation of Trigonella foenum-graecum extract in combination with swimming exercise compared to glibenclamide consumption on type 2 Diabetic rodents. Food Nutr Res. 2015;59(1):29717. | ||
Zorofchian Moghadamtousi S, Karimian H, Khanabdali R, et al. Anticancer and antitumor potential of fucoidan and fucoxanthin, two main metabolites isolated from brown algae. ScientificWorldJournal. 2014;2014(3):1–10. | ||
Niswender KD, Schwartz MW. Insulin and leptin revisited: adiposity signals with overlapping physiological and intracellular signaling capabilities. Front Neuroendocrinol. 2003;24(1):1–10. | ||
Mayer CM, Belsham DD. Insulin directly regulates NPY and AgRP gene expression via the MAPK MEK/ERK signal transduction pathway in mHypoE-46 hypothalamic neurons. Mol Cell Endocrinol. 2009;307(1–2):99–108. | ||
Bjørbaek C, Kahn BB. Leptin signaling in the central nervous system and the periphery. Recent Prog Horm Res. 2004;59(1):305–331. | ||
Plum L, Schubert M, Brüning JC. The role of insulin receptor signaling in the brain. Trends Endocrinol Metab. 2005;16(2):59–65. | ||
Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005;115(4):951–958. | ||
Li F, Yin Y, Tan B, Kong X, Wu G. Leucine nutrition in animals and humans: mTOR signaling and beyond. Amino Acids. 2011;41(5):1185–1193. | ||
Lauzon MA, Daviau A, Marcos B, Faucheux N. Growth factor treatment to overcome Alzheimer’s dysfunctional signaling. Cell Signal. 2015;27(6):1025–1038. | ||
Shafi O. Inverse relationship between Alzheimer’s disease and cancer, and other factors contributing to Alzheimer’s disease: a systematic review. BMC Neurol. 2016;16(1):236. | ||
Williams AJ, Umemori H. The best-laid plans go oft awry: synaptogenic growth factor signaling in neuropsychiatric disease. Front Synaptic Neurosci. 2014;6(6):4. | ||
Arya A, Gangwar A, Singh SK, et al. Cerium oxide nanoparticles promote neurogenesis and abrogate hypoxia-induced memory impairment through AMPK-PKC-CBP signaling cascade. Int J Nanomedicine. 2016;11:1159. | ||
Abbott MA, Wells DG, Fallon JR. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J Neurosci. 1999;19(17):7300–7308. | ||
Maiese K, Chong ZZ, Wang S, Shang YC. Oxidant stress and signal transduction in the nervous system with the PI 3-K, Akt, and mTOR cascade. Int J Mol Sci. 2012;13(11):13830–13866. | ||
Mendez P, Wandosell F, Garcia-Segura LM. Cross-talk between estrogen receptors and insulin-like growth factor-I receptor in the brain: cellular and molecular mechanisms. Front Neuroendocrinol. 2006;27(4):391–403. | ||
Papazoglou I. Cross-talk between insulin and serotonin signaling in the brain: involvement of the PI3K/Akt pathway and behavioral consequences in models of insulin resistance. Paris 11; 2013. | ||
Nelson TJ, Sun MK, Hongpaisan J, Alkon DL. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur J Pharmacol. 2008;585(1):76–87. | ||
Martínez MC, Andriantsitohaina R. Reactive nitrogen species: molecular mechanisms and potential significance in health and disease. Antioxid Redox Signal. 2009;11(3):669–702. | ||
Afanas’ev I. Signaling of reactive oxygen and nitrogen species in diabetes mellitus. Oxid Med Cell Longev. 2010;3(6):361–373. | ||
Cole GM, Frautschy SA. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer’s disease. Exp Gerontol. 2007;42(1–2):10–21. | ||
Adam SH, Giribabu N, Kassim N, et al. Protective effect of aqueous seed extract of Vitis Vinifera against oxidative stress, inflammation and apoptosis in the pancreas of adult male rats with diabetes mellitus. Biomed Pharmacother. 2016;81:439–452. | ||
Stukenbrock H, Mussmann R, Geese M, et al. 9-cyano-1-azapaullone (cazpaullone), a glycogen synthase kinase-3 (GSK-3) inhibitor activating pancreatic beta cell protection and replication. J Med Chem. 2008;51(7):2196–2207. | ||
Rachana ST, Basu S. Oxidative stress and diabetes. In: Rani V, Yadav UCS, editors. Free Radicals in Human Health and Disease. Springer; 2015:241–257. | ||
Karasu C. Glycoxidative stress and cardiovascular complications in experimentally-induced diabetes: effects of antioxidant treatment. Open Cardiovasc Med J. 2010;4(1):240–256. | ||
Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058–1070. | ||
Tiong SH, Looi CY, Hazni H, et al. Antidiabetic and antioxidant properties of alkaloids from Catharanthus roseus (L.) G. Don. Molecules. 2013;18(8):9770–9784. | ||
Taha H, Arya A, Paydar M, et al. Upregulation of insulin secretion and downregulation of pro-inflammatory cytokines, oxidative stress and hyperglycemia in STZ-nicotinamide-induced type 2 diabetic rats by Pseuduvaria monticola bark extract. Food Chem Toxicol. 2014;66:295–306. | ||
Arya A, Looi C, Wong W, et al. In vitro antioxidant, PTP-1B inhibitory effects and in vivo hypoglycemic potential of selected medicinal plants. Int J Pharmacol. 2013;9:50–57. | ||
Albers RW, Siegel GJ, Stahl W. Membrane transport. In: Siegel GJ, Agranoff BW, Albers RW, Molinoff PB, editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. New York: Raven Press; 1994:49–74. | ||
Rorsman P, Berggren P-O, Bokvist K. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature. 1989;341(6239):233–236. | ||
Ahrén B, Taborsky GJ. The mechanism of vagal nerve stimulation of glucagon and insulin secretion in the dog. Endocrinology. 1986;118(4):1551–1557. | ||
Ahrén B, Veith RC, Taborsky GJ. Sympathetic nerve stimulation versus pancreatic norepinephrine infusion in the dog: 1). Effects on basal release of insulin and glucagon. Endocrinology. 1987;121(1):323–331. | ||
Srivastava AK, Pandey SK. Potential mechanism(s) involved in the regulation of glycogen synthesis by insulin. Mol Cell Biochem. 1998;182(1–2):135–141. | ||
Nikoulina SE, Ciaraldi TP, Mudaliar S, Carter L, Johnson K, Henry RR. Inhibition of glycogen synthase kinase 3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes. 2002;51(7):2190–2198. | ||
Ferrer JC, Favre C, Gomis RR, et al. Control of glycogen deposition. FEBS Lett. 2003;546(1):127–132. | ||
Semiz S. Effects of diabetes, insulin, and vanadium on regulation of glycogen synthesis: roles of glycogen synthase kinase-3 and protein phosphatase-1, University of British Columbia; 2001. Available from: https://open.library.ubc.ca/cIRcle/collections/ubctheses/831/items/1.0103833. Accessed November 13, 2018. | ||
Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144(12):5179–5183. | ||
Wan M, Leavens KF, Hunter RW, et al. A noncanonical, GSK3-independent pathway controls postprandial hepatic glycogen deposition. Cell Metab. 2013;18(1):99–105. | ||
Hooper C, Markevich V, Plattner F, et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur J Neurosci. 2007;25(1):81–86. | ||
Hernández F, Borrell J, Guaza C, Avila J, Lucas JJ. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J Neurochem. 2002;83(6):1529–1533. | ||
Arthur JS, Fong AL, Dwyer JM, et al. Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J Neurosci. 2004;24(18):4324–4332. | ||
Johannessen M, Moens U. Multisite phosphorylation of the cAMP response element-binding protein (CREB) by a diversity of protein kinases. Front Biosci. 2007;12(12):e32. | ||
Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J Neurochem. 2008;104(6):1433–1439. | ||
Gao C, Hölscher C, Liu Y, Li L. GSK3: a key target for the development of novel treatments for type 2 diabetes mellitus and Alzheimer disease. Rev Neurosci. 2012;23(1):1–11. | ||
Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169–178. | ||
Looi CY, Arya A, Mustafa MA. Anti-inflammatory and anti-diabetic activities of the methanolic fraction of Centratherum anthelminticum seed in STZ nicotinamide-induced type 2 diabetic rat. Paper presented at: Frontiers in Immunology. Conference Abstract: 15th International Congress of Immunology (ICI). MiCo - Milano Congressi, Milan, Italy. | ||
Arya A, Cheah SC, Looi CY, Taha H, Mustafa MR, Mohd MA. The methanolic fraction of Centratherum anthelminticum seed downregulates pro-inflammatory cytokines, oxidative stress, and hyperglycemia in STZ-nicotinamide-induced type 2 diabetic rats. Food Chem Toxicol. 2012;50(11):4209–4220. | ||
Pisar MM. The Role of Kynurenine Metabolism in the Development of the Central Nervous System [PhD thesis]. Glasgow: University of Glasgow; 2014. | ||
Ando K, Oka M, Ohtake Y, et al. Tau phosphorylation at Alzheimer’s disease-related Ser356 contributes to tau stabilization when PAR-1/MARK activity is elevated. Biochem Biophys Res Commun. 2016;478(2):929–934. | ||
Verdile G, Fuller SJ, Martins RN. The role of type 2 diabetes in neurodegeneration. Neurobiol Dis. 2015;84:22–38. | ||
Martin L, Latypova X, Wilson CM, et al. Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res Rev. 2013;12(1):289–309. | ||
Barghorn S, Davies P, Mandelkow E. Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry. 2004;43(6):1694–1703. | ||
Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15(3):112–119. | ||
Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33(1):95–130. | ||
Anderton B, Hanger D, Ward M, Byers H, inventors; King’s College London Proteome Sciences PLC., assignee. Methods for Screening Inhibitors of Tau Phosphorylation By Casein Kinase I. United States patent US8822171B1. 2014. | ||
Chi S, Yu J-T, Tan M-S, Tan L. Depression in Alzheimer’s disease: epidemiology, mechanisms, and management. J Alzheimer’s Dis. 2014;42(3):739–755. | ||
Hanger DP, Byers HL, Wray S, et al. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem. 2007;282(32):23645–23654. | ||
Moszczynski AJ, Gohar M, Volkening K, Leystra-Lantz C, Strong W, Strong MJ. Thr175-phosphorylated tau induces pathologic fibril formation via GSK3β-mediated phosphorylation of Thr231 in vitro. Neurobiol Aging. 2015;36(3):1590–1599. | ||
Wang D, Fu Q, Zhou Y, et al. β2 adrenergic receptor, protein kinase A (PKA) and c-Jun N-terminal kinase (JNK) signaling pathways mediate tau pathology in Alzheimer disease models. J Biol Chem. 2013;288(15):10298–10307. | ||
Killick R, Ribe EM, Al-Shawi R, et al. Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway. Mol Psychiatry. 2014;19(1):88–98. | ||
de Felice FG, Ferreira ST. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes. 2014;63(7):2262–2272. | ||
Verdile G, Keane KN, Cruzat VF, et al. Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediators Inflamm. 2015;2015(6):1–17. | ||
Wang JH, Kelly PT. Postsynaptic injection of CA2+/CaM induces synaptic potentiation requiring CaMKII and PKC activity. Neuron. 1995;15(2):443–452. | ||
Wei Y, Han C, Wang Y, et al. Ribosylation triggering Alzheimer’s disease-like Tau hyperphosphorylation via activation of CaMKII. Aging Cell. 2015;14(5):754–763. | ||
Erickson JR, Pereira L, Wang L, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502(7471):372–376. | ||
Lopes JP, Oliveira CR, Agostinho P. Role of cyclin-dependent kinase 5 in the neurodegenerative process triggered by amyloid-Beta and prion peptides: implications for Alzheimer’s disease and prion-related encephalopathies. Cell Mol Neurobiol. 2007;27(7):943–957. | ||
Patrick GN, Zhou P, Kwon YT, Howley PM, Tsai LH. p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J Biol Chem. 1998;273(37):24057–24064. | ||
Zhang Y, Li H, Hao J, Zhou Y, Liu W. High glucose increases Cdk5 activity in podocytes via transforming growth factor-β1 signaling pathway. Exp Cell Res. 2014;326(2):219–229. | ||
Patrick GN, Zukerberg L, Nikolic M, de La Monte S, Dikkes P, Tsai L-H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402(6762):615–622. | ||
Choi SS, Kim ES, Koh M, et al. A novel non-agonist peroxisome proliferator-activated receptor γ (PPARγ) ligand UHC1 blocks PPARγ phosphorylation by cyclin-dependent kinase 5 (CDK5) and improves insulin sensitivity. J Biol Chem. 2014;289(38):26618–26629. | ||
Choi JH, Banks AS, Estall JL, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466(7305):451–456. | ||
Granic I, Dolga AM, Nijholt IM, van Dijk G, Eisel UL. Inflammation and NF-kappaB in Alzheimer’s disease and diabetes. J Alzheimers Dis. 2009;16(4):809–821. | ||
Binukumar BK, Zheng YL, Shukla V, Amin ND, Grant P, Pant HC. TFP5, a peptide derived from p35, a Cdk5 neuronal activator, rescues cortical neurons from glucose toxicity. J Alzheimers Dis. 2014;39(4):899. | ||
Lau L-Fui, Schachter J, Seymour P, Sanner M. Tau protein phosphorylation as a therapeutic target in Alzheimer’s disease. Curr Top Med Chem. 2002;2(4):395–415. | ||
Sadik G, Tanaka T, Kato K, et al. Phosphorylation of tau at Ser214 mediates its interaction with 14-3-3 protein: implications for the mechanism of tau aggregation. J Neurochem. 2009;108(1):33–43. | ||
Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K. Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis. 2013;33(Suppl 1s1):S123–S139. | ||
Hernandez F, Lucas JJ, Avila J. GSK3 and tau: two convergence points in Alzheimer’s disease. J Alzheimers Dis. 2013;33(Suppl 1s1):S141–S144. | ||
Sathanoori R, Olde B, Erlinge D, Göransson O, Wierup N. Cocaine- and amphetamine-regulated transcript (CART) protects beta cells against glucotoxicity and increases cell proliferation. J Biol Chem. 2013;288(5):3208–3218. | ||
Yan S, Zhang Q, Zhong X, et al. I prostanoid receptor-mediated inflammatory pathway promotes hepatic gluconeogenesis through activation of PKA and inhibition of AKT. Diabetes. 2014;63(9):2911–2923. | ||
Corrêa SA, Eales KL. The role of p38 MAPK and its substrates in neuronal plasticity and neurodegenerative disease. J Signal Transduct. 2012;2012(6):1–12. | ||
Dias-Santagata D, Fulga TA, Duttaroy A, Feany MB. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J Clin Invest. 2007;117(1):236–245. | ||
Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem Int. 2011;58(4):458–471. | ||
Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000;59(10):880–888. | ||
Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38(12):3549–3558. | ||
Drewes G, Lichtenberg-Kraag B, Döring F, et al. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. Embo J. 1992;11(6):2131–2138. | ||
Sun C, Tian L, Nie J, Zhang H, Han X, Shi Y. Inactivation of MARK4, an AMP-activated protein kinase (AMPK)-related kinase, leads to insulin hypersensitivity and resistance to diet-induced obesity. J Biol Chem. 2012;287(45):38305–38315. | ||
Marsh SE, Blurton-Jones M. Examining the mechanisms that link β-amyloid and α-synuclein pathologies. Alzheimers Res Ther. 2012;4(2):1:11. | ||
Muntané G, Dalfó E, Martinez A, Ferrer I. Phosphorylation of tau and alpha-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease, and in Parkinson’s disease and related alpha-synucleinopathies. Neuroscience. 2008;152(4):913–923. | ||
Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31(21):7604–7618. | ||
Lebouvier T, Scales TM, Hanger DP, et al. The microtubule-associated protein tau is phosphorylated by Syk. Biochim Biophys Acta (BBA)-Mol Cell Res. 1783;2008(2):188–192. | ||
Tremblay MA, Acker CM, Davies P. Tau phosphorylated at Tyrosine 394 is found in Alzheimer’s disease tangles and can be a product of the Abl-related kinase, Arg. J Alzheimer’s Dis. 2010;19(2):721–733. | ||
Scales TME, Derkinderen P, Leung K-Y, et al. Tyrosine phosphorylation of tau by the SRC family kinases lck and fyn. Mol Neurodegener. 2011;6(1):1. | ||
Wang P, Alvarez-Perez J-C, Felsenfeld DP, et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med. 2015;21(4):383–388. | ||
Shen W, Taylor B, Jin Q, et al. Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat Commun. 2015;6(1):8372. | ||
Liu F, Grundke-Iqbal I, Iqbal K, Gong C-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22(8):1942–1950. | ||
Gong C-X, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem. 2000;275(8):5535–5544. | ||
Pei J-J, An W-L, Zhou X-W, et al. P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett. 2006;580(1):107–114. | ||
An WL. Role of P70 S6 kinase in the formation of tau pathologies in Alzheimer´s disease: Institutionen för klinisk neurovetenskap, arbetsterapi och äldrevårdsforskning (NEUROTEC)/Department of Clinical Neuroscience, Occupational Therapy and Elderly Care Research (NEUROTEC). 2005. | ||
Zhou X-W, Tanila H, Pei J-J. Parallel increase in p70 kinase activation and tau phosphorylation (S262) with Aβ overproduction. FEBS Lett. 2008;582(2):159–164. | ||
Kojro E, Fahrenholz F. The non-amyloidogenic pathway: structure and function of α-secretases. In: Harris R, Fahrenholz F, editors. Alzheimer’s Disease: Cellular and Molecular Aspects of Amyloid β. New York: Springer; 2005:105–127. | ||
Tang BL. Alzheimer’s disease: channeling APP to non-amyloidogenic processing. Biochem Biophys Res Commun. 2005;331(2):375–378. | ||
Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. | ||
Guo Q, Xie J, Chang X, du H. Prostate apoptosis response-4 enhances secretion of amyloid beta peptide 1–42 in human neuroblastoma IMR-32 cells by a caspase-dependent pathway. J Biol Chem. 2001;276(19):16040–16044. | ||
Tang BL. Neuronal protein trafficking associated with Alzheimer disease: from APP and BACE1 to glutamate receptors. Cell Adh Migr. 2009;3(1):118–128. | ||
Fu Y, Zhao D, Yang L. Protein-based biomarkers in cerebrospinal fluid and blood for Alzheimer’s disease. J Mol Neurosci. 2014;54(4):739–747. | ||
de La Monte SM, Suzanne M. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs. 2012;72(1):49–66. | ||
Abdul-Hay SO, Lane AL, Caulfield TR, et al. Optimization of peptide hydroxamate inhibitors of insulin-degrading enzyme reveals marked substrate-selectivity. J Med Chem. 2013;56(6):2246–2255. | ||
Watson GS, Cholerton BA, Reger MA, et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13(11):950–958. | ||
El-Mir MY, Detaille D, R-Villanueva G, et al. Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J Mol Neurosci. 2008;34(1):77–87. | ||
Mielke JG, Taghibiglou C, Wang YT. Endogenous insulin signaling protects cultured neurons from oxygen-glucose deprivation-induced cell death. Neuroscience. 2006;143(1):165–173. | ||
Gupta A, Bisht B, Dey CS. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology. 2011;60(6):910–920. | ||
Hwang IK, Kim IY, Joo EJ, et al. Metformin normalizes type 2 diabetes-induced decrease in cell proliferation and neuroblast differentiation in the rat dentate gyrus. Neurochem Res. 2010;35(4):645–650. | ||
Hsu C-C, Wahlqvist ML, Lee M-S, Tsai H-N. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimer’s Dis. 2011;24(3):485–493. | ||
Imfeld P, Bodmer M, Jick SS, Meier CR. Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: a population-based case-control study. J Am Geriatr Soc. 2012;60(5):916–921. | ||
Moore E, Mander A, Ames D, et al. Martins R, Szoeke C, Rowe C, Watters DA, the AIBL Investigators. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care. 2013;36:2981–2987. | ||
Khanfar MA, Abukhader MM, Alqtaishat S, Taha MO. Pharmacophore modeling, homology modeling, and in silico screening reveal mammalian target of rapamycin inhibitory activities for sotalol, glyburide, metipranolol, sulfamethizole, glipizide, and pioglitazone. J Mol Graphics Model. 2013;42:39–49. | ||
Lamkanfi M, Mueller JL, Vitari AC, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187(1):61–70. | ||
Heneka MT, Landreth GE, Feinstein DL. Role for peroxisome proliferator-activated receptor-gamma in Alzheimer’s disease. Ann Neurol. 2001;49(2):276. | ||
de Felice FG, Vieira MN, Bomfim TR. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc Nat Acad Sci. 2009:pnas. 0809158106. | ||
Pathan AR, Viswanad B, Sonkusare SK, Ramarao P. Chronic administration of pioglitazone attenuates intracerebroventricular streptozotocin induced-memory impairment in rats. Life Sci. 2006;79(23):2209–2216. | ||
Risner ME, Saunders AM, Altman JF, et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6(4):246–254. | ||
Tzimopoulou S, Cunningham VJ, Nichols TE, et al. A multi-center randomized proof-of-concept clinical trial applying [18F]FDG-PET for evaluation of metabolic therapy with rosiglitazone XR in mild to moderate Alzheimer’s disease. J Alzheimers Dis. 2010;22(4):1241–1256. | ||
Hanyu H, Sato T, Kiuchi A, Sakurai H, Iwamoto T. Pioglitazone improved cognition in a pilot study on patients with Alzheimer’s disease and mild cognitive impairment with diabetes mellitus. J Am Geriatr Soc. 2009;57(1):177–179. | ||
Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord. 2010;30(2):131–146. | ||
Bomfim TR, Forny-Germano L, Sathler LB, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Aβ oligomers. J Clin Invest. 2012;122(4):1339–1353. | ||
Yang Y, Zhang J, Ma D, et al. Subcutaneous administration of liraglutide ameliorates Alzheimer-associated tau hyperphosphorylation in rats with type 2 diabetes. J Alzheimers Dis. 2013;37(3):637–648. | ||
Gould GW, Brant AM, Kahn BB, Shepherd PR, Mccoid SC, Gibbs EM. Expression of the brain-type glucose transporter is restricted to brain and neuronal cells in mice. Diabetologia. 1992;35(4):304–309. | ||
Schubert M, Brazil DP, Burks DJ, et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci. 2003;23(18):7084–7092. | ||
Könner AC, Brüning JC. Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol Metabol. 2011;22(1):16–23. | ||
Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci. 2002;5(6):566–572. | ||
Langlais PR, Mandarino LJ, Garvey WT. Mechanisms of insulin signal transduction. In: DeFronzo RA, editor. International Textbook of Diabetes Mellitus. Chichester: Wiley Blackwel; 2015. | ||
Jackson WF. Boosting the signal: Endothelial inward rectifier K+ channels. Microcirculation. 2017;24(3):e12319. | ||
Inoue H. Central insulin-mediated regulation of hepatic glucose production [Review]. Endocr J. 2016;63(1):1–7. | ||
Cotero VE, Routh VH. Insulin blunts the response of glucose-excited neurons in the ventrolateral-ventromedial hypothalamic nucleus to decreased glucose. Am J Physiol Endocrinol Metab. 2009;296(5):E1101–E1109. | ||
Lee C-C, Huang C-C, Wu M-Y, Hsu K-S. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem. 2005;280(18):18543–18550. | ||
Filosa A, Klein R. Chapter 34: Ephrins and Eph receptors–synaptogenesis and synaptic function. In: Rubenstein J, Rakic P, editors. Comprehensive Developmental Neuroscience: Cellular Migration and Formation of Neuronal Connections. Amsterdam: Elsevier Inc; 2013. | ||
Geddes JW, Wilson MC, Miller FD. 1Div. Neurosurgery and 2Dept. Psychobiology Univ. Calif., Irvine, CA 92717: 3Dept. Neuropharmacology Scripps Clinic & Res. Foundation, La Jolla, CA 92037 and 4Dept. Anatomy & Cell Biology, Univ. Alberta. Excitatory Amino Acids and Neuronal Plasticity. 2013;268:425. | ||
Wang Z, Xiong L, Wang G, Wan W, Zhong C, Zu H. Insulin-like growth factor-1 protects SH-SY5Y cells against β-amyloid-induced apoptosis via the PI3K/Akt-Nrf2 pathway. Exp Gerontol. 2017;87(Pt A):23–32. | ||
Rhee YH, Choi M, Lee HS, et al. Insulin concentration is critical in culturing human neural stem cells and neurons. Cell Death Dis. 2013;4(8):e766. | ||
Gilgun-Sherki Y, Melamed E, Offen D. Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology. 2001;40(8):959–975. | ||
Cusi K, Consoli A, Defronzo RA. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1996;81(11):4059–4067. | ||
Levin BE, Israel P, Lattemann DP, Lattemann DF. Insulin selectively downregulates alpha2-adrenoceptors in the arcuate and dorsomedial nucleus. Brain Res Bull. 1998;45(2):179–181. | ||
Kasahara Y, Sato K, Takayanagi Y, et al. Oxytocin receptor in the hypothalamus is sufficient to rescue normal thermoregulatory function in male oxytocin receptor knockout mice. Endocrinology. 2013;154(11):4305–4315. | ||
Huang CC, Lee CC, Hsu KS. An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. J Neurochem. 2004;89(1):217–231. | ||
Chater TE, Goda Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front Cell Neurosci. 2014;8:401. | ||
Ghasemi R, Haeri A, Dargahi L, Mohamed Z, Ahmadiani A. Insulin in the brain: sources, localization and functions. Mol Neurobiol. 2013;47(1):145–171. | ||
Murthy R, Kim J, Sun X, Giger RJ, Fink DJ, Mata M. Post-transcriptional regulation of GABAB receptor and GIRK1 channels by Nogo receptor 1. Mol Brain. 2013;6(1):1. | ||
Zhou Y, Danbolt NC. GABA and glutamate transporters in brain. Front. Endocrinol. 2014;4:165. | ||
Qosa H, Miller DS, Pasinelli P, Trotti D. Regulation of ABC efflux transporters at blood-brain barrier in health and neurological disorders. Brain Res. 2015;1628(Pt B):298–316. | ||
Collins B, Hoffman J, Martinez K, et al. A polyphenol-rich fraction obtained from table grapes decreases adiposity, insulin resistance and markers of inflammation and impacts gut microbiota in high-fat-fed mice. J Nutr Biochem. 2016;31:150–165. | ||
Yang C, Zhu X, Liu N, et al. Lactoferrin up-regulates intestinal gene expression of brain-derived neurotrophic factors BDNF, UCHL1 and alkaline phosphatase activity to alleviate early weaning diarrhea in postnatal piglets. J Nutr Biochem. 2014;25(8):834–842. | ||
Wtl L, Chen C, Lim Y-A. Effects of neuroaid II (MLC901) on app processing and tau phosphorylation. Alzheimer’s Dementia: J Alzheimer’s Assoc. 2016;12(7):P620–P621. | ||
Tenreiro S, Eckermann K, Outeiro TF. Protein phosphorylation in neurodegeneration: friend or foe? Front Mol Neurosci. 2014;7:42. | ||
Bergeron M, Motter R, Tanaka P, et al. In vivo modulation of polo-like kinases supports a key role for PLK2 in Ser129 α-synuclein phosphorylation in mouse brain. Neuroscience. 2014;256:72–82. | ||
Hernandez P, Lee G, Sjoberg M, Maccioni RB. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25–35): involvement of lipid rafts. J Alzheimers Dis. 2009;16(1):149–156. | ||
Ubhi K, Masliah E. Alzheimer’s disease: recent advances and future perspectives. J Alzheimers Dis. 2013;33(Suppl 1):S185–S194. | ||
Ward M, Byers H, Anderton BH, Derkinderen P, Reynolds CH, Williamson R, inventors; Proteome Sciences PLC King’s College London., assignee. Method of screening for inhibitors of tau protein phosphororylation by tyrosine kinase c-Abl. United States patent US8367360B2. 2013. | ||
Kanaan NM, Morfini G, Pigino G, et al. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol Aging. 2012;33(4):826.e15–826826. | ||
Anderton B, Hanger D, Ward M, Byers H, inventors; King’s College London, Proteome Sciences PLC., assignee. Methods for screening for inhibitors of tau phosphorylation by casein kinase I. United States patent US8822171B1. 2014. | ||
Lau DHW, Hogseth M, Phillips EC. Critical residues involved in tau binding to fyn: implications for tau phosphorylation in Alzheimer’s disease. Acta Neuropathol Commun. 2016;4(1):1. | ||
Kyoung Pyo H, Lovati E, Pasinetti GM, Ksiezak-Reding H. Phosphorylation of tau at THR212 and SER214 in human neuronal and glial cultures: the role of AKT. Neuroscience. 2004;127(3):649–658. | ||
Hong XP, Peng CX, Wei W, et al. Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus. 2010;20(12):1339–1349. | ||
Hong Y, Chan CB, Kwon IS, et al. SRPK2 phosphorylates tau and mediates the cognitive defects in Alzheimer’s disease. J Neurosci. 2012;32(48):17262–17272. | ||
Reynolds CH, Garwood CJ, Wray S, et al. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J Biol Chem. 2008;283(26):18177–18186. | ||
Cheung ZH, Ip NY, Ny I. Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012;22(3):169–175. | ||
Moll L, Schubert M. The role of insulin and insulin-like growth factor-1/FoxO-mediated transcription for the pathogenesis of obesity-associated dementia. Curr Gerontol Geriatr Res. 2012;2012:1–13. | ||
Leugers CJ, Koh J, Hong W, Lee G. Tau in MAPK activation. Front Neurol. 2013;4:161. | ||
Liu F, Jin N, Yin X, et al. Regulation of alternative splicing of tau exon 10 by dual-specificity tyrosine-phosphorylation-regulated kinase 1A. Alzheimer’s Dementia. 2015;11(7):P203. | ||
Schwalbe M, Kadavath H, Biernat J, et al. Structural impact of tau phosphorylation at threonine 231. Structure. 2015;23(8):1448–1458. | ||
Deng J, Habib A, Obregon DF, et al. Soluble amyloid precursor protein alpha inhibits tau phosphorylation through modulation of GSK3β signaling pathway. J Neurochem. 2015;135(3):630–637. | ||
Yamamoto H, Hiragami Y, Murayama M, Ishizuka K, Kawahara M, Takashima A. Phosphorylation of tau at serine 416 by Ca2+/calmodulin-dependent protein kinase II in neuronal soma in brain. J Neurochem. 2005;94(5):1438–1447. | ||
Köhler C, Dinekov M, Götz J. Active glycogen synthase kinase-3 and tau pathology-related tyrosine phosphorylation in pR5 human tau transgenic mice. Neurobiol Aging. 2013;34(5):1369–1379. | ||
Sun LH, Ban T, Liu CD, et al. Activation of Cdk5/p25 and tau phosphorylation following chronic brain hypoperfusion in rats involves microRNA-195 down-regulation. J Neurochem. 2015;134(6):1139–1151. | ||
Sahara N, Murayama M, Higuchi M, Suhara T, Takashima A. Biochemical distribution of tau protein in synaptosomal fraction of transgenic mice expressing human P301L tau. Front Neurol. 2014;5:26. | ||
Kerr F, Augustin H, Piper MDW, et al. Erratum to “Dietary restriction delays aging, but not neuronal dysfunction, in Drosophila models of Alzheimer’s disease.” [Neurobiol Aging 32 (2011) 1977–1989]. Neurobiol Aging. 2015;36(7):2331. | ||
Hajiaghaalipour F, Khalilpourfarshbafi M, Arya A. Modulation of glucose transporter protein by dietary flavonoids in type 2 diabetes mellitus. Int J Biol Sci. 2015;11(5):508–524. | ||
Yamagishi S, Imaizumi T. Diabetic vascular complications: pathophysiology, biochemical basis and potential therapeutic strategy. Curr Pharm Des. 2005;11(18):2279–2299. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.