")
Back to Archived Journals » Cell Health and Cytoskeleton » Volume 7
Mechanism and regulation of epithelial–mesenchymal transition in cancer
Authors Guttilla Reed I
Received 27 June 2015
Accepted for publication 22 July 2015
Published 20 August 2015 Volume 2015:7 Pages 155—166
DOI https://doi.org/10.2147/CHC.S73822
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Denis Wirtz
Irene K Guttilla Reed
Department of Biology, University of Saint Joseph, West Hartford, CT, USA
Abstract: During development and the pathogenesis of certain diseases, including cancer, the epithelial–mesenchymal transition (EMT) program is activated. It is hypothesized that EMT plays a major role in tumor invasion and the establishment of distant metastases. Metastatic disease is responsible for the vast majority of cancer-related deaths, which provides a precedent for elucidating pathways that regulate EMT. EMT is defined as the transition of cells with an epithelial phenotype into cells with a mesenchymal phenotype through a series of genetic and environmental events. This leads to the repression of epithelial-associated markers, upregulation of mesenchymal-associated markers, a loss of cell polarity and adhesion, and increased cell motility and invasiveness. EMT is a reversible and dynamic process, and can be regulated by signals from the microenvironment such as inflammation, hypoxia, and growth factors or epigenetically via microRNAs. These signals modulate key EMT-associated transcription factors and effector proteins that control cellular phenotype and regulate tumor plasticity in response to changing conditions in the microenvironment and the progressive nature of cancer. Understanding the complex regulatory networks controlling EMT can provide insight into tumor progression and metastasis.
Keywords: EMT, metastasis, microRNA, transcription factor, growth factor, tumor progression
Introduction: epithelial–mesenchymal transition and its role in cancer plasticity
Cancer is a heterogeneous disease regulated by complex mechanisms that promote both tumor initiation and progression. However, metastatic disease still causes over 90% of cancer-related deaths regardless of diverse cancer phenotypes.1 It is hypothesized that epithelial–mesenchymal transition (EMT) initiates the metastatic cascade. EMT is a normal cellular process that regulates embryogenesis and wound healing; however, it can be exploited during tumor progression to generate an invasive cellular phenotype.2 EMT has primarily been characterized in carcinomas as well as in some mesenchymal tumors, such as sarcomas, gastrointestinal stromal tumors, and a subtype of glioblastomas.3 Factors involved in initiating or maintaining the EMT program have been identified in a wide variety of carcinomas including breast, prostate, colorectal, head and neck, ovarian, lung, and more recently endometrial.4,5
In cancer, EMT is a multistep and reversible process defined by the loss of epithelial characteristics (such as cell–cell junctions, adhesion, and apical–basal polarity) and the gain of mesenchymal characteristics (such as increased motility, invasive properties, and a spindle-like morphology).6,7 These phenotypic changes result from the rearrangement of cytoskeletal proteins (particularly F-actin) due to increased mesenchymal markers. In addition to morphological changes and increased invasive potential, cells that have undergone EMT display increased resistance to apoptosis, attenuation of cell-cycle progression, and stem cell-like properties.6,7 Initiation of the EMT cascade leads to local invasion, followed by the dissemination of circulating tumor cells (CTCs), and eventually the establishment of distant metastases. This process is dependent on the plasticity of EMT since evidence suggests that establishment of metastases is a consequence of mesenchymal–epithelial transition (MET).8 Clinically detectable macrometastases are largely epithelial in nature, and recent studies suggest that the downregulation of EMT-promoting factors is crucial for the establishment and proliferation of metastatic colonies.9
Due to the dynamic and transient nature of EMT, clinical relevance of this process in metastasis has been debated.10,11 However, strong evidence exists for the involvement of EMT regulatory mechanisms in various stages of tumor initiation, development, and progression. Further characterizing a partial-EMT phenotype may be crucial to understand the intricacies of the EMT program in vivo. Cells may reside in a state where they express newly acquired mesenchymal markers while still possessing epithelial markers as they transition from one phenotypic state to another. Indeed, circulating mesenchymal cells have been shown to work in concert with epithelial cells to protect them from anoikis, conferring a heterogeneous cell population with representation from both transition states.12
The aim of this review is to summarize the prominent regulatory mechanisms involved in activation and maintenance of the EMT program in cancer progression and metastasis. EMT is a complex and multilayered process that involves the coordination of transcription factors (TFs), effector proteins, and cellular regulators such as microRNAs. Adding to the complexity is the role of the microenvironment in facilitating EMT, as this process can be initiated by myriad signals such as hypoxia, growth factors, and inflammation. Elucidating the orchestration among these pathways will provide insight into the plastic and heterogeneous nature of tumor cells, and the cellular events leading to metastasis.
The EMT cascade and molecular players
Induction of EMT
A variety of microenvironmental cues can initiate the EMT program including growth factor signaling, hypoxia, and inflammatory pathways, and these pathways are not mutually exclusive. For example, the well-characterized EMT-inducing growth factor TGF-β (transforming growth factor beta) is directly activated by hypoxia via hypoxia-inducible factor 1 alpha (HIF1α).13 HIF1α can also induce epigenetic regulation of EMT by transcriptionally targeting HDAC3, which cooperates with the EMT-associated TF Snail1 to mediate gene repression of epithelial-specific promoters.14 EMT also occurs during wound healing; however, chronic inflammation at a particular site can lead to persistent EMT and fibrosis.5 During wound healing, EMT is mediated by inflammatory immune cells and fibroblasts.15 This process parallels EMT in cancer since tumor-associated macrophages and the tumor stroma have been shown to play a role in invasion and metastasis of breast cancer cells.16 However, it should be noted that mesenchymal markers are also expressed in CTCs of both early and metastatic breast cancer patients regardless of hormone status and tumor grade, suggesting that EMT may also play a role in tumor initiation.17 These upstream signals initiate EMT primarily through the direct activation or epigenetic regulation of EMT-inducing TFs,6 which exert phenotypic effects on the cell by modulating downstream epithelial and mesenchymal effector molecules (Figure 1).
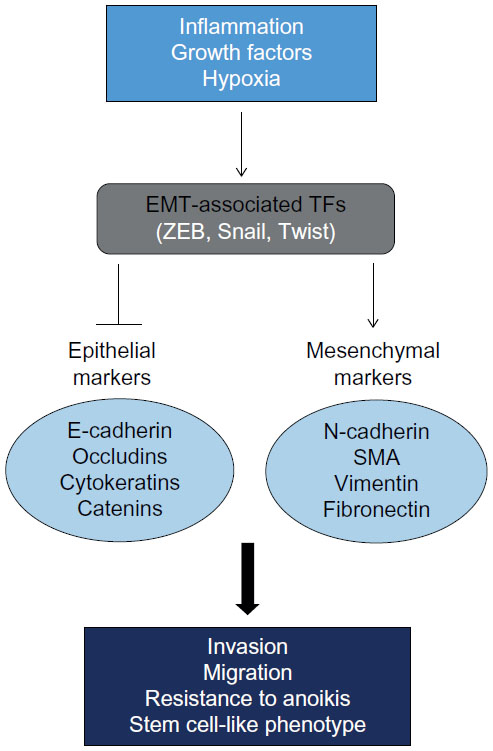 | Figure 1 Overview of key molecular players in epithelial–mesenchymal transition. |
The EMT cascade
In order for tumor cells to escape from the primary site and travel to distant organs, they need to become more motile and degrade the basement membrane from the extracellular matrix. This step initiates local invasion and eventually leads to intravasation (cellular infiltration of endothelium and blood and/or lymphatic vessels), which generates CTCs (Figure 2).9 A small subset of these CTCs may undergo extravasation and colonize micrometastases via MET (Figure 2).9 Additional signals are required for these colonies to proliferate into macrometastases that can be clinically detected.9 Traditionally, EMT is thought to play a role in these later stage events in tumor progression (local invasion and maintenance of CTCs); however, recent evidence suggests involvement of EMT in malignant transformation and tumor initiation.9 Expression of the EMT-inducing TF Twist1 was detected in patients with atypical breast ductal hyperplasia, a very early-stage neoplastic disease.18
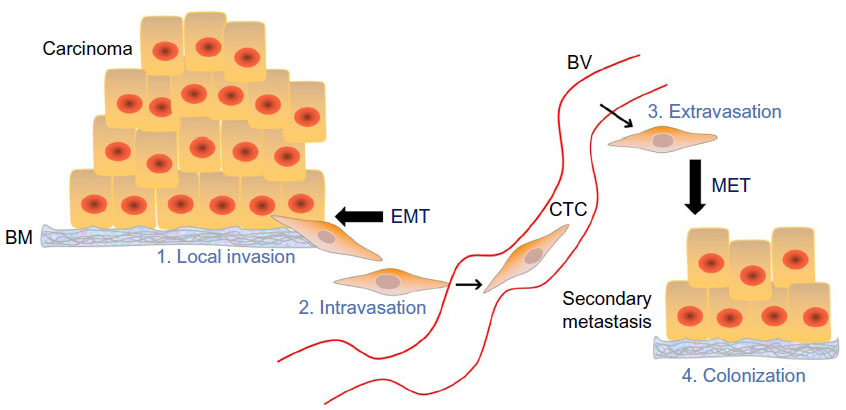 | Figure 2 The metastatic cascade. |
Induction of the EMT program is not governed by one singular cascade of molecular events. As a result, many studies have focused on characterization of various EMT markers in progressive stages of tumor development. Clusters of cells on the invasive front often display decreased levels of E-cadherin, a hallmark of the epithelial phenotype.6 Similarly, high-grade tumors associated with a poor prognosis often display molecular signatures associated with the EMT program; however, these signatures can vary between tumor type, and do not always correlate with disease-free survival.19–22 Nevertheless, Snail1/2 and Twist-induced EMT has been associated with an increased population of CTCs that maintain mesenchymal markers and characteristics.23,24 While the functional relevance of the expression of mesenchymal markers such as vimentin is unclear, recent evidence suggests that maintenance of the mesenchymal phenotype may protect CTCs from natural killer cells, increasing the probability that they will survive circulation and continue onto extravasation.25
The final step in metastasis is colonization, which is dependent on both genetic and environmental cues. Metastatic lesions are commonly epithelial in nature, suggesting that the reversion from a mesenchymal phenotype to an epithelial phenotype (MET) is favorable for establishing a metastatic niche. Tsai et al demonstrated that although Twist1 activation was required for the early steps of metastasis (invasion and intravasation), loss of this EMT-inducing signal was essential for proliferation and colonization.23 However, the exact mechanisms governing this phenomenon remain unclear. One explanation is the ability of Twist1 to downregulate E-cadherin,26 since the restoration of E-cadherin can induce MET in several systems.27 Alternatively, microRNAs have also been shown to regulate MET by targeting (and repressing) EMT TFs such as ZEB1, ZEB2, and Snail2, leading to an increase in the expression of epithelial markers.28 Interestingly, MET and E-cadherin-mediated cell–cell adhesion are crucial for reprogramming fibroblasts into induced-pluripotent stem cells,29,30 which eludes to the parallel between EMT and a stem cell-like phenotype in both normal and neoplastic cells.31 Though it has been demonstrated that EMT generates CTCs, it is plausible that this process could also be responsible for local recurrence if these cells do not invade and migrate but retain cancer stem cell-like (tumor-initiating) properties.
Molecular players
EMT is characterized by a loss of epithelial markers and a gain of mesenchymal markers (Figure 1). In concert, these effector proteins modulate cytoskeletal rearrangement leading to changes in cellular adhesion and motility. Epithelial markers include cell junction proteins (ie, E-cadherin and claudins), tight junction proteins (ie, ZO and occludins), cytokeratins, and catenins. E-cadherin in particular is a hallmark of EMT, and interacts with intercellular adhesion networks to maintain cell polarity, differentiation, migration, and signaling in proliferation pathways. The downregulation of E-cadherin is sufficient to induce EMT in some, but not in all experimental cancer models.8,32 E-cadherin loss can be due to inactivating gene mutations,33 however, most often downregulation is due to epigenetic or transcriptional silencing.3,34,35 A study by Onder et al suggests that even though loss of E-cadherin protein expression is sufficient for metastasis, active β-catenin is necessary for invasion of cells in culture and an experimental metastasis model.36 Together, these findings suggest that even though E-cadherin is an integral player in maintaining the epithelial phenotype, it is likely that additional factors act in concert with the loss of this protein to induce a full EMT.
Upregulation of mesenchymal markers such as vimentin, fibronectin, N-cadherin, and smooth muscle actin leads to the detachment of tumor cells, proteolytic digestion of the basement membrane, and the generation of CTCs (Figure 1). One of the most well-characterized mesenchymal effector proteins is vimentin, a type III intermediate filament that plays a role in cell attachment and migration by controlling cytoskeletal dynamics.37 Vimentin is highly expressed in high-grade ductal carcinomas and is a predictive biomarker for lymph-node metastasis and poor prognosis in colorectal cancer.20,38,39 Though it is unclear whether vimentin expression is sufficient to induce a mesenchymal phenotype, it has been shown to correlate with increased Snail2 (Slug) expression.40 A related factor, Snail1, has also been shown to increase matrix metalloproteinase-2 (MMP-2) expression by binding to its promoter in squamous cell carcinoma.41 MMPs are proteolytic enzymes involved in remodeling the extracellular matrix and dismantling the epithelial membrane, and several MMPs have been implicated in the EMT cascade including MMP-2, MMP-3, and MMP-9.42 Additional genes such as ETS1, FLT1, VSIG, stremelysin-3, FOXC2, HOXB7, and EMT-inducing TFs such as Twist, ZEB1/2, Snail1/2 are also expressed at the invasive front.42 Many of these proteins are downstream effector molecules of various signaling pathways, including Wnt/β-catenin signaling, Notch signaling, and the Sonic Hedgehog pathway.42
Transcriptional regulators of EMT
The EMT program can be regulated by a multitude of TFs; however, three families are primarily responsible for the onset and maintenance of EMT. Members of the Snail, ZEB, and Twist families of TFs are integral to the regulation of embryonic development, and a comprehensive review has shown that these factors are elevated in a wide variety of invasive tumors.43 ZEB, Snail, and Twist TFs facilitate EMT by downregulating epithelial genes (such as E-cadherin) and upregulating mesenchymal genes.43 Though these TFs are not typically expressed in normal epithelium, introduction of ZEB, Snail, Twist, or the homeobox TF Goosecoid induces EMT in human mammary epithelial cells.6 Recent studies have also shown that the majority of these TFs can regulate other hallmarks of cancer such as angiogenesis induction, apoptosis/anoikis resistance, and immortalization in addition to invasion and metastasis.43
Snail1 and Snail2 (also known as Slug) are zinc finger TFs that can bind directly to the promoter of E-cadherin. Though these two factors do have overlapping functions, they are capable of interacting with different target genes and can be activated via independent mechanisms. For instance, Snail2 also inhibits epithelial desmosomal markers such as desmoplakin and desmoglein.44 In addition to the E-cadherin repressing abilities of Snail1, this factor also enhances invasive and angiogenic properties in vitro and in vivo, presumably through the activation of vimentin, fibronectin, and MMPs.45,46 These TFs also interact with several signaling pathways involved in the induction and maintenance of EMT including Wnt/β-catenin, serine/threonine receptor signaling, and the PI3K/AKT/mTOR axis.47 Both Snail1 and Snail2 are overexpressed in a variety of cancers including breast, lung, ovarian, pancreatic, colorectal, and esophageal,43 and Snail1 is also associated with breast cancer recurrence.48 Moody et al showed that Snail1 was sufficient to promote mammary tumor recurrence in an inducible HER2/Neu mouse model, and high levels of Snail1 predicted decreased relapse-free survival in breast cancer patients.48 Interestingly, the recurrent tumors generated displayed a mesenchymal as opposed to an epithelial phenotype, suggesting that the mechanisms for recurrence and metastasis are independent. It is possible that in addition to generating CTCs, EMT may also be responsible for local recurrence due to the failure of mesenchymal cells to migrate and invade surrounding tissue. Indeed, tumor cells surviving after hormonal and chemotherapeutic treatments in breast cancer patients displayed an EMT gene signature and tumor-initiating properties.49
ZEB1 and ZEB2 (also known as SIP1) are also zinc finger TFs that directly bind to and repress E-cadherin, and induce invasion and metastasis in vitro and in animal models.50 These TFs can be activated by multiple EMT-inducing signals including TGF-β, hypoxia, and inflammatory cytokines.51 Expression and repression of ZEB factors results in a rapid EMT and MET, respectively. These factors are also regulated by the miR-200 family of microRNAs, and reciprocally repress the expression of miR-200.52 This double negative feedback loop between ZEB factors and miR-200 further exemplifies the dynamic nature of EMT control, and suggests a mechanism for plasticity depending on external signals. Recent studies have also shown that hypoxia in glioblastoma cells induced ZEB1 and fibronectin (but not Snail1, Snail2, or Twist) and conferred a mesenchymal phenotype, and the invasive phenotype of these cells was inhibited by siRNA targeting ZEB1.53 This effect is directly mediated by HIF1α, which binds to the ZEB1 promoter.54 In addition to repressing E-cadherin, ZEB2 can induce several mesenchymal genes, and specifically transcriptionally upregulates vimentin.55
Twist1 and Twist2 are basic helix–loop–helix TFs that do not bind to the promoter of E-cadherin but decrease its transcription indirectly.26 Twist1 is strongly associated with the metastatic cascade, and is upregulated in cells that can invade, intravasate, extravasate, and metastasize, but is not required for primary tumor formation.26 Inhibition of Twist1 significantly decreased the formation of metastatic nodules in a mouse mammary carcinoma model, and elevated Twist1 expression was strongly associated with invasive lobular breast carcinomas in patients (in contrast to ductal carcinomas or normal tissue).26 Furthermore, the loss of FOXF2, which directly represses Twist1, promotes metastasis in triple negative breast cancer.56 The expression of Twist can also induce EMT in Madin-Darby canine kidney and human mammary epithelial cells, leading to decreased E-cadherin and α, β, and γ-catenin expression, and elevated vimentin, N-cadherin, and smooth muscle actin expression coupled with increased invasion in vitro.26
Though members of the ZEB, Snail, and Twist families of TFs are considered master regulators of EMT, little is known about the hierarchy of these factors since they are often studied individually or in limited groups.6 However, redundancy and crosstalk are prevalent within this transcriptional network. For example, the expression of both ZEB1 and ZEB2 is regulated by Snail1 in certain contexts, and Snail2 activates ZEB1 by directly binding to its promoter.57,58 Snail1 also increases the stability of Twist1, which then leads to the activation of Snail2.59 These interactions are likely context dependent and suggest spatiotemporal regulation of these factors. Further elucidation of these pathways will help to establish functional relationships among these factors in the context of EMT, and may identify specific regulatory mechanisms driving the invasion and metastasis of cancer subtypes.
Role of microRNAs in EMT
MicroRNAs are small, 21–24 nucleotide single-stranded RNA molecules that negatively regulate target messenger RNA (mRNA) transcripts by direct sequence interaction and subsequent alteration of mRNA stability or translation.60 The mode of microRNA-directed mRNA silencing appears to be tissue specific, and many microRNAs work in conjunction to fine tune protein expression on a global level.61 MicroRNAs are involved in the regulation of many normal biological processes, such as development, differentiation, and stem cell maintenance.60 However, dysregulation of microRNAs has been shown to contribute to many diseases, including cancer.60 Various microRNAs have been shown to play a role in the regulation of EMT either through repression of EMT-inducing TFs or genes involved in the maintenance of the epithelial phenotype (Table 1). These pathways are further complicated by reciprocal feedback loops between microRNAs and TFs, or the induction/repression of microRNAs by EMT regulators such as TGF-β, hypoxia, and p53 (Table 2).
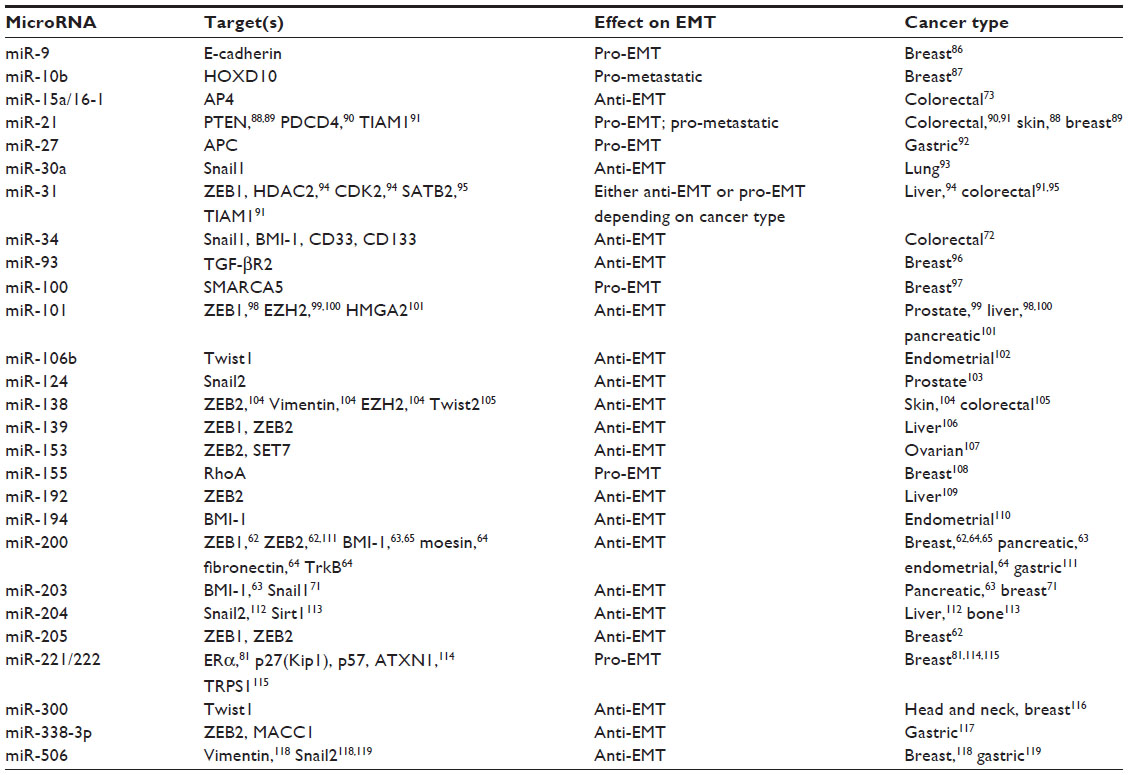 | Table 1 Select microRNAs that regulate EMT in cancer |
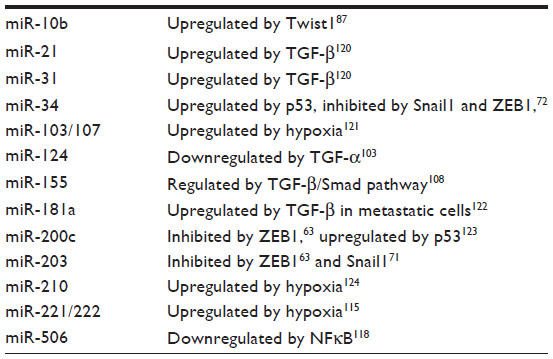 | Table 2 Regulators of EMT-associated microRNAs |
The miR-200 and ZEB1/2 feedback loop
The miR-200 microRNA family consists of miR-200a, miR-200b, miR-200c, miR-141, and miR-429.62 These microRNAs work in concert to repress EMT by targeting ZEB1 and ZEB2, which are direct repressors of E-cadherin. ZEB1 and ZEB2 also transcriptionally repress miR-200c in a double negative feedback loop, facilitating the maintenance of a mesenchymal state (Figure 3).63 Other targets of the miR-200 family include moesin, fibronectin, and TrkB which are involved in cytoskeletal reorganization, cell motility, and resistance to anoikis, respectively, and restoration of miR-200c suppresses anoikis resistance in breast and endometrial cancer cell lines.64 Downregulation of miR-200 family members has been observed in invasive breast cancers that lack E-cadherin and present with a mesenchymal phenotype.62 MiR-200c also suppresses stem cell function and tumor initiating capacity by targeting BMI-1, which plays a role in the maintenance of EMT by targeting the PI3K/Akt inhibitor PTEN, promoting the activation of Snail.65
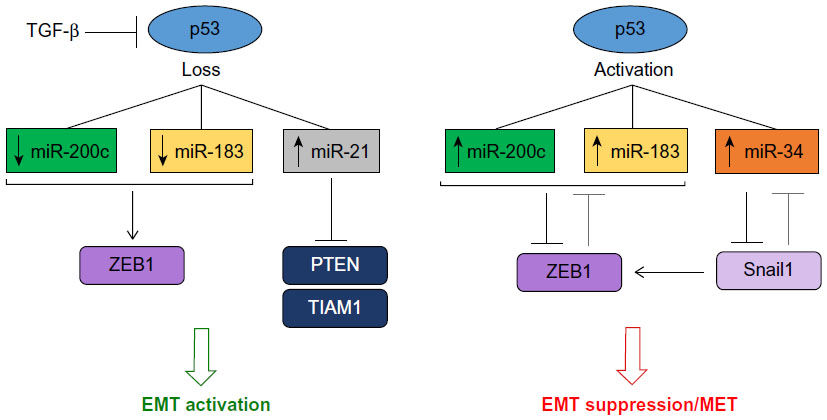 | Figure 3 The interaction of p53 and microRNAs controls regulatory networks that modulate EMT. |
Interestingly, recent studies have shown that the miR-200 family members can be overexpressed in certain cancer types such as endometrial, pancreatic, and ovarian, and higher expression levels are associated with a poor prognosis.66 Though this seems contradictory to the maintenance of a mesenchymal state by a miR-200/ZEB feedback loop, re-expression of miR-200 may be required for the induction of MET and colonization. Indeed, metastatic mouse 4T1 cells displayed high levels of miR-200 as did the metastatic lesions they formed, and the overexpression of miR-200 in cells that were previously unable to colonize led to the formation of lung metastases.67 This observation is further supported by miR-200 regulation of Sec23a, which mediates the secretion of metastasis-suppressing proteins.68 A recent study by Madhavan et al demonstrated that expression of miR-200 family members, especially miR-200b, could be utilized as a prognostic marker for CTCs. Circulating miR-200b levels predicted over 80% of CTC-positive patients.69 These data suggest that the miR-200/ZEB feedback loop acts as a fine tuning mechanism to switch cells between epithelial and mesenchymal states depending on the microenvironment. While downregulation of miR-200 may be initially required for local invasion, it appears that this microRNA needs to be reactivated in order for cells to successfully colonize and generate metastases in other tissues.
Regulation of the miR-200 family has primarily focused on the role of ZEB1/2; however, other factors have recently emerged that may modulate this microRNA in the context of EMT. Members of the miR-200 family are regulated by p53, and loss of p53 function is associated with the induction of EMT and the associated stem cell-like phenotype (Figure 3).6 Growth factors such as TGF-β and platelet-derived growth factor (PDGF)-D also repress miR-200 via indirect or direct mechanisms (ie, methylation of the miR-200 promoter in the case of TGF-β).70 Stable overexpression of PDGF-D in prostate cancer cells as well as treatment with pure active PDGF-D resulted in decreased levels of miR-200 and the induction of EMT; however, it is unclear whether this growth factor acts directly on miR-200 transcription.28
Additional regulatory feedback loops
Following the discovery of the miR-200/ZEB feedback loop, other microRNA/TF relationships were also uncovered. Two microRNAs, miR-34 and miR-203, directly target and inhibit Snail1, and conversely Snail inhibits the transcription of these microRNAs.71,72 Like ZEB, Snail is also associated with stemness markers, and regulates the expression BMI-1, CD33, and CD133 in colorectal cancer cells. Activation of p53 can also regulate miR-34 expression, and the loss of miR-34 is required for TGF-β-induced EMT.72 Considering that TGF-β negatively regulates p53, these recent studies provide a mechanism for EMT activation via p53 loss as a result of TGF-β signaling and the silencing of miR-34, allowing for the expression of EMT-promoting TFs such as Snail1 (Figure 3). p53 also induces the expression of miR-15a/16-1, which represses the EMT-inducing TF AP4 in colorectal cancer.73 The interaction of AP4 with other characterized TFs such ZEB, Snail, and Twist (if any) is currently unknown, however should be explored further to elucidate the p53-microRNA-EMT cascade. These complex networks bring into question the tissue specificity of these relationships, as well as the relevance of these regulatory axes during normal development.
Small molecules that modulate EMT
TGF-β
TGF-β is a well-characterized growth factor that induces EMT in a variety of cellular contexts, including cancer. Stimulation of TGF-β production can occur via hypoxia, autocrine (produced by the tumor itself), and/or paracrine (produced by the tumor stroma) mechanisms.42 TGF-β exhibits a dualistic nature during tumor progression, acting as a tumor suppressor in early stages of tumorigenesis by inhibiting cell-cycle progression or apoptosis, and an oncogenic growth factor later in tumor development during metastatic events.42 TGF-β binds to its receptor (TGF-βRII) and activates multiple pathways, including Ras-MAPK and Smad-dependent signaling pathways. Activation of the Ras-MAPK pathway induces expression of Snail1 and Snail2, leading to the repression of E-cadherin.42 In brief, Smad signaling results in nuclear translocation of an activated Smad2/3 and Smad4 complex, which induces the transcription of key EMT target genes such as ZEB1.42 TGF-β can also exert gene expression changes through activation of the Notch and Wnt pathways, which lead to EMT induction via degradation of intracellular catenins, Snail1 and Snail2 activation, and expression of mesenchymal markers such as vimentin.42 TGF-β-induced EMT has also been implicated in chemoresistance, specifically of platinum therapies in ovarian cancer and tamoxifen in breast cancer.74
Other growth factors
In addition to TGF-β, a variety of other growth factors have been shown to play a role in the induction or maintenance of EMT by regulating TFs or effector proteins. Relevant factors include epidermal growth factor (EGF), fibroblast growth factor, hepatocyte growth factor, PDGF, and insulin-like growth factor. Many of these growth factors are present in normal epithelium and contribute to the differentiation and maintenance of the epithelial phenotype. However, aberrant signaling of these factors in cancer can lead to activation of the EMT cascade. For example, EGF signaling can disrupt desmosome and adherins junctions, which can lead to the dissociation of tumor cells and increased motility via upregulation of MMP-2 and MMP-9.75 Both EGF and fibroblast growth factor can activate Snail through independent pathways; however, mesenchymal cells can also attenuate their dependence on growth factors such as EGF by upregulating other growth factor receptors such as PDGFR.76 In prostate cancer cells, high levels of PDGF-D resulted in loss of E-cadherin and zonula occludins as well as a gain of vimentin expression and rapid tumor growth in immunodeficient mice.77 Insulin-like growth factor can also act on EMT effector molecules such as β-catenin by sequestering and degrading E-cadherin, leading to the nuclear relocation of β-catenin and activation of target genes.78
Interleukins
Inflammatory cytokines also modulate EMT, presumably due to their role in wound healing and tumorigenesis. In breast cancer, IL-6 has been shown to induce EMT in epithelial-type cancer cell lines, via either ectopic expression or exposure in cell culture media.79,80 Likewise, we and others have found that mammosphere culture conditions induce EMT and expression of IL-6 was concomitantly upregulated.80,81 Exposure of breast cancer cells to IL-6 also generates breast cancer stem cells (characterized by a CD44+/CD24−/low phenotype), which is consistent with activation of the EMT program. Some interleukins, such as IL-17, also act directly on EMT-inducing TFs such as ZEB1.82 Exposure of lung cancer cells to IL-17 not only upregulated gene expression of ZEB1, but also facilitated its translocation to the nucleus.82 The EMT-inducing effects of both IL-6 and another cytokine, IL-32, were dependent on Stat3 activation, which contributes to increased motile and invasive properties.83,84
Conclusion and closing remarks
The EMT program plays a significant role in tumor progression and metastasis, and is modulated by a wide variety of regulatory mechanisms including TFs, microRNAs, and growth factors. EMT should be viewed as a dynamic and reversible process that is dependent on the primary tumor and metastatic microenvironment. Complex regulatory mechanisms modulate the plasticity of EMT, allowing for this program to be fine-tuned in response to the cellular environment. This plasticity also poses challenges due to the adaptive nature of cancer cells as well as the role of EMT in normal cellular processes such as wound healing and inflammation. Drugs that promote an epithelial shift may consequently induce MET and colonization of metastases. Even if localized treatments could be administered, the mechanisms controlling the switch between an epithelial and mesenchymal state are complex. It has been proposed that the epithelial differentiation program is a default pathway for cells in a mesenchymal state, and that the absence of signals that sustain the mesenchymal phenotype may force cells to revert back to an epithelial state.85 This hypothesis brings into question whether the best approach to modulate EMT is through targeting epithelial or mesenchymal effector molecules. Activation of the EMT program also increases stem-like characteristics and consequently chemoresistance in cell populations, and it is unclear whether cells in a permanent mesenchymal state are synonymous with cancer stem-like cells, or if these are distinct populations. Further elucidating EMT (and MET) promoting pathways in specific cancer types and clinical samples will provide a stronger framework for tissue specificity and the role of the microenvironment in modulating the EMT cascade.
Acknowledgments
Apologies to those researchers whose studies were omitted or only cited indirectly through reviews due to space constraints.
Disclosure
The author reports no conflicts of interest in this work.
References
Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–695. | |
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. | |
Gurzu S, Turdean S, Kovecsi A, Contac AO, Jung I. Epithelial-mesenchymal, mesenchymal-epithelial, and endothelial-mesenchymal transitions in malignant tumors: an update. World J Clin Cases. 2015; 3(5):393–404. | |
Colas E, Pedrola N, Devis L, et al. The EMT signaling pathways in endometrial carcinoma. Clin Transl Oncol. 2012;14(10):715–720. | |
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | |
De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. | |
Guttilla IK, Adams BD, White BA. ERalpha, microRNAs, and the epithelial-mesenchymal transition in breast cancer. Trends Endocrinol Metab. 2012;23(2):73–82. | |
Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–454. | |
Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27(20):2192–2206. | |
Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005;65(14):5996–6000; discussion 6000–5991. | |
Thompson EW, Newgreen DF, Tarin D. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res. 2005;65(14):5991–5995; discussion 5995. | |
Barriere G, Fici P, Gallerani G, Fabbri F, Rigaud M. Epithelial mesenchymal transition: a double-edged sword. Clin Transl Med. 2015;4:14. | |
Moustakas A, Heldin CH. Induction of epithelial-mesenchymal transition by transforming growth factor beta. Semin Cancer Biol. 2012;22(5–6):446–454. | |
Wu MZ, Tsai YP, Yang MH, et al. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell. 2011;43(5):811–822. | |
Ogunwobi OO, Liu C. Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via Akt and COX-2 pathways. Clin Exp Metastasis. 2011; 28(8):721–731. | |
Su S, Liu Q, Chen J, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. 2014;25(5):605–620. | |
Kallergi G, Papadaki MA, Politaki E, Mavroudis D, Georgoulias V, Agelaki S. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res. 2011;13(3):R59. | |
Husemann Y, Geigl JB, Schubert F, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13(1):58–68. | |
Kolijn K, Verhoef EI, van Leenders GJ. Morphological and immunohistochemical identification of epithelial-to-mesenchymal transition in clinical prostate cancer. Oncotarget. Epub 2015 May 19. | |
Liu T, Zhang X, Shang M, et al. Dysregulated expression of Slug, vimentin, and E-cadherin correlates with poor clinical outcome in patients with basal-like breast cancer. J Surg Oncol. 2013;107(2):188–194. | |
Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68(4):989–997. | |
Ipekci T, Ozden F, Unal B, Saygin C, Uzunaslan D, Ates E. Epithelial-mesenchymal transition markers beta-catenin, Snail, and E-Cadherin do not predict disease free survival in prostate adenocarcinoma: a prospective study. Pathol Oncol Res. Epub 2015 Jun 4. | |
Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22(6):725–736. | |
Bonnomet A, Syne L, Brysse A, et al. A dynamic in vivo model of epithelial-to-mesenchymal transitions in circulating tumor cells and metastases of breast cancer. Oncogene. 2012;31(33):3741–3753. | |
Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–590. | |
Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. | |
Yao D, Dai C, Peng S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res. 2011;9(12):1608–1620. | |
Kong D, Li Y, Wang Z, et al. miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells. 2009;27(8):1712–1721. | |
Chen T, Yuan D, Wei B, et al. E-cadherin-mediated cell-cell contact is critical for induced pluripotent stem cell generation. Stem Cells. 2010;28(8):1315–1325. | |
Li R, Liang J, Ni S, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7(1):51–63. | |
Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. | |
Chen A, Beetham H, Black MA, et al. E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial-mesenchymal transition. BMC Cancer. 2014;14:552. | |
Becker KF, Atkinson MJ, Reich U, et al. E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res. 1994;54(14):3845–3852. | |
Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JG. Methylation patterns of the E-cadherin 5′ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem. 2000;275(4):2727–2732. | |
Lombaerts M, van Wezel T, Philippo K, et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br J Cancer. 2006;94(5):661–671. | |
Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68(10):3645–3654. | |
Liu CY, Lin HH, Tang MJ, Wang YK. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget. 2015;6(18):15966–15983. | |
Sommers CL, Walker-Jones D, Heckford SE, et al. Vimentin rather than keratin expression in some hormone-independent breast cancer cell lines and in oncogene-transformed mammary epithelial cells. Cancer Res. 1989;49(15):4258–4263. | |
Toiyama Y, Yasuda H, Saigusa S, et al. Increased expression of Slug and Vimentin as novel predictive biomarkers for lymph node metastasis and poor prognosis in colorectal cancer. Carcinogenesis. 2013;34(11):2548–2557. | |
Vuoriluoto K, Haugen H, Kiviluoto S, et al. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011;30(12):1436–1448. | |
Yokoyama K, Kamata N, Fujimoto R, et al. Increased invasion and matrix metalloproteinase-2 expression by Snail-induced mesenchymal transition in squamous cell carcinomas. Int J Oncol. 2003;22(4):891–898. | |
Said NA, Williams ED. Growth factors in induction of epithelial-mesenchymal transition and metastasis. Cells Tissues Organs. 2011; 193(1–2):85–97. | |
Sanchez-Tillo E, Liu Y, de Barrios O, et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69(20):3429–3456. | |
Savagner P, Yamada KM, Thiery JP. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol. 1997;137(6):1403–1419. | |
Peinado H, Marin F, Cubillo E, et al. Snail and E47 repressors of E-cadherin induce distinct invasive and angiogenic properties in vivo. J Cell Sci. 2004;117(Pt 13):2827–2839. | |
Jethwa P, Naqvi M, Hardy RG, et al. Overexpression of Slug is associated with malignant progression of esophageal adenocarcinoma. World J Gastroenterol. 2008;14(7):1044–1052. | |
Sipos F, Galamb O. Epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions in the colon. World J Gastroenterol. 2012;18(7):601–608. | |
Moody SE, Perez D, Pan TC, et al. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005;8(3):197–209. | |
Creighton CJ, Li X, Landis M, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106(33):13820–13825. | |
Vandewalle C, Van Roy F, Berx G. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci. 2009;66(5):773–787. | |
Samatov TR, Tonevitsky AG, Schumacher U. Epithelial-mesenchymal transition: focus on metastatic cascade, alternative splicing, non-coding RNAs and modulating compounds. Mol Cancer. 2013;12(1):107. | |
Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop – a motor of cellular plasticity in development and cancer? EMBO Rep. 2010; 11(9):670–677. | |
Joseph JV, Conroy S, Pavlov K, et al. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1alpha-ZEB1 axis. Cancer Lett. 2015;359(1):107–116. | |
Zhang W, Shi X, Peng Y, et al. HIF-1alpha promotes epithelial-mesenchymal transition and metastasis through direct regulation of ZEB1 in colorectal cancer. PLoS One. 2015;10(6):e0129603. | |
Bindels S, Mestdagt M, Vandewalle C, et al. Regulation of vimentin by SIP1 in human epithelial breast tumor cells. Oncogene. 2006; 25(36):4975–4985. | |
Wang QS, Kong PZ, Li XQ, Yang F, Feng YM. FOXF2 deficiency promotes epithelial-mesenchymal transition and metastasis of basal-like breast cancer. Breast Cancer Res. 2015;17:30. | |
Wels C, Joshi S, Koefinger P, Bergler H, Schaider H. Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. J Invest Dermatol. 2011;131(9):1877–1885. | |
Dave N, Guaita-Esteruelas S, Gutarra S, et al. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem. 2011; 286(14):12024–12032. | |
Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011;71(1):245–254. | |
Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4(3):143–159. | |
Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455(7209):64–71. | |
Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. | |
Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–1495. | |
Howe EN, Cochrane DR, Richer JK. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011;13(2):R45. | |
Shimono Y, Zabala M, Cho RW, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009; 138(3):592–603. | |
Brabletz T. To differentiate or not – routes towards metastasis. Nat Rev Cancer. 2012;12(6):425–436. | |
Dykxhoorn DM, Wu Y, Xie H, et al. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One. 2009;4(9):e7181. | |
Korpal M, Ell BJ, Buffa FM, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17(9):1101–1108. | |
Madhavan D, Zucknick M, Wallwiener M, et al. Circulating miRNAs as surrogate markers for circulating tumor cells and prognostic markers in metastatic breast cancer. Clin Cancer Res. 2012;18(21):5972–5982. | |
Gregory PA, Bracken CP, Smith E, et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22(10):1686–1698. | |
Moes M, Le Bechec A, Crespo I, et al. A novel network integrating a miRNA-203/SNAI1 feedback loop which regulates epithelial to mesenchymal transition. PLoS One. 2012;7(4):e35440. | |
Siemens H, Jackstadt R, Hunten S, et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011;10(24):4256–4271. | |
Shi L, Jackstadt R, Siemens H, Li H, Kirchner T, Hermeking H. p53-induced miR-15a/16-1 and AP4 form a double-negative feedback loop to regulate epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res. 2014;74(2):532–542. | |
Helleman J, Jansen MP, Burger C, van der Burg ME, Berns EM. Integrated genomics of chemotherapy resistant ovarian cancer: a role for extracellular matrix, TGFbeta and regulating microRNAs. Int J Biochem Cell Biol. 2010;42(1):25–30. | |
Hugo H, Ackland ML, Blick T, et al. Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213(2):374–383. | |
Thomson S, Petti F, Sujka-Kwok I, Epstein D, Haley JD. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin Exp Metastasis. 2008;25(8):843–854. | |
Kong D, Wang Z, Sarkar SH, et al. Platelet-derived growth factor-D overexpression contributes to epithelial-mesenchymal transition of PC3 prostate cancer cells. Stem Cells. 2008;26(6):1425–1435. | |
Morali OG, Delmas V, Moore R, Jeanney C, Thiery JP, Larue L. IGF-II induces rapid beta-catenin relocation to the nucleus during epithelium to mesenchyme transition. Oncogene. 2001;20(36):4942–4950. | |
Sullivan NJ, Sasser AK, Axel AE, et al. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28(33):2940–2947. | |
Xie G, Yao Q, Liu Y, et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int J Oncol. 2012;40(4):1171–1179. | |
Guttilla IK, Phoenix KN, Hong X, Tirnauer JS, Claffey KP, White BA. Prolonged mammosphere culture of MCF-7 cells induces an EMT and repression of the estrogen receptor by microRNAs. Breast Cancer Res Treat. 2012;132(1):75–85. | |
Gu K, Li MM, Shen J, et al. Interleukin-17-induced EMT promotes lung cancer cell migration and invasion via NF-kappaB/ZEB1 signal pathway. Am J Cancer Res. 2015;5(3):1169–1179. | |
Miao JW, Liu LJ, Huang J. Interleukin-6-induced epithelial-mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int J Oncol. 2014;45(1):165–176. | |
Park JS, Choi SY, Lee JH, et al. Interleukin-32beta stimulates migration of MDA-MB-231 and MCF-7cells via the VEGF-STAT3 signaling pathway. Cell Oncol (Dordr). 2013;36(6):493–503. | |
Frisch SM. The epithelial cell default-phenotype hypothesis and its implications for cancer. Bioessays. 1997;19(8):705–709. | |
Ma L, Young J, Prabhala H, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12(3):247–256. | |
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007; 449(7163):682–688. | |
Bornachea O, Santos M, Martinez-Cruz AB, et al. EMT and induction of miR-21 mediate metastasis development in Trp53-deficient tumours. Sci Rep. 2012;2:434. | |
Han M, Liu M, Wang Y, et al. Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS One. 2012;7(6):e39520. | |
Asangani IA, Rasheed SA, Nikolova DA, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27(15):2128–2136. | |
Cottonham CL, Kaneko S, Xu L. miR-21 and miR-31 converge on TIAM1 to regulate migration and invasion of colon carcinoma cells. J Biol Chem. 2010;285(46):35293–35302. | |
Zhang Z, Liu S, Shi R, Zhao G. miR-27 promotes human gastric cancer cell metastasis by inducing epithelial-to-mesenchymal transition. Cancer Genet. 2011;204(9):486–491. | |
Kumarswamy R, Mudduluru G, Ceppi P, et al. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer. 2012;130(9):2044–2053. | |
Kim HS, Lee KS, Bae HJ, et al. MicroRNA-31 functions as a tumor suppressor by regulating cell cycle and epithelial-mesenchymal transition regulatory proteins in liver cancer. Oncotarget. 2015; 6(10):8089–8102. | |
Yang MH, Yu J, Chen N, et al. Elevated microRNA-31 expression regulates colorectal cancer progression by repressing its target gene SATB2. PLoS One. 2013;8(12):e85353. | |
Liu S, Patel SH, Ginestier C, et al. MicroRNA93 regulates proliferation and differentiation of normal and malignant breast stem cells. PLoS Genet. 2012;8(6):e1002751. | |
Chen D, Sun Y, Yuan Y, et al. miR-100 induces epithelial-mesenchymal transition but suppresses tumorigenesis, migration and invasion. PLoS Genet. 2014;10(2):e1004177. | |
Zhao S, Zhang Y, Zheng X, et al. Loss of microRNA-101 promotes epithelial to mesenchymal transition in hepatocytes. J Cell Physiol. 2015;230(11):2706–2717. | |
Varambally S, Cao Q, Mani RS, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322(5908):1695–1699. | |
Zheng F, Liao YJ, Cai MY, et al. Systemic delivery of microRNA-101 potently inhibits hepatocellular carcinoma in vivo by repressing multiple targets. PLoS Genet. 2015;11(2):e1004873. | |
Jiang W, Gu W, Qiu R, et al. miRNA-101 suppresses epithelial-to-mesenchymal transition by targeting HMGA2 in pancreatic cancer cells. Anticancer Agents Med Chem. Epub 2015 May 7. | |
Dong P, Kaneuchi M, Watari H, Sudo S, Sakuragi N. MicroRNA-106b modulates epithelial-mesenchymal transition by targeting TWIST1 in invasive endometrial cancer cell lines. Mol Carcinog. 2014;53(5):349–359. | |
Qin W, Pan Y, Zheng X, et al. MicroRNA-124 regulates TGF-alpha-induced epithelial-mesenchymal transition in human prostate cancer cells. Int J Oncol. 2014;45(3):1225–1231. | |
Liu X, Wang C, Chen Z, et al. MicroRNA-138 suppresses epithelial- mesenchymal transition in squamous cell carcinoma cell lines. Biochem J. 2011;440(1):23–31. | |
Long L, Huang G, Zhu H, Guo Y, Liu Y, Huo J. Down-regulation of miR-138 promotes colorectal cancer metastasis via directly targeting TWIST2. J Transl Med. 2013;11:275. | |
Qiu G, Lin Y, Zhang H, Wu D. miR-139-5p inhibits epithelial-mesenchymal transition, migration and invasion of hepatocellular carcinoma cells by targeting ZEB1 and ZEB2. Biochem Biophys Res Commun. 2015;463(3):315–321. | |
Zhou J, Xie M, Shi Y, et al. MicroRNA-153 functions as a tumor suppressor by targeting SET7 and ZEB2 in ovarian cancer cells. Oncol Rep. 2015;34(1):111–120. | |
Kong W, Yang H, He L, et al. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008;28(22):6773–6784. | |
Kim T, Veronese A, Pichiorri F, et al. p53 regulates epithelial- mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208(5):875–883. | |
Dong P, Kaneuchi M, Watari H, et al. MicroRNA-194 inhibits epithelial to mesenchymal transition of endometrial cancer cells by targeting oncogene BMI-1. Mol Cancer. 2011;10:99. | |
Kurashige J, Kamohara H, Watanabe M, et al. MicroRNA-200b regulates cell proliferation, invasion, and migration by directly targeting ZEB2 in gastric carcinoma. Ann Surg Oncol. 2012;19(Suppl 3):S656–S664. | |
Qiu YH, Wei YP, Shen NJ, et al. miR-204 inhibits epithelial to mesenchymal transition by targeting slug in intrahepatic cholangiocarcinoma cells. Cell Physiol Biochem. 2013;32(5):1331–1341. | |
Shi Y, Huang J, Zhou J, et al. MicroRNA-204 inhibits proliferation, migration, invasion and epithelial-mesenchymal transition in osteosarcoma cells via targeting Sirtuin 1. Oncol Rep. 2015;34(1):399–406. | |
Ke J, Zhao Z, Hong SH, et al. Role of microRNA221 in regulating normal mammary epithelial hierarchy and breast cancer stem-like cells. Oncotarget. 2015;6(6):3709–3721. | |
Stinson S, Lackner MR, Adai AT, et al. miR-221/222 targeting of trichorhinophalangeal 1 (TRPS1) promotes epithelial-to-mesenchymal transition in breast cancer. Sci Signal. 2011;4(186):pt5. | |
Yu J, Xie F, Bao X, Chen W, Xu Q. miR-300 inhibits epithelial to mesenchymal transition and metastasis by targeting Twist in human epithelial cancer. Mol Cancer. 2014;13:121. | |
Huang N, Wu Z, Lin L, et al. MiR-338-3p inhibits epithelial-mesenchymal transition in gastric cancer cells by targeting ZEB2 and MACC1/Met/Akt signaling. Oncotarget. 2015;6(17):15222–15234. | |
Arora H, Qureshi R, Park WY. miR-506 regulates epithelial mesenchymal transition in breast cancer cell lines. PLoS One. 2013; 8(5):e64273. | |
Sakimura S, Sugimachi K, Kurashige J, et al. The miR-506-induced epithelial-mesenchymal transition is involved in poor prognosis for patients with gastric cancer. Ann Surg Oncol. Epub 2015 Feb 24. | |
Slaby O, Svoboda M, Fabian P, et al. Altered expression of miR-21, miR-31, miR-143 and miR-145 is related to clinicopathologic features of colorectal cancer. Oncology. 2007;72(5–6):397–402. | |
Chen HY, Lin YM, Chung HC, et al. miR-103/107 promote metastasis of colorectal cancer by targeting the metastasis suppressors DAPK and KLF4. Cancer Res. 2012;72(14):3631–3641. | |
Taylor MA, Sossey-Alaoui K, Thompson CL, Danielpour D, Schiemann WP. TGF-beta upregulates miR-181a expression to promote breast cancer metastasis. J Clin Invest. 2013;123(1):150–163. | |
Chang CJ, Chao CH, Xia W, et al. p53 regulates epithelial- mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13(3):317–323. | |
Li L, Huang K, You Y, et al. Hypoxia-induced miR-210 in epithelial ovarian cancer enhances cancer cell viability via promoting proliferation and inhibiting apoptosis. Int J Oncol. 2014;44(6):2111–2120. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.