")
Back to Journals » Drug Design, Development and Therapy » Volume 11
MC-PPEA as a new and more potent inhibitor of CLP-induced sepsis and pulmonary inflammation than FK866
Authors Huang P, Lee Jr MW , Sadrerafi K , Heruth DP, Zhang LQ, Maulik D, Ye SQ
Received 21 October 2016
Accepted for publication 10 January 2017
Published 3 March 2017 Volume 2017:11 Pages 629—641
DOI https://doi.org/10.2147/DDDT.S125349
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Frank Boeckler
Peixin Huang,1 Mark W Lee Jr,2 Keivan Sadrerafi,2 Daniel P Heruth,1 Li Q Zhang,1 Dev Maulik,3,4 Shui Qing Ye1,4
1Division of Experimental and Translational Genetics, Department of Pediatrics, The Children’s Mercy Hospital, University of Missouri Kansas City School of Medicine Kansas City, 2Department of Chemistry, University of Missouri, Columbia, MO, 3Department of Biomedical and Health Informatics, University of Missouri Kansas City School of Medicine, 4Department of Obstetrics and Gynecology, Truman Medical Center, Kansas City, MO, USA
Abstract: Our previous study indicated that overexpression of nicotinamide phosphoribosyltransferase (NAMPT) aggravated acute lung injury, while knockdown of NAMPT expression attenuated ventilator-induced lung injury. Recently, we found that meta-carborane-butyl-3-(3-pyridinyl)-2E-propenamide (MC-PPEA, MC4), in which the benzoylpiperidine moiety of FK866 has been replaced by a carborane, displayed a 100-fold increase in NAMPT inhibition over FK866. Here, we determined the effects of MC4 and FK866 on cecal ligation and puncture (CLP) surgery-induced sepsis in C57BL/6J mice. MC4 showed stronger inhibitory effects than FK866 on CLP-induced mortality, serum tumor necrosis factor α (TNFα) levels, pulmonary myeloperoxidase activity, alveolar injury, and interleukin 6 and interleukin1β messenger RNA levels. In vitro cell permeability and electric cell–substrate impedance sensing assays demonstrated that MC4 inhibited TNFα- and thrombin-mediated pulmonary endothelial cell permeability better than FK866. MC4 also exerted more potent effects than FK866, at concentrations as low as 0.3 nM, to attenuate TNFα-mediated intracellular cytokine expression, nicotinamide adenine dinucleotide (NAD+) and its reduced form NADH levels, and nuclear factor kappa B p65 phosphorylation and nuclear translocation in A549 cells. Our results strongly suggest that the newly developed MC4 is a more potent suppressor of CLP-induced pulmonary inflammation and sepsis than FK866, with potential clinical application as a new treatment agent for sepsis and inflammation.
Keywords: NAMPT, pulmonary inflammation, sepsis
Introduction
Nicotinamide phosphoribosyltransferase (NAMPT) catalyzes the synthesis of nicotinamide mononucleotide (NMN) from nicotinamide (NM) and 5′-phosphoribosyl-1′-pyrophosphate (PRPP). It catalyzes the key rate-limiting step in the nicotinamide adenine dinucleotide (NAD+) synthetic salvage pathway, plays a central role in cellular bioenergetics,1,2 and may indirectly control a number of signaling pathways that depend on NAD levels, such as poly (ADP-ribose) polymerase (PARP) and sirtuin activation.2 A secreted form of NAMPT was originally identified as an extracellular proinflammatory cytokine that could induce cellular expression of inflammatory cytokines, such as tumor necrosis factor α (TNFα), interleukin (IL)1β and IL6, and promote pre-B-cell colony formation.3,4 Lymphocytes, dendritic cells, monocytes, and macrophages express NAMPT when presented with inflammatory stimuli.1,5,6 Thus, NAMPT, either via the NAD salvage pathway or by other unknown mechanisms relating to its secreted form, may modulate innate or acquired immune functions.
Our previous study indicated that overexpression of NAMPT aggravated acute lung injury,7 while knockdown of NAMPT expression attenuated ventilator-induced lung injury.8 Emerging data have implicated NAMPT in the pathogenesis of a number of different human diseases, particularly in the field of cancer and inflammation.9 NAMPT was upregulated in immune cells, including monocytes, macrophages, dendritic cells, T cells, and B cells, as well as in hematopoietic precursors during granulocyte colony-stimulating factor (G-CSF)-induced granulocytic differentiation.1,10,11 NAMPT levels have also been found being elevated in the systemic circulation of patients suffering from diseases with inflammatory components, such as type 2 diabetes,12 acute pancreatitis,13 osteoarthritis,14 sepsis,15 atherosclerosis,16 and psoriasis.17
The search for novel antitumor drugs has led to the identification of inhibitors of NAMPT. FK866 (also known as APO866) is the first known specific and highly potent small-molecule inhibitor of NAMPT. Inhibition of NAMPT by administration of FK866 ameliorates inflammatory symptoms in animal models of arthritis, endotoxic shock, autoimmune encephalitis, and spinal cord injury.10,11,18,19 Unfortunately, Phase II clinical trials have identified severe dose-limiting adverse effects of FK866, such as thrombocytopenia, lymphopenia, and anemia.20 Thus, development of more potent NAMPT inhibitors remains an area of strong interest in pharmaceutical research. In our previous study, the new NAMPT inhibitor analog, meta-carborane-butyl-3-(3-pyridinyl)-2E-propenamide (MC-PPEA, MC4), was synthesized by replacing the benzoylpiperidine moiety in FK866 with meta-carborane.21 The carborane moiety proved superior to benzoylpiperidine, owing to the size and hydrophobicity of carboranes, as well as their potential to form strong dihydrogen bonds with available amino acid residues. This new inhibitor, MC4, exhibits up to 10-fold greater anti-proliferative activity against cancer cells in vitro and a 100-fold increase in NAMPT inhibition than FK866.21 This new carborane-based agent is among the most potent inhibitors reported to date for NAMPT. However, this new inhibitor has not yet been applied to suppress inflammation via its inhibition of NAMPT function. We hypothesize that MC4 would be a much stronger suppressor of inflammation than FK866 since MC4 is a much stronger inhibitor of NAMPT, a known novel inflammatory cytokine.22,23 To test the hypothesis, we have engaged in this comparative study between MC4 and FK866 to evaluate their therapeutic efficacies in the treatment of both cecal ligation and puncture (CLP)-induced sepsis and pulmonary inflammation and to investigate the molecular mechanisms underlying their differences both in vivo and in vitro.
Materials and methods
Mice, CLP-induced sepsis, myeloperoxidase (MPO) activity assay, and histological examination of mouse lungs
Eight- to 12-week-old male C57BL/6J mice (body weight [BW]: 25–31 g; The Jackson Laboratory, Catalog#: 000664) were housed in accordance with the institutional animal care regulations of the University of Missouri Kansas City Institutional Animal Care and Use Committee (IACUC) with an open access to standard chow and water. Animal experiments were approved by the University of Missouri Kansas City IACUC. FK866 and MC4 were dissolved in dimethyl sulfoxide (DMSO) and diluted further with phosphate-buffered saline (PBS) before being applied to mice. In the preventative study, mice received three intraperitoneal injections of 10 mg/kg FK866 or MC4 at 24, 16, and 2 h prior to CLP surgery according to Esposito et al.19 For the therapeutic study, mice were administered once 10 mg/kg FK866 or MC4 by intraperitoneal injection immediately after the CLP surgery. CLP surgery was performed as described by Rittirsch et al.24 Briefly, mice were anesthetized by 60 mg/kg BW (IP) pentobarbital. Under sterile conditions, a 1.5 cm incision was made in the lower abdominal region to expose the cecum. The distal portion of the cecum was ligated 1.5 cm from the end with a 4–0 silk suture and punctured once with a 16-G needle. The cecum was squeezed to place a small portion of its content (bacteria and feces) into the peritoneum before it was replaced in the peritoneal cavity. The peritoneal wall and skin were closed with double sutures. The mice were resuscitated with a subcutaneous injection of sterile saline (1 mL). As a control, mice were sham-operated as described earlier, except that the cecum was neither ligated nor perforated. After the procedure, the mice had an accessible source of water and food ad libitum. The mice were either observed for 6 days for survival experiment or euthanized 24 h after CLP for TNFα and histology analyses. Serum was extracted from drawn blood and stored at −80°C, and TNFα levels were measured using a commercial kit (Catalog#: MTA00B; R&D Systems Inc., Minneapolis, MN, USA). The right lobe of the lung was isolated, snap frozen in liquid nitrogen, and stored at −80°C for RNA isolation and MPO activity assay (Catalog#: MAK068; Sigma-Aldrich, St Louis, MO, USA) according to the manufacturer’s procedure. The left lung lobe was removed, preserved in formalin, embedded in paraffin, cut at 5 μm thickness, and stained with hematoxylin and eosin (H&E). Lung histopathology was examined in a blinded manner as to what group/mouse was reviewed using a modification of a histologic scoring system described by Londhe et al.25 In each section, 10 fields were examined at 400× magnification and randomly scored for 1) interstitial edema, 2) alveolar edema, 3) hemorrhage, and 4) leukocytes and neutrophil infiltrates. The scores from each field were averaged and presented on a scale of 0–4 (0 being none present; 4 being severe and diffuse throughout the chosen field). The histological lung injury scores present the sum of the mean injury subtype scores for each condition on a scale of 0–16.26 Two sections of each lung and four lungs for each group were scored.
Chemicals and antibodies
FK866 was obtained from Cayman Chemical (Ann Arbor, MI, USA; Catalog#: 13287). MC-PPEA (MC4) was synthesized as described previously.21 Rabbit anti-human nuclear factor kappa B (NFκB) p65 (D14E12) XP monoclonal antibody (Catalog#: 8242) and phospho-NFκB p65 (Ser536) (93H1) rabbit monoclonal antibody (Catalog#: 3033) were purchased from Cell Signaling Technology (Beverly, MA, USA). Rabbit anti-human GAPDH monoclonal antibody (Catalog#: G9545) was from Sigma-Aldrich. Rabbit anti-human PBEF/NAMPT polyclonal antibody was from Bethyl Laboratories, Inc. (Montgomery, TX, USA; Catalog#: A300-372A). Recombinant human TNFα was from R&D Systems Inc. (Catalog#: 210-TA). Superscript III Reverse Transcriptase (Catalog#: 18080044) and Platinum Taq DNA Polymerase (Catalog#: 10966018) were from Thermo Fisher Scientific (Waltham, MA, USA). Sources of other key reagents are specified in the relevant text.
Cell culture
Human A549 cells, a lung carcinomatous type II alveolar epithelial cell line, were obtained from ATCC (Manassas, VA, USA; Catalog#: CCL-185™) and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM glutamine, and 1% penicillin/streptomycin. Human lung microvascular endothelial cells (HMVEC-L) were obtained from Lonza (Walkersville, MD, USA; Catalog#: CC-2527) and maintained in EBM-2 endothelial media (Catalog#: CC-3162; Lonza). All cells were cultured at 37°C in a humidified atmosphere of 5% CO2. Cells (passage: 15–23) from each primary flask were detached with 0.05% trypsin, resuspended in fresh culture medium, and seeded into 6-well plates for various experimental conditions followed by Western blot and reverse transcription-polymerase chain reaction (RT-PCR) analysis or seeded into the culture inserts for in vitro cell permeability assays.
In vitro cell permeability assays
In vitro cell permeability assays were carried out according to the protocol of the CHEMICON In Vitro Vascular Permeability Assay kit (Catalog#: ECM644; Millipore, Billerica, MA, USA). Briefly, cells (5×105) were seeded into the culture inserts of permeability chambers that were coated with collagen and incubated at 37°C for 24 h until a monolayer was formed. After the cells were starved for serum for 2 h, TNFα (50 ng/mL), TNFα with MC4 (30, 3, 0.3 nM), or TNFα with FK866 (30, 3, 0.3 nM) was added, cells were incubated for another 18 h at 37°C, followed by addition of 150 μL of FITC-Dextran to each insert for 5 min at room temperature (RT), and then 100 μL of the solution in the bottom chamber was transferred to a 96-well plate. Absorbance at 485 and 530 nm was measured in a TriStar Multimode Reader (LB 941, Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany). Reagent control wells were treated with basal medium or growth medium only. Blank inserts without plated cells were also included as controls.
Measurement of transendothelial electrical resistance (TER)
Cellular barrier properties were measured using an electrical cell–substrate impedance sensing system (ECIS Ztheta; Applied Biophysics, Troy, NY, USA). HMVEC-L were seeded onto array chambers containing 40 gold electrodes per well (8W10E+, Applied Biophysics) pretreated with 10 mM cysteine and coated with fibronectin (20 μg) according to the manufacturers’ specifications. The experiments were initiated when the cells reached confluence, as determined by a capacitance of <10 at 32,000 Hz. HMVEC-L were starved for 2 h, then pretreated with MC4 (0.3 nM) or FK866 (0.3 nM) for 2 h prior to treatment with 0.5 units/mL of thrombin (Sigma-Aldrich; Catalog#: T4393). The data are presented as normalized resistance vs time.
Isolation of RNA, RT-PCR analysis, and Western blot analysis
Total RNA was isolated from A549 cells with a mirVana™ miRNA Isolation Kit (Catalog#: AM1561; Thermo Fisher Scientific) as described previously.27 RT-PCR was performed with Superscript III (Catalog#: 11752-250; Thermo Fisher Scientific) with specific primers (Table 1) synthesized by Integrated DNA Technologies (Coralville, IA, USA). PCR products were separated on 3% agarose gels and stained with ethidium bromide (0.5 μg/mL). The gel image was acquired using an Alpha Imager and analyzed by the AlphaEase™ Stand Alone Software (Alpha Innotech Corporation, San Leandro, CA, USA). Western blots were performed as described previously.28 Band images were acquired using an Alpha Imager and analyzed by the AlphaEase Stand Alone Software as a measure of assayed protein level.
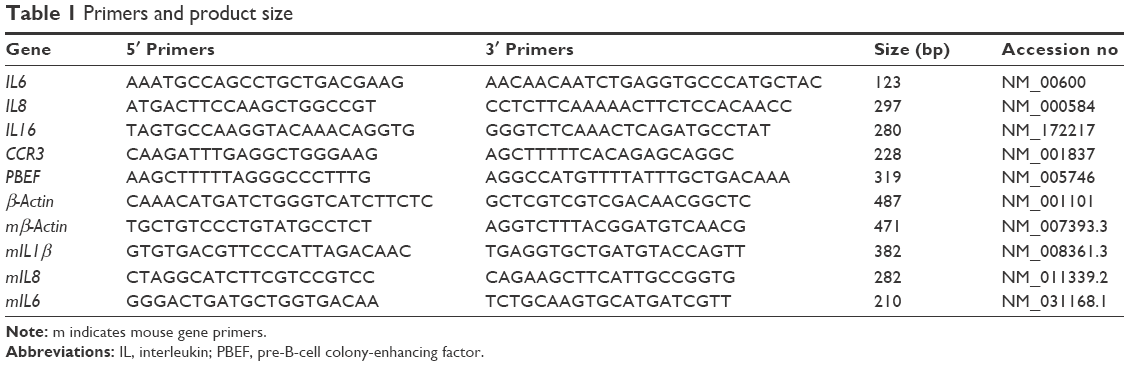 | Table 1 Primers and product size |
Enzyme-linked immunosorbent assay (ELISA) for cell-based human IL6
Intracellular human IL6 levels were determined using a Human IL-6 ELISA Kit (Catalog#: RAB0307; Sigma-Aldrich). Briefly, A549 cells (3×105) were seeded into 6-well plates and incubated overnight at 37°C. After incubation in serum-free media for 2 h, the cells were treated with or without TNFα (50 ng/mL), TNFα with MC4 (30, 3, 0.3 nM), or TNFα with FK866 (30, 3, 0.3 nM) for another 6 h, at which time the cells were lysed in preparation for the ELISA.
Immunofluorescence imaging of nuclear translocation of NFκB in A549 cells
A549 cells were seeded on poly-L-lysine-coated glass slides (Catalog#: p0425; Sigma-Aldrich) and cultured in a 35 mm Petri dish with 3 mL of media at 37°C in a humidified atmosphere of 5% CO2. After 24 h, cells were pre-incubated with or without 0.3 nM MC4 or FK866 for 2 h. Then, cell layers were treated with TNFα (10 ng/mL), TNFα plus 0.3 nM of MC4, or TNFα plus 0.3 nM of FK866 for an additional 20 min. Cell layers were washed three times with 5 mL PBS for 5 min each and fixed in 4% paraformaldehyde for 15 min at RT. Afterward, the slides were rinsed three times in 2 mL of PBS for 5 min each, followed by 60 min in blocking buffer (PBS/5% normal goat serum; Catalog#: 5245; Cell Signaling Technology). The slides were incubated overnight at 4°C with NFκB antibody (1:500) in antibody dilution buffer (PBS/1% BSA/0.3% Triton™ X-100) followed by three washes with PBS for 5 min each. Incubation of the slides in diluted anti-rabbit IgG (H+L), F(ab′)2 fragment (Alexa Fluor® 488 Conjugate) with 1:50 antibody dilution buffer for 1 h at RT in the dark. The slides were then washed (3×) with PBS, prior to attaching coverslips with Prolong Gold Antifade Reagent (Catalog#: 9071; Cell Signaling Technology) with 4,6-diamidino-2-phenylindole (DAPI). The slides were cured for 24 h at RT prior to microscopy (Olympus, U-TV0.5xC-3, Center Valley, PA, USA).
Intracellular NAD+ plus NADH-level assays and MPO activity assay
Intracellular NAD+ plus NADH (NAD/NADH) concentration assays were performed according to the manufacturer’s protocol for an NAD+/NADH Cell-Based Assay Kit (Catalog#: 600480; Cayman Chemical) with minor modifications. Briefly, A549 cells were seeded in 96-well plates (3×104 cells/well in 120 μL of culture medium) and incubated overnight in a CO2 incubator at 37°C. The cells were starved in serum-free media for 2 h, treated with or without TNFα (50 ng/mL) or TNFα plus indicated concentrations of MC4 or FK866 for another 6 h, prior to the measurement of NAD+/NADH levels.
Statistics
All data were expressed as mean ± standard deviation (SD). Differences among treatments were assessed by the one-way analysis of variance (ANOVA) followed by the Holm–Sidak post hoc test. IBM SPSS 23 was used to perform logrank and Breslow tests for survival analysis. Differences between groups were considered statistically significant at P<0.05.
Results
Inhibitory effects of MC4 and FK866 on CLP-induced mouse mortality, serum TNFα levels, pulmonary MPO activities, pulmonary IL6 and IL1β mRNA expressions
The NAMPT inhibitor, FK866, has been shown previously to have an anti-inflammatory function in vivo. In this study, we investigated the effects of FK866 and our newly designed NAMPT inhibitor, MC4, on CLP surgery-induced sepsis in C57BL/6J mice. We first compared the ability of FK866 and MC4 to protect the CLP-induced mortality in mice. C57BL/6J mice (8–12 weeks old, male; n=16 per group) were pre-treated with three doses of 10 mg/kg BW of either FK866 or MC4 or an equal volume of DMSO (1 mL/kg BW) as a vehicle control prior to CLP surgery. We performed sham surgery on three mice to serve as surgical controls. As shown in Figure 1A, 18.8% of mice survived in vehicle (DMSO)-treated animals, while 25% and 43.8% of mice survived in FK866- and MC4-treated mice (n=16), respectively. The survival rate, as analyzed by the Breslow test (Kaplan–Meier method), was significantly higher in MC4-treated CLP mice than vehicle-treated CLP mice (P=0.043). Although FK866 treatment also improved the mouse survival, the difference was not significant by logrank (P=0.24) and Breslow (P=0.23) tests, when compared to the vehicle-treated CLP mice (Figure 1A).
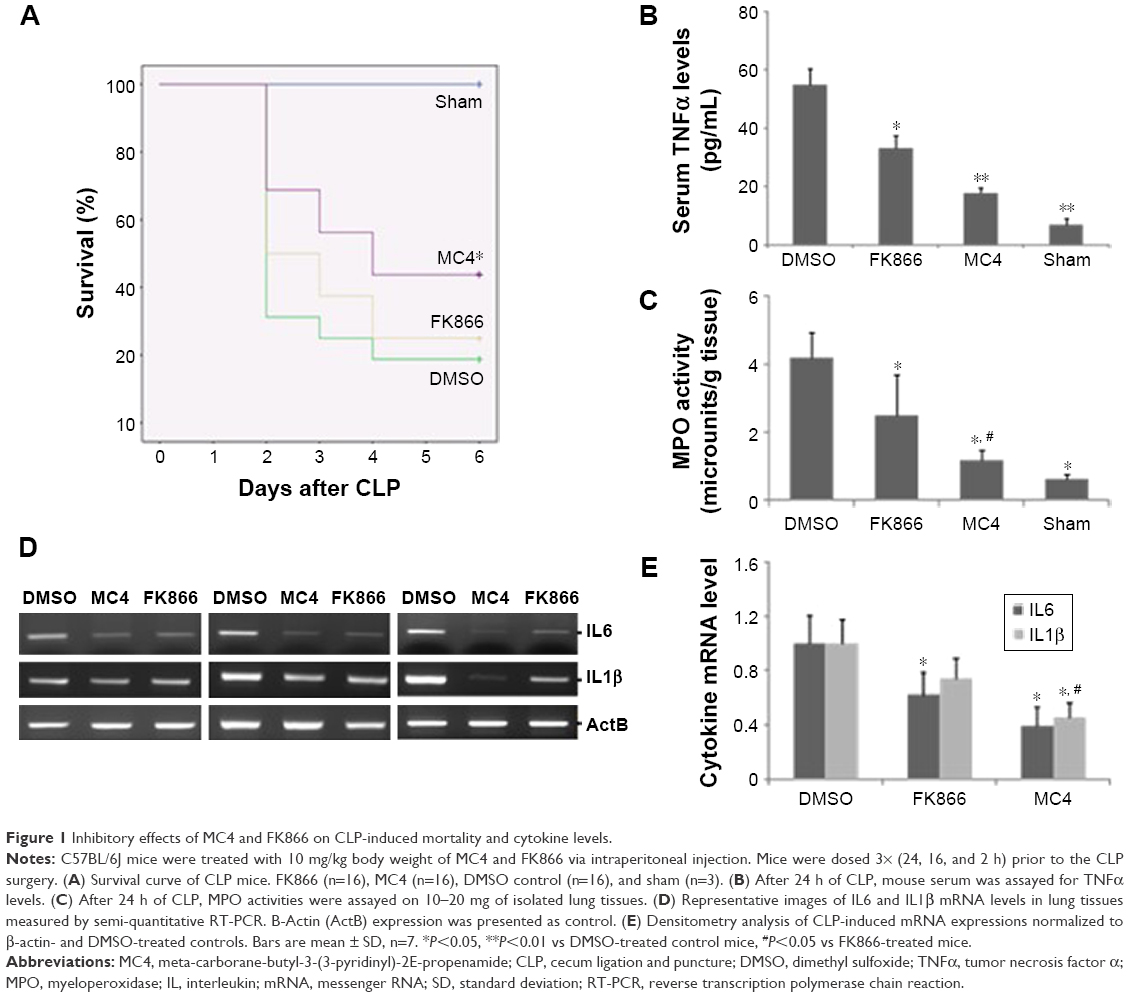 | Figure 1 Inhibitory effects of MC4 and FK866 on CLP-induced mortality and cytokine levels. |
To investigate the mechanisms of protect effects of MC4 and FK866, we detected the CLP-induced TNFα levels in mouse sera, a key inflammatory marker. In sham mice, the mean serum TNFα level was 4.13±5.1 pg/mL. In mice administrated with DMSO, CLP increased serum levels to 54.7±5.6 pg/mL, while in mice administrated with FK866 or MC4, CLP-induced TNFα levels in serum decreased significantly to 32.99±4.5 pg/mL (P=0.039) and 17.81±3.2 pg/mL (P=0.007), respectively (Figure 1B).
Pulmonary tissue MPO activity is an indicator of neutrophil infiltrates into lung parenchyma during pulmonary inflammation, a frequent sequelae of CLP-induced sepsis. We determined lung MPO activity after the treatment of either FK866 or MC4 in CLP-operated mice (Figure 1C). The mean MPO activities were 4.20±0.7, 2.49±1.2, and 1.19±0.3 microunits/g tissue for treatments with DMSO, FK866, and MC4, respectively. This finding suggested that MC4 was a more potent inhibitor of neutrophil infiltrates in the inflammatory mouse lungs than FK866 (P=0.03). In sham mice, the MPO activity (0.47±0.14 microunits/g) was much lower than CLP mice.
We also directly compared the inhibitory effects of FK866 and MC4 on the expression of pulmonary inflammatory cytokines. Compared with DMSO-treated control mice, the relative mRNA levels of IL6 were decreased significantly to 0.62±0.2 (P=0.047) and 0.39±0.2 (P=0.021) in FK866- and MC4-treated mice, respectively (Figures 1D and 1E). IL1β mRNA levels also decreased upon FK866 (0.74±0.2, P=0.081) and MC4 (0.44±0.1, P=0.037) treatment. The IL1β mRNA levels in MC4-treated mice were decreased significantly when compared to both the DMSO-treated and FK866-treated mice. IL6 and IL1β mRNA levels were not detectable in sham-operated mice (data not shown).
Therapeutic effects of FK866 and MC4 on CLP-induced serum TNFα levels and lung injury in mice
To test the potential therapeutic effect of both FK866 and MC4 on CLP-induced sepsis, mice were administered with FK866 and MC4 (10 mg/kg) via a single intraperitoneal injection immediately following CLP. After 24 h, serum TNFα levels were significantly lower in the MC4 treatment group (39.8±7.1 pg/mL) compared to the vehicle control (83.6±32.3; P=0.028) and FK866 (63.6±18.8 pg/mL; P=0.043) groups, respectively (Figure 2A). The TNFα levels in FK866-treated mice were also lower than the control group, but without statistical significance (P=0.21).
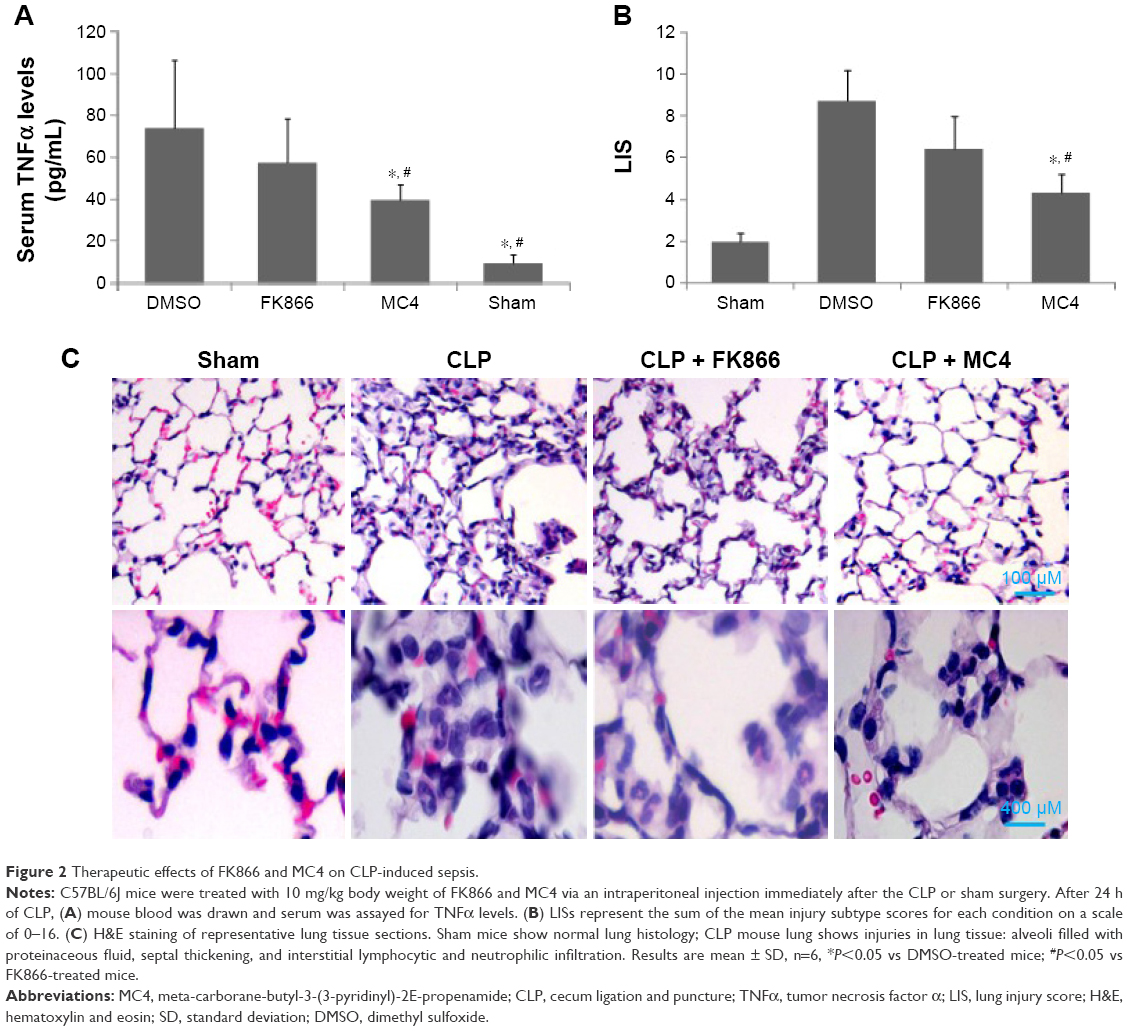 | Figure 2 Therapeutic effects of FK866 and MC4 on CLP-induced sepsis. |
Histological examination by H&E staining of sections from vehicle control lung tissue after CLP showed increased damage of the lung tissue, including alveoli filled with proteinaceous fluid, septal thickening, and interstitial neutrophilic and lymphocytic infiltration relative to the sham-operated control mice that presented with normal lung tissue structure (Figures 2B and 2C). FK866 and MC4 treatment attenuated the CLP-induced injury in lung tissues, with MC4 treatment demonstrating more potent therapeutic effects than FK866 (Figure 2C). Mortality was not observed in any of the four treatment groups (sham, CLP + DMSO, CLP + FK866, and CLP + MC4) within the 24 h time frame and the selected dosages (10 mg/kg) of the drug treatments.
The lung injury score (LIS) was used to evaluate neutrophil infiltration, hemorrhage, interstitial edema, and alveolar wall damage in histological sections of lungs from mice treated therapeutically with DMSO, FK866, and MC4 following CLP. Consistent with the observation in H&E staining in lung tissue (Figure 2C), the CLP + DMSO mice presented with the highest LIS (8.7±1.9) compared to the CLP + FK866 (6.4±2.3), CLP + MC4 (4.3±1.7), and sham mice (1.9±0.6) (Figure 2B). MC4 treatment following CLP resulted in significantly lowered LIS scores compared to either CLP + DMSO (P=0.017) or CLP + FK866 (P=0.031). Although FK866 treatment following CLP also lowered the LIS compared with the CLP + DMSO control group, this treatment only approached statistical significance (P=0.067).
Inhibitory effects of MC4 and FK866 on TNFα- or thrombin-induced cell permeability in HMVEC-L
To investigate further the mechanisms by which MC4 and FK866 inhibit inflammation, we performed in vitro monolayer cell permeability assays in HMVEC-L. In line with our previous observation,29 treating cells with 50 ng/mL TNFα for 22 h increased cell permeability to 2.4-fold of controls (Figure 3A). MC4 showed an inhibitory effect on TNFα-stimulated cell permeability at all concentrations of 30, 3, and 0.3 nM, and at the lowest concentration of 0.3 nM, MC4 exerted the best inhibitory effect, which was 1.2±0.2-fold of untreated cells (P=0.005). Although FK866 also showed an inhibitory effect, the permeability at the lowest level of 0.3 nM was 1.57±0.29-fold of untreated cells, which was much higher than MC4 at the same concentration (P=0.053). Our data suggested that both MC4 and FK866 decreased the TNFα-induced HMVEC-L cell permeability, but MC4 showed more potent inhibitory effect.
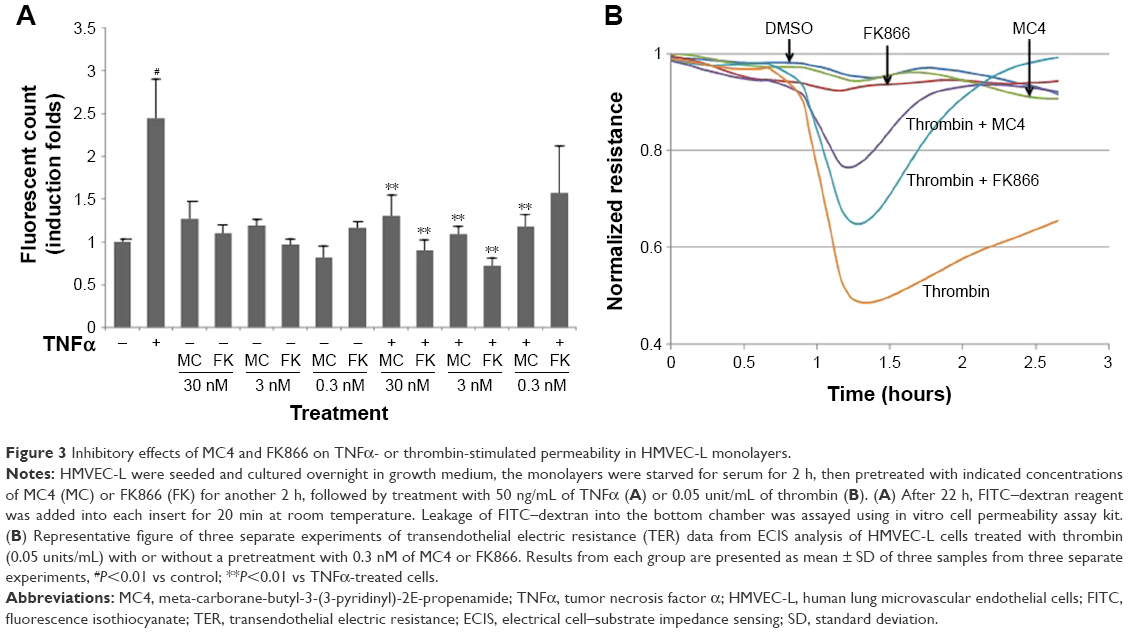 | Figure 3 Inhibitory effects of MC4 and FK866 on TNFα- or thrombin-stimulated permeability in HMVEC-L monolayers. |
To strengthen the findings that MC4 at 0.3 nM is a stronger inhibitor than FK866 on TNFα-induced cell permeability, we measured the effects of MC4 and FK866 on transendothelial electrical resistance (TER) in thrombin-treated HMVEC-L (Figure 3B). Pretreatment with either MC4 (0.3 nM) or FK866 (0.3 nM) attenuated HMVEC-L from the thrombin (0.05 units/mL)-induced reduction in TER (Figure 3B). While both MC4 and FK866 were effective, MC4 was more potent in blocking the effect of thrombin.
Effects of MC4 and FK866 on TNFα-induced cytokine expressions in A549 cells
Since our previous data demonstrated that NAMPT plays a key role in pulmonary epithelial barrier dysfunction via its regulation of cytokines,29 we measured the effects of MC4 and FK866 on TNFα-induced cytokine expressions in A549 cells by performing semi-quantitative RT-PCR analyses of IL6, IL8, IL16, NAMPT, and CCR3 expression in A549 cells. Serum-starved A549 cells were pre-treated with three different concentrations of MC4 and FK866 prior to a 5 h treatment with TNFα (50 ng/mL) (Figure 4). MC4 and FK866 showed inhibitory effects on TNFα-induced cytokine expression at the mRNA level (Figures 4A and 4B). MC4 showed inhibitory effects on TNFα-induced cytokine expression in a concentration-dependent manner. FK866 showed inhibitory effects only at the higher concentrations (3 and 30 nM, P<0.05), while no inhibitory effect was observed at the lowest concentration (0.3 nM) (Figures 4A and 4B). No significant inhibition was observed for NAMPT expression by MC4 or FK866 treatment.
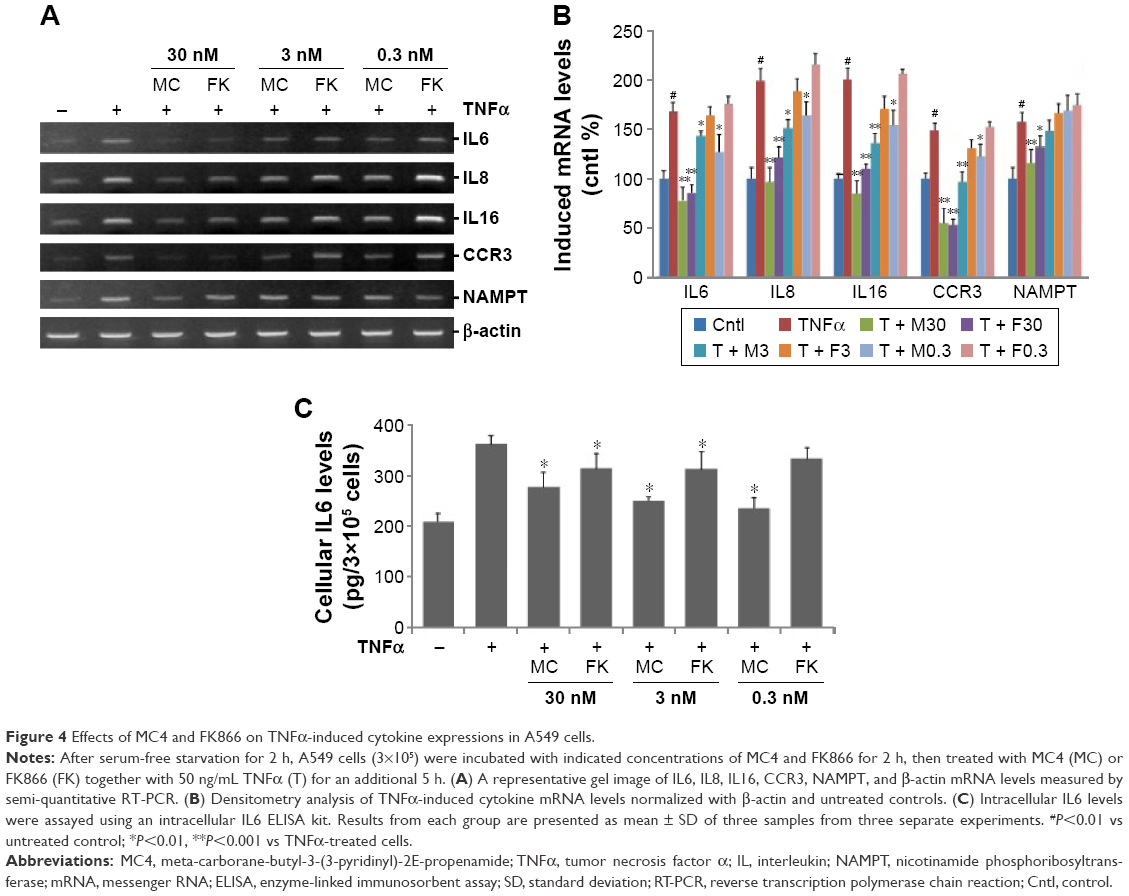 | Figure 4 Effects of MC4 and FK866 on TNFα-induced cytokine expressions in A549 cells. |
To corroborate our RT-PCR results (Figures 4A and 4B), ELISA was conducted to assay IL6 expression at the protein level in A549 cells. As expected, compared with untreated controls, TNFα (50 ng/mL) treatment elevated the intracellular IL6 level from 207±16.3 to 361±17.2 pg/mL (P=0.013). Pretreatment with MC4 for 2 h, at concentrations of 30, 3, and 0.3 nM prior to TNFα treatment, decreased the upregulation of IL6 levels to 275±30.4, 248±8.1, and 234±21.4 pg/mL, respectively, which are higher than untreated control, but significantly lower than cells treated with TNFα alone, P=0.037, P=0.014, and P=0.027, respectively. The IL6 levels in the cells pretreated with three concentrations of FK866 prior to treatment with TNFα were 312±29.9, 311±34.8, and 332±22.4 pg/mL, which were lower than cells only treated with TNFα (P=0.041, P=0.047, and P=0.42, respectively), but much higher than cells pretreated with MC4 prior to TNFα, suggesting that MC4 showed stronger inhibitory effects on TNFα-induced IL6 expression in A549 cells than FK866 (Figure 4C).
Inhibitory effects of MC4 and FK866 on TNFα-induced intracellular NAD/NADH levels in A549 cells
Since MC4 and FK866 are inhibitors of NAMPT, a key enzyme in the mammalian salvage pathway for NAD synthesis, we assayed the total intracellular NAD plus NADH concentrations in treated A549 cells. TNFα at a concentration of 50 ng/mL increased NAD/NADH levels, while MC4 and FK866 showed inhibitory effects in this process (Figure 5). Treatment of A549 cells with TNFα for 20 h increased NAD/NADH levels 1.49±0.2-fold relative to untreated cells. Pretreatment with both small-molecule inhibitors at 3 and 30 nM resulted in NAD/NADH levels significantly below non-TNFα-treated control cells. At 0.3 nM, MC4 was able to significantly decrease the NAD/NADH levels (0.67±0.1-fold) relative to the TNFα-treated controls (P=0.007). Although FK866 also inhibited NAD/NADH levels (0.81±0.71-fold, P=0.04), it was not a strong inhibitor as MC4 at this concentration (Figure 5).
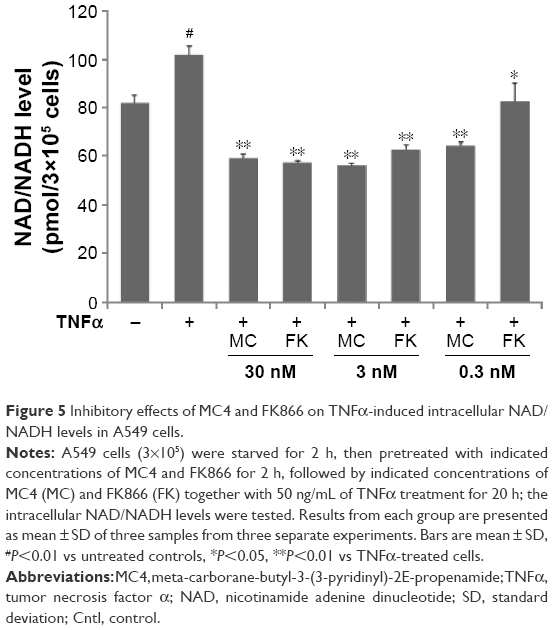 | Figure 5 Inhibitory effects of MC4 and FK866 on TNFα-induced intracellular NAD/NADH levels in A549 cells. |
Inhibitory effects of MC4 and FK866 on TNFα-induced phosphorylation and translocation of NFκB
TNFα activates the NFκB pathway by regulating the phosphorylation of NFκB, the key step in the initiation of the translocation of NFκB from the cytoplasm to the nucleus.30 Therefore, we explored the inhibitory effects of MC4 and FK866 on TNFα-induced phosphorylation of NFκB and translocation of NFκB from cytoplasm into nuclei. Western blot analysis showed that TNFα increased phosphorylation of NFκB (Figures 6A and 6B). Phospho-NFκB was elevated by 4.7-fold relative to the untreated control. However, pretreatment with MC4 and FK866 at higher concentrations (3 and 30 nM) inhibited TNFα-induced phosphorylation of NFκB. MC4 at a concentration as low as 0.3 nM also inhibited TNFα-induced phospho-NFκB, which was 0.54±0.1-fold of TNFα-treated controls (P=0.004), while the phospho-NFκB level in cells pretreated with 0.3 nM FK866 was 0.72±0.2-fold of TNFα-treated controls (P=0.03). There was no obvious change observed in NFκB expression in treated and untreated cells (Figures 6A and 6B).
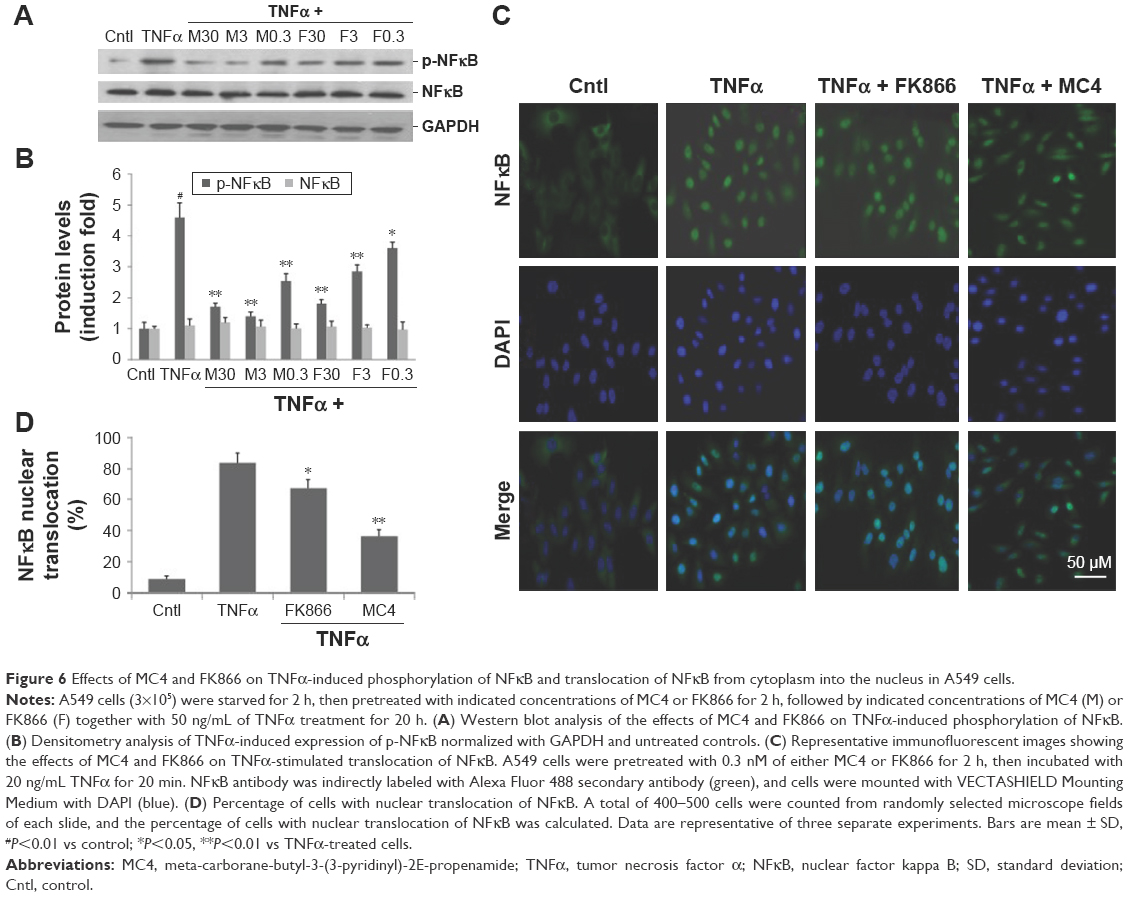 | Figure 6 Effects of MC4 and FK866 on TNFα-induced phosphorylation of NFκB and translocation of NFκB from cytoplasm into the nucleus in A549 cells. |
The block of TNFα-induced NFκB translocation in A549 cells by MC4 and FK866 was also observed by an immunofluorescent assay (Figure 6C). In untreated A549 cells, nuclear NFκB was detected in only 9%±1.4% of the cells, while after treatment with 20 ng/mL of TNFα for 20 min, nuclear NFκB was detected in 83%±4.3% of cells, suggesting that TNFα greatly induced the nuclear translocation of NFκB. When the cells were pretreated with 0.3 nM MC4 and FK866 for 2 h, TNFα-induced translocation was decreased to 67.2±4.1 (P=0.037) and 36.4±2.7 (P=0.007), respectively (Figure 6C), with MC4 providing greater inhibition of NFκB translocation than FK866 (0.3 nM).
Discussion and conclusion
In this study, we have provided new and robust evidence that our newly developed NAMPT inhibitor, MC4, had much stronger anti-inflammatory effects than FK866. In vivo studies supported that MC4 was a stronger inhibitor of CLP-induced sepsis in C57 BL/6J mice than FK866. MC4-treated mice had lower mortality, lower serum TNFα levels, lower lung MPO activity, lower lung expression of inflammatory cytokines (IL1β and IL6), and milder lung injuries than the FK866-treated group. In vitro cell culture experimental results dovetail well with the in vivo findings that MC4 displayed more potent inhibitory effects on the TNFα-induced upregulation of inflammatory cytokines such as IL6, IL8, CCR3, and IL16 in A549 cells, a lung alveolar type II epithelial cell line, than FK866. Our finding represents the first study to evaluate NAMPT inhibition-based preclinical therapy of CLP-induced sepsis in mice. Our results support MC4 as a potential therapeutic agent for sepsis.
Both FK866 and MC4 decreased CLP-induced mortality (Figure 1A). Statistical analysis by the Kaplan–Meier method demonstrated that MC4 significantly decreased mortality (Breslow test, P=0.043), while testing logrank showed an approaching significance (P=0.077). Both the Breslow and logrank tests did not show a significant difference in survival between FK866 and control groups.
When comparing between prophylactic and therapeutic treatments of both FK866 and MC4, we found that the magnitude of the difference in the inhibitory effect on CLP-induced sepsis was larger in prophylactic experiments (Figures 1B and 2A). That may be due to the frequency and dose of drug administration. The prophylactic treatment involved administration of FK866 or MC4 three times prior to CLP, while the therapeutic treatment was a single dose of the drugs. Nevertheless, histological examination solidified our initial findings that MC4 was a more potent inhibitor of mouse lung injury induced by CLP than FK866. MC4, as shown by H&E staining, more strongly attenuated CLP-induced acute lung injuries, including less alveoli filled with proteinaceous fluid, milder septal thickening, and less interstitial lymphocytic infiltrates, than FK866 compared to the control DMSO-treated mice (Figures 2B and 2C).
Mechanistically, we found that MC4 inhibited NFκB phosphorylation and translocation from cytoplasm to nucleus better than FK866 (Figure 6). NFκB is central to the innate immunity response.31 The vertebrate inflammatory response is the most obvious manifestation of innate immunity.31 Deregulation of NFκB is a hallmark of chronic inflammatory diseases.32 In most cell types, the inactive p50/p65 NFκB heterodimer is located in the cytoplasm, complexed to its IκB inhibitory unit. Stimulation of cells by various reagents, such as bacterial endotoxins or cytokines, phosphorylates NFκB, which leads to a dissociation of NFκB from IκB and a rapid translocation of free NFκB to the nucleus where it induces hundreds of genes, including many inflammatory cytokines.31 Thus, it can be safely reasoned that the inhibitory effect by MC4 on CLP-induced sepsis, pulmonary inflammation, and pulmonary cell cytokine expressions was due to its inhibition of NFκB activation. Our comparative study also revealed that MC4 inhibits TNFα-mediated NAD+ and NADH levels better than FK866 in A549 cells (Figure 5). It has been well established that NAMPT is a key enzyme in the eukaryotic cell NAD synthesis of mammalian salvage pathway.33,34 Our findings in this study are consistent with our previous finding that MC4 exhibits 100-fold increase in NAMPT inhibition than FK866.21 This explains the stronger inhibitory effect of MC4 on cell NAD synthesis than FK866. Van Gool et al18 reported that intracellular NAD levels regulate TNF protein synthesis via Sirt6, a member of the NAD-dependent sirtuin family. We surmise that TNFα induced upregulation of cell NAD synthesis via its upregulation of NAMPT. Because MC4 had a stronger inhibitory effect on NAMPT, it led to a greater inhibition of TNFα-mediated NAD+ and NADH levels in A549 cells than FK866. It is recognized that NAD+ biosynthesis through NAMPT holds potential as a target for the treatment of inflammatory disorders due to the role of NAD+ in immune cell signaling and metabolism. Thus, the attenuation of MC4 of CLP-induced sepsis, pulmonary inflammation, and pulmonary cell cytokine expressions may lie at its more effective inhibition of NAD synthesis catalyzed by NAMPT. However, this may not be the whole picture. Our previous study reported the regulation of inflammatory cytokine expression in pulmonary epithelial cells by pre-B-cell colony-enhancing factor (PBEF, another name of NAMPT) via a non-enzymatic and AP-1-dependent mechanism.35 Li et al36 also reported that extracellular PBEF protected macrophages from endoplasmic reticulum stress-induced apoptosis by activating an IL6/STAT3 signaling pathway via a non-enzymatic mechanism of NAMPT. Malam et al37 demonstrated that priming for neutrophil respiratory burst by PBEF (NAMPT) occurred independently of its NAD-generating capacity because neither NM nor NAD could recapitulate the effects and, furthermore, that FK866 could not inhibit priming. Based on these observations, we posit that MC4 may have a higher binding affinity to NAMPT than FK866, which leads to a more severe distorted conformation, resulting in structural constraints not favorable to the interaction of NAMPT with its elusive receptor or other key proteins. The engendered consequence would be a diminished cytokine activity of NAMPT. Whether this non-enzymatic mechanism is operative here warrants further investigation.
Furthermore, our experiments demonstrated that MC4 was a more potent inhibitor of TNFα-induced HMVEC-L permeability and thrombin-induced decreases of transendothelial electric resistance in HMVEC-L, a cell barrier function indicator, than FK866 (Figure 3). TNFα and thrombin are known inducers of vascular endothelial permeability.29,38,39 Presumably, MC4 attenuated the stimuli-induced permeability better than FK866 via its stronger inhibition of NAMPT functions and hence, corresponding suppression of stimuli-induced signal transduction pathways. These observations can also explain fewer lung neutrophil infiltrates (Figure 2C) as indicated by lower lung MPO activity (Figure 1C) in the MC4-treated group compared with the FK866-treated group.
Both MC4 and FK866 decreased pro-inflammation cytokine mRNA expression in A549 cells but did not downregulate NAMPT mRNA significantly (Figure 4B). This may be explained by the feature that both chemicals bind the two active sites of NAMPT homodimers, leading to the inhibition of its enzymatic activity21 rather than the inhibition of NAMPT expression. Less neutrophil infiltration and milder lung injury in the MC4-treated mice than in the FK866-treated mice (Figure 2) may be in part ascribed to the stronger inhibitory effect of MC4 on NAMPT, causing more apoptosis of neutrophils and hence less numbers and duration of neutrophil infiltrates into the lung tissues.
Sepsis is a major cause of death in intensive care units worldwide, with mortality rates ranging from 20% for sepsis, through 40% for severe sepsis, to >60% for septic shock.40,41 This is because the molecular pathogenesis of sepsis is still incompletely understood and its specific and effective therapies are lacking. NAMPT is emerging as a potential new therapeutic target in sepsis. Jia et al15 first reported that NAMPT mRNA in neutrophils from critically ill septic patients was expressed at higher levels than those in controls. Our previous study discovered that a susceptible haplotype GC in the promoter of human NAMPT gene had a 4.84-fold higher risk of sepsis, while a potential protective haplotype TT had a lower risk of sepsis in a Caucasian patient population.7 Bajwa et al replicated and extended our findings that the NAMPT-1001T>G variant allele and related haplotype were associated with increased odds of developing acute respiratory distress syndrome (ARDS), which is frequently associated with severe sepsis, and increased hazard of intensive care unit mortality among at-risk patients, whereas the −1535C>T (originally labeled as −1543C>T) variant allele and related haplotype are associated with decreased odds of ARDS among patients with septic shock and better outcomes among patients with ARDS.42 These results suggest that NAMPT is a biochemical and genetic marker to sepsis. Thus, NAMPT may represent a viable therapeutic target to sepsis. Although there are some limitations on this study, such as failing to compare the side effects between two inhibitors in mice, failing to explore the best option of drug administration routes in mice, and failing to gain systematically into underlying molecular mechanisms, our study has provided nine lines of solid evidence, including serum TNFα levels, pulmonary cytokine expression, lung histology, and MPO activity in a mouse model of sepsis, as well as pulmonary A549 cell cytokine expression, HMVEC-L cell permeability, HMVEC-L TER, NFκB phosphorylation, and nuclear translocation, which all support that small chemical NAMPT inhibitors MC4 and FK866 can attenuate sepsis-induced systematic and pulmonary inflammation and that MC4 is a significantly more potent inhibitor than FK866. Considering that FK866 displayed dose-limiting side effects of thrombopenia and bleeding in clinical trials,20 MC4, in which a carborane has replaced the benzoylpiperidine moiety of FK866, may reduce or attenuate this potential side effects of thrombopenia and bleeding since the benzoylpiperidine could be a toxic-causing component.21 Previous works have demonstrated that carboranes are potentially valuable pharmacophores owing to their hydrophobicity, extensively developed chemistry, and high biostability.43 Our study prompts us to reason that MC4 could be a viable therapeutic alternative for clinical application because of its lower effective dosage and presumed fewer side effects, which will be verified by in vivo animal studies.
Acknowledgments
We thank Ms Marianne Nsumu for excellent technical assistance. This work is in part supported by National Institutes of Health Grant HL 080042 (Ye, SQ) and the start-up fund and William R Brown/Missouri Endowment of The Children’s Mercy Hospital, University of Missouri Kansas City (Ye, SQ).
Disclosure
The authors report no conflicts of interest in this work.
References
Rongvaux A, Shea RJ, Mulks MH, et al. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol. 2002;32(11):3225–3234. | ||
Revollo JR, Korner A, Mills KF, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6(5):363–375. | ||
Brentano F, Schorr O, Ospelt C, et al. Pre-B cell colony-enhancing factor/visfatin, a new marker of inflammation in rheumatoid arthritis with proinflammatory and matrix-degrading activities. Arthritis Rheum. 2007;56(9):2829–2839. | ||
Moschen AR, Kaser A, Enrich B, et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007;178(3):1748–1758. | ||
Huang Q, Liu D, Majewski P, et al. The plasticity of dendritic cell responses to pathogens and their components. Science. 2001;294(5543):870–875. | ||
Iqbal J, Zaidi M. TNF regulates cellular NAD+ metabolism in primary macrophages. Biochem Biophys Res Commun. 2006;342(4):1312–1318. | ||
Ye SQ, Simon BA, Maloney JP, et al. Pre-B-cell colony-enhancing factor as a potential novel biomarker in acute lung injury. Am J Respir Crit Care Med. 2005;171(4):361–370. | ||
Hong SB, Huang Y, Moreno-Vinasco L, et al. Essential role of pre-B-cell colony enhancing factor in ventilator-induced lung injury. Am J Respir Crit Care Med. 2008;178(6):605–617. | ||
Garten A, Petzold S, Korner A, Imai S, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab. 2009;20(3):130–138. | ||
Busso N, Karababa M, Nobile M, et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PLoS One. 2008;3(5):e2267. | ||
Bruzzone S, Fruscione F, Morando S, et al. Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS One. 2009;4(11):e7897. | ||
Kang YS, Song HK, Lee MH, Ko GJ, Cha DR. Plasma concentration of visfatin is a new surrogate marker of systemic inflammation in type 2 diabetic patients. Diabetes Res Clin Pract. 2010;89(2):141–149. | ||
Daniel P, Lesniowski B, Mokrowiecka A, Jasinska A, Pietruczuk M, Malecka-Panas E. Circulating levels of visfatin, resistin and pro-inflammatory cytokine interleukin-8 in acute pancreatitis. Pancreatology. 2010;10(4):477–482. | ||
Duan Y, Hao D, Li M, et al. Increased synovial fluid visfatin is positively linked to cartilage degradation biomarkers in osteoarthritis. Rheumatol Int. 2012;32(4):985–990. | ||
Jia SH, Li Y, Parodo J, et al. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J Clin Invest. 2004;113(9):1318–1327. | ||
Sethi AA, Amar M, Shamburek RD, Remaley AT. Apolipoprotein AI mimetic peptides: possible new agents for the treatment of atherosclerosis. Curr Opin Investig Drugs. 2007;8(3):201–212. | ||
Koczan D, Guthke R, Thiesen HJ, et al. Gene expression profiling of peripheral blood mononuclear leukocytes from psoriasis patients identifies new immune regulatory molecules. Eur J Dermatol. 2005;15(4):251–257. | ||
Van Gool F, Galli M, Gueydan C, et al. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med. 2009;15(2):206–210. | ||
Esposito E, Impellizzeri D, Mazzon E, et al. The NAMPT inhibitor FK866 reverts the damage in spinal cord injury. J Neuroinflammation. 2012;9:66. | ||
Holen K, Saltz LB, Hollywood E, Burk K, Hanauske AR. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest New Drugs. 2008;26(1):45–51. | ||
Lee MW Jr, Sevryugina YV, Khan A, Ye SQ. Carboranes increase the potency of small molecule inhibitors of nicotinamide phosphoribosyltranferase. J Med Chem. 2012;55(16):7290–7294. | ||
Ognjanovic S, Bryant-Greenwood GD. Pre-B-cell colony-enhancing factor, a novel cytokine of human fetal membranes. Am J Obstet Gynecol. 2002;187(4):1051–1058. | ||
Moschen AR, Gerner R, Schroll A, Fritz T, Kaser A, Tilg H. A key role for Pre-B cell colony-enhancing factor in experimental hepatitis. Hepatology. 2011;54(2):675–686. | ||
Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4(1):31–36. | ||
Londhe VA, Belperio JA, Keane MP, Burdick MD, Xue YY, Strieter RM. CXCR2/CXCR2 ligand biological axis impairs alveologenesis during dsRNA-induced lung inflammation in mice. Pediatr Res. 2005;58(5):919–926. | ||
Makena PS, Gorantla VK, Ghosh MC, et al. Lung injury caused by high tidal volume mechanical ventilation and hyperoxia is dependent on oxidant-mediated c-Jun NH2-terminal kinase activation. J Appl Physiol (1985). 2011;111(5):1467–1476. | ||
Grigoryev DN, Cheranova DI, Heruth DP, et al. Meta-analysis of molecular response of kidney to ischemia reperfusion injury for the identification of new candidate genes. BMC Nephrol. 2013;14:231. | ||
Huang P, Riordan SM, Heruth DP, Grigoryev DN, Zhang LQ, Ye SQ. A critical role of nicotinamide phosphoribosyltransferase in human telomerase reverse transcriptase induction by resveratrol in aortic smooth muscle cells. Oncotarget. 2015;6(13):10812–10824. | ||
Li H, Liu P, Cepeda J, et al. Augmentation of pulmonary epithelial cell IL-8 expression and permeability by pre-B-cell colony enhancing factor. J Inflamm (Lond). 2008;5:15. | ||
Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995;14(12):2876–2883. | ||
Baltimore D. NF-kappaB is 25. Nat Immunol. 2011;12(8):683–685. | ||
Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30(1):43–52. | ||
Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279(49):50754–50763. | ||
Zhang LQ, Heruth DP, Ye SQ. Nicotinamide phosphoribosyltransferase in human diseases. J Bioanal Biomed. 2011;3:13–25. | ||
Liu P, Li H, Cepeda J, et al. Regulation of inflammatory cytokine expression in pulmonary epithelial cells by pre-B-cell colony-enhancing factor via a nonenzymatic and AP-1-dependent mechanism. J Biol Chem. 2009;284(40):27344–27351. | ||
Li Y, Zhang Y, Dorweiler B, et al. Extracellular Nampt promotes macrophage survival via a nonenzymatic interleukin-6/STAT3 signaling mechanism. J Biol Chem. 2008;283(50):34833–34843. | ||
Malam Z, Parodo J, Waheed F, Szaszi K, Kapus A, Marshall JC. Pre-B cell colony-enhancing factor (PBEF/Nampt/visfatin) primes neutrophils for augmented respiratory burst activity through partial assembly of the NADPH oxidase. J Immunol. 2011;186(11):6474–6484. | ||
Ye SQ, Zhang LQ, Adyshev D, et al. Pre-B-cell-colony-enhancing factor is critically involved in thrombin-induced lung endothelial cell barrier dysregulation. Microvasc Res. 2005;70(3):142–151. | ||
Liu P, Li H, Cepeda J, et al. Critical role of PBEF expression in pulmonary cell inflammation and permeability. Cell Biol Int. 2009;33(1):19–30. | ||
Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348(16):1546–1554. | ||
Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med. 2007;35(5):1244–1250. | ||
Bajwa EK, Yu CL, Gong MN, Thompson BT, Christiani DC. Pre-B-cell colony-enhancing factor gene polymorphisms and risk of acute respiratory distress syndrome. Crit Care Med. 2007;35(5):1290–1295. | ||
Issa F, Kassiou M, Rendina LM. Boron in drug discovery: carboranes as unique pharmacophores in biologically active compounds. Chem Rev. 2011;111(9):5701–5722. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.