")
Back to Journals » International Journal of Women's Health » Volume 7
Mayer-Rokitansky-Kuster-Hauser syndrome: a review
Authors Londra L, Chuong F, Kolp L
Received 15 June 2015
Accepted for publication 30 July 2015
Published 2 November 2015 Volume 2015:7 Pages 865—870
DOI https://doi.org/10.2147/IJWH.S75637
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Elie Al-Chaer
Laura Londra, Farah S Chuong, Lisa Kolp
Division of Reproductive Endocrinology and Infertility, Department of Gynecology and Obstetrics, Johns Hopkins University, Baltimore, MD, USA
Abstract: The congenital aplasia or severe hypoplasia of mullerian structures is infrequent. However, the features of normal female endocrine function paired with the absence of a functional uterus and vagina makes it a fascinating entity. The diagnosis and work-up in these patients has become very efficient, thanks to the use of imaging, and there are multiple successful procedures for the creation of a neovagina. In recent years, infertility treatment options through in vitro fertilization have also become available as part of the long-term care of these patients.
Keywords: vaginal agenesis, neovagina, MRKH, mullerian agenesis
Introduction
Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome refers to the congenital aplasia or severe hypoplasia of the structures that derive from the mullerian ducts, including the upper vagina, uterus, and fallopian tubes. It is estimated to occur in one in 4,000 to 5,000 births.1 Developmental abnormalities of some of these structures can be found in other entities, but they have a central role in MRKH. Although a plausible explanation for the classic findings of a rudimentary or absent uterus and vagina in an individual with an XX karyotype would be the abnormal activation of mullerian-inhibiting substance, which would be further conducive to the inhibition of the development of paramesonephric structures in females, there has not been molecular evidence of this so far.2,3
There are multiple genes implicated in the normal development of the mullerian, renal, and bone structures, but two groups appear to be the strongest candidates: the HOXA genes and the WNT4 genes.4–6 Since HOXA10 represents the area of the developing uterus, HOXA11 the lower uterine segment and cervix, and HOXA13 the vagina, it is biologically plausible that altered expression of these genes would result in the anomalies found in MRKH. Interestingly, the HOX genes are also associated with the normal development of the kidneys, bone, and vascular structures, which would reinforce the hypothesis of dysregulation of developmental genes involved in the embryonic origin of the female reproductive tract.4–6
Due to the difficulty in classifying the various clinical presentations, multiple authors have proposed systems that either reflect the embryologic correlate of the abnormality7 or the predominance of particular clinical findings.8 In general, it is accepted the existence of a typical form (fallopian tubes, ovaries, and renal system normally developed), atypical form (with malformations in the ovary or renal system), and the MURCS association (mullerian, renal, and cervico thoracic somite malformations).2,9 The latter refers to associated anomalies in the renal system and in the axial skeleton, although vascular anomalies have also been described.10 In this review, we will describe the most commonly recommended diagnostic modalities and management options, and summarize the current data regarding treatment results in terms of sexual function and social and reproductive issues.
Clinical presentation and work-up
The typical initial presentation in MRKH is primary amenorrhea in an otherwise normally developed adolescent female. When physical examination findings are consistent with absent or hypoplastic vagina, the immediate differential diagnosis includes MRKH and complete androgen insensitivity syndrome, which is due to an inactivating mutation in the androgen receptor. Differences that can help differentiate these entities are summarized in Table 1.
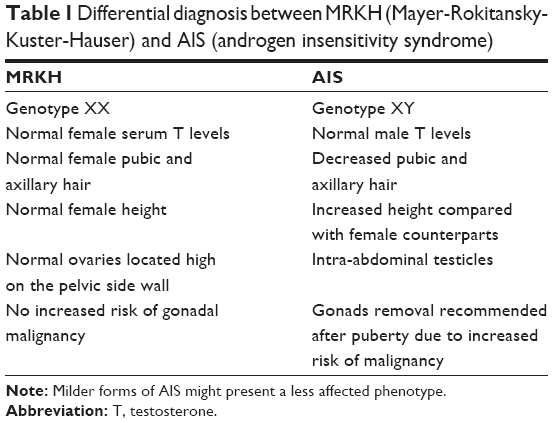 | Table 1 Differential diagnosis between MRKH (Mayer-Rokitansky-Kuster-Hauser) and AIS (androgen insensitivity syndrome) |
Once the diagnosis of MRKH is suspected, imaging studies have a central role in unveiling the degree and extension of gynecologic and extra-gynecologic abnormalities. In a large review of cases, Oppelt et al had found that associated malformations were present in almost half of patients, with the renal and skeletal systems as the most frequent.2,11 Renal anomalies were present in 30% of cases, and among those, renal agenesis was present in more than half of them.
The main options among imaging studies are ultrasound and magnetic resonance imaging (MRI). Ultrasound is easily accessible and readily available in many settings, but it is not always effective in identifying underdeveloped mullerian structures and ovaries, which are usually located high in the pelvis, often at the level of the pelvic brim. The presence of extra-pelvic ovaries has been reported in 16%–19% of the patients.12,13 For surgical planning, MRI is the most useful method, but it is more expensive than ultrasound.14 There is agreement in multiple studies that MRI alone is the modality of choice for further evaluation of all uterine anomalies, and this includes MRKH.2,11,15–17
An overall correlation above 95% between MRI and laparoscopic findings has been reported in a case series of 214 patients with MRKH,18 which included 75% patients with bilateral uterine rudiments, 15% with unilateral uterine rudiments, and 10% with complete uterine agenesis. In 85% of cases where uterine rudiments were removed, the presence of endometrial tissue was adequately diagnosed by MRI. Additionally, MRI was able to diagnose the presence of normal ovaries in more than 97% of patients. Likewise, during laparoscopic evaluation in patients undergoing the Vechietti procedure for treatment, there were mullerian remnants in 87% of cases, of which 26% had some endometrial tissue.
In settings where MRI is not readily accessible, clinical exam with ultrasound has been found to be almost equivalent in the ability to make the initial diagnosis, with the caveat that ureteral anomalies and skeleton anomalies may not be adequately diagnosed.16,19 Computed tomography is rather avoided because it rarely offers any advantage over MRI in these cases and includes radiation.
Laparoscopy is sometimes necessary, particularly when there are pelvic symptoms due to the presence of uterine horns and mullerian remnants with functional endometrium; however, it is not preferred as a diagnostic tool because it is invasive and requires general anesthesia. Additionally, if surgical management is planned for treatment, there might be an opportunity to do further assessments at that time.
Non-surgical and surgical approaches to the creation of a neovagina
Following the diagnosis of MRKH, these young women often experience anxiety and psychological distress surrounding their diagnosis. It is imperative that the physician adequately counsels the patient prior to embarking on any treatment options. The sensitivity and compassion with which these patients are initially treated with will have lasting effects on them. The timing of the creation of a neovagina is elective, but treatment should be deferred until late adolescence to allow informed consent and compliance.20 There is agreement among pediatric surgeons, pediatric urologists, and gynecologists about refraining from creating a vagina for girls with MRKH during childhood. Long-term follow-up has shown that vaginas created during childhood have high failure rates and require additional procedures for the creation of a functional vagina. Even in rare cases where parents of girls with MRKH may seek consultation for surgical correction during childhood to “resolve” the anomaly, it is recommended that any technique for creation of a functional vagina be postponed until the mid to late teens, when the patient can comfortably decide for herself, and is willing to be compliant with her role in the process.21 Multiple web-based resources are available for helping patients and families, such as www.MRKH.org and www.youngwomenshealth.org.
The range of treatment options includes both non-surgical and surgical approaches (Table 2). Vaginal dilation therapy is widely considered as the first line treatment.22 Because of the physically low complication rate and an overall success rate of 75%–85%, vaginal dilation as first choice treatment seems to be justified.23–27 The most commonly used non-surgical method includes Frank’s dilator method and the Ingram method. The Frank method was published by Frank in 1938 and includes an initial demonstration of the introduction of a vaginal mold as a dilator device by the physician, which is then done by the patient for 20 minutes daily, progressively increasing the length and width of the dilator. This method requires the presence of a short vaginal dimple to start, and typically takes 6 months to reach a functional depth and width. Some commonly cited barriers to success have been identified such as cramping and fatigue of the patient, lack of comfort, privacy issues, and lack of time to dilate daily.23
 | Table 2 Treatment options for the creation of neovagina |
In the early 1980s, Ingram sought to overcome these obstacles by using the patient’s own body weight and gravity to assist with the dilation, describing a method with progressively increasing dilators attached to a bicycle seat, where patients provide perineal pressure by sitting and slightly leaning forward. The patients are asked to do this in 15–30-minute intervals for at least 2 hours per day.28 In a similar fashion as the Frank method, the dilators increase in size progressively. The advantages of any of these methods include no hospitalization, patient control, cost-effectiveness, and minimal morbidity and complications. Furthermore, if these methods prove ineffective or the patient is unable to complete the treatment, the option of surgical intervention would still be available. The fact that the patient may become more familiar with the use of a mold is regarded to be an advantage, because the mold must also be used extensively after surgery. The patient’s skills and motivation to use a mold can be assessed during the period of dilation; if a surgical option needs to be considered, the lack of the patient’s postoperative cooperation may lead to failure of almost any further therapy.24 The same cannot be said of surgical approaches, since the presence of scar tissue may not allow for enough dilatation and appropriate size of the neovagina in cases of failure or complications. Although there are clear advantages with the non-surgical methods, disadvantages include the length of time required to achieve a functional vagina, discomfort, and increased risk of vaginal prolapse.23
The various surgical methods are divided into subcategories for ease of discussion: traction methods and graft-based methods. Surgical traction methods have been described and thoroughly used, such as the Vecchietti procedure.29 With laparoscopic assistance, an acrylic “olive” is attached to the vaginal dimple and a thread that courses through the female’s vesicorectal space and into the pelvis then through the anterior abdominal wall with attachment to a traction device. The tension is increased on the device to increase the stretch on the vagina every other day as an outpatient.30,31 After a functional length of 7–8 cm is obtained, the bead is removed and dilators are used to maintain the length. This approach uses surgical methods to create a vagina through tissue stretch, much like the Frank and Ingram methods, but because it requires careful dissection, it is only recommended for surgically naïve tissue.
A variety of tissues including skin grafting, use of a bowel segment, or more recently the use of bioengineered tissue have been described for the creation of a neovagina. The Abbe-McIndoe procedure is the most well-known among these procedures,32 and utilizes a vaginal approach with a split thickness tissue graft taken from the anterior thigh or buttock. The graft is then placed over a vaginal stent and introduced into the previously dissected space between the bladder and rectum. The vaginal stent that serves as a mold for the graft stays in place for the first 7 postoperative days. After the initial healing from the surgery, the patient has a functional vagina. This approach requires continued dilation or frequent intercourse to prevent stricture formation. An interesting modification of the original McIndoe procedure has been reported with the use of autologous in vitro cultured vaginal tissue, then neovaginoplasty with the autologous vaginal tissue as the graft material.33 In a report of 23 cases, all patients completed the Female Sexual Function Index questionnaire at 12 months after surgery, with scores consistent with a satisfactory quality of sexual life. There is ongoing, promising research on development of autologous cell lines derived from the vaginal mucosa for autologous transplant in the treatment of patients with vaginal agenesis.34
An intestinal vaginoplasty can be performed using a segment of sigmoid colon, ileum, or jejunum. With this approach, there is a low risk of tissue shrinkage and little need for long-term vaginal dilation. The tissue produces “lubrication”; however, the discharge at times can be excessive. Harvesting the segment of bowel usually requires a laparotomy – although laparoscopic approaches have been reported with bowel resection and anastomosis which can be associated with increased morbidity.35,36
The Davydov procedure relies on epithelization of the vagina by using an autologous peritoneal grafting technique. The concept was first described by Ott in 1897, but it underwent several modifications, including the use of laparoscopy for the dissection and mobilization of the peritoneum of the Douglas pouch. Vaginally, a space is created similarly as for the other grafting procedures, until the peritoneum is reached. The mobilized peritoneal sac is then opened and pulled downward to connect with the vaginal epithelium. The peritoneum is finally closed abdominally, over a vaginal stent that stays in place for 7 days. The postoperative care is similar to the other grafting techniques, requiring regular use of vaginal dilators to prevent obliteration. A recent review of the long-term results of this approach in a comparison with the Frank non-surgical treatment showed that the most common complications of the surgical approach were rectal injury and obliteration of the neovagina.24
In one of few prospective randomized controlled trials comparing different techniques, it was found that although both the intestinal graft and the Davydov procedure achieve a similar vaginal length, the latter is associated with less discomfort and less vaginal secretion complaints than the bowel graft procedure.37
The decision of which surgical method to offer is often based on the surgeon’s personal experience and preference. Referrals to centers with expertise should be sought and recommended to the patient because the primary surgery is the most likely to succeed.21 Multiple studies have shown that subsequent surgeries increased the chance of operative morbidity with injury to surrounding organs and poor functional outcome. Interestingly, outcomes of surgical treatment were not changed by the previous use of non-surgical techniques or attempted intercourse.24
Reproductive issues and psychological well-being
The vast majority of the literature suggests that patients with MRKH, although subjected to social and personal distress due to the inability to have normal intercourse and bear children, can have a satisfactory sexual function once treated, and can have the option of building a genetically related family with the use of in vitro fertilization (IVF) and a gestational carrier.38–41 Because of their different embryologic origin, ovaries are usually normal and it is possible to obtain oocytes with ovarian hyperstimulation and subsequent transfer of the embryos to a woman who is willing to be a gestational carrier.38,40,42 Since there is no need to coordinate the embryo transfer with the endometrial dating, ovarian hyperstimulation can be conducted with random start protocols, in a similar way as urgent oocyte cryopreservation protocols for oncological patients.43 Previously, there have been reports on the use of ovulation monitoring to ascertain the menstrual cycle timing and allow the use of conventional protocols for ovarian hyperstimulation.42 The response to treatment in terms of number of oocytes retrieved, fertilization rate, and embryo quality has been reported to be slightly lower than average; likewise, pregnancy rates reported so far have been below the average for infertile patients. Additionally, the unique anatomy of the pelvis in MRKH might require the retrieval of oocytes by a transabdominal route instead of the usual transvaginal approach.40 In summary, the number of reported pregnancies after IVF in MRKH patients is still small but has emerged as an attractive option for women who were previously hopeless about having children. Although these children of patients with MRKH are typically normal, in the setting of the relative uncertainty of the etiology of MRKH, it would be important to follow-up on the health of the offspring born after such procedures.44
A recent, extensive review on particular quality of life issues in MRKH patients,45 found that in patients who are able to undergo treatment with creation of a neovagina, there is a restoration of self-confidence. Still, others had found that, compared to matched controls, patients with MRKH scored significantly worse in questionnaires measuring phobic anxiety and self-esteem, as well as in inventories reflecting eating disorders.46 Although these results were found in a small study, the fact that patients were well past the time of the initial diagnosis indicated that the use of brief assessments of psychological distress should ideally be done at regular intervals after the time of diagnosis and after treatment.
The successful creation of a functional neovagina addresses what often manifests as “sex role insecurity”, due to the initial question that the absence of uterus and vagina poses about the ability to fulfill the female sex role, personally and socially. Interestingly, there is scant guidance for practitioners and patients about how to best manage information disclosure issues to the patient and, as often appropriate, to the family, which might be important in patients who are typically very young at the time of diagnosis and treatment consideration. As a general rule, once the diagnosis is made, it is important to emphasize to patients that, with treatment, it is possible for them to have sexual intercourse and build healthy sexual relationships. The American College of Obstetrics and Gynecology recommends that patients be given a brief, written explanation of their condition, including a description of additional organ anomalies. This information could be very useful in cases where patients need to undergo urgent care by practitioners who might not be familiar with MRKH.22 Although a successful treatment of the anatomical abnormality is central to the achievement of sexual well-being, the addition of psychological support and adequate information contribute to the fulfillment of the complexities of female sexual response.47,48 Of note, studies that had questioned the partner’s perception of the newly created vagina have found that there’s a high level of satisfaction and that partners were not able to tell whether the vagina was artificially created or not.49 Additionally, it has been reported that the length of the neovagina is not generally correlated with sexual satisfaction from the patient’s perspective.24 Many articles in the literature of the treatment for MRKH associate sexual outcomes with vaginal length, but as an example of the dissociation between this measure and patient satisfaction, the bowel vaginoplasty offers the longest overall average vaginal length and, nevertheless, has the lowest overall subjective sexual satisfaction scores.50 In the same review of cases, authors found that the full-thickness flap method has been reported with some of the highest subjective sexual satisfaction at 97.8% (69 out of 72) in patients currently sexually active; however, this method had the least number of respondents engaging in sexual activity posttreatment (69.2%). Overall, these results underscore the difficulty in assessing the results of different approaches, and reconciling measures of treatment success with patient satisfaction.
Summary
Although there is still much to learn about the etiology of MRKH, steady progress has been made in the last decades regarding efficient diagnostic modalities and appropriate medical management. Non-surgical approaches for the creation of a neovagina are at the center of therapeutic options, and should be recommended also for most patients who undergo surgical treatment, in order to preserve functional results. Treatment in childhood or early adolescence is not recommended, because of unacceptable complication rate and because full understanding and engagement from the patient is required for optimal results. Fertility options through IVF using autologous oocytes and a surrogate gestational carrier are increasingly available at referral centers. Continued surveillance of psychological well-being of these patients should be considered. Finally, because this condition is rare, there are limitations to obtaining data from long-term follow-up studies, which in turn hampers the ability to offer evidence-based options when counseling MRKH patients. Efforts toward the creation of international centers of excellence for the care of women with complex congenital anomalies with development of associated databases may help to facilitate more accurate comparisons of current management options.
Disclosure
The authors report no conflict of interest in this work.
References
Evans TN, Poland ML, Boving RL. Vaginal malformations. Am J Obstet Gynecol. 1981;141(8):910–920. | ||
Oppelt P, Renner SP, Kellermann A, et al. Clinical aspects of Mayer-Rokitansky-Kuester-Hauser syndrome: recommendations for clinical diagnosis and staging. Hum Reprod. 2006;21(3):792–797. | ||
Ekici AB, Strissel PL, Oppelt PG, et al. HOXA10 and HOXA13 sequence variations in human female genital malformations including congenital absence of the uterus and vagina. Gene. 2013;518(2):267–272. | ||
Nodale C, Ceccarelli S, Giuliano M, et al. Gene expression profile of patients with Mayer-Rokitansky-Kuster-Hauser syndrome: new insights into the potential role of developmental pathways. PloS One. 2014;9(3):e91010. | ||
Sultan C, Biason-Lauber A, Philibert P. Mayer-Rokitansky-Kuster-Hauser syndrome: recent clinical and genetic findings. Gynecol Endocrinol. 2009;25(1):8–11. | ||
Bombard DS 2nd, Mousa SA. Mayer-Rokitansky-Kuster-Hauser syndrome: complications, diagnosis and possible treatment options: a review. Gynecol Endocrinol. 2014;30(9):618–623. | ||
Acien P, Acien M, Sanchez-Ferrer M. Complex malformations of the female genital tract. New types and revision of classification. Hum Reprod. 2004;19(10):2377–2384. | ||
Oppelt P, Renner SP, Brucker S, et al. The VCUAM (Vagina Cervix Uterus Adnex-associated Malformation) classification: a new classification for genital malformations. Fertil Steril. 2005;84(5):1493–1497. | ||
Duncan PA, Shapiro LR, Stangel JJ, Klein RM, Addonizio JC. The MURCS association: Mullerian duct aplasia, renal aplasia, and cervicothoracic somite dysplasia. J Pediatr. 1979;95(3):399–402. | ||
Londra L, Tobler K, Wu J, Kolp L. Mayer-rokitansky-kuster-hauser syndrome associated with severe inferior vena cava stenosis. Case Rep Obstet Gynecol. 2014;2014:745658. | ||
Oppelt PG, Lermann J, Strick R, et al. Malformations in a cohort of 284 women with Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH). Reprod Biol Endocrinol. 2012;10:57. | ||
Fedele L, Bianchi S, Frontino G, Ciappina N, Fontana E, Borruto F. Laparoscopic findings and pelvic anatomy in Mayer-Rokitansky-Kuster-Hauser syndrome. Obstet Gynecol. 2007;109(5):1111–1115. | ||
Raziel A, Vaknin Z, Schachter M, et al. Ultrasonographic-guided percutaneous transabdominal puncture for oocyte retrieval in a rare patient with Rokitansky syndrome in an in vitro fertilization surrogacy program. Fertil Steril. 2006;86(6):1760–1763. | ||
Fedele L, Dorta M, Brioschi D, Giudici MN, Candiani GB. Magnetic resonance imaging in Mayer-Rokitansky-Kuster-Hauser syndrome. Obstet Gynecol. 1990;76(4):593–596. | ||
Pompili G, Munari A, Franceschelli G, et al. Magnetic resonance imaging in the preoperative assessment of Mayer-Rokitansky-Kuster-Hauser syndrome. Radiol Med. 2009;114(5):811–826. | ||
Lermann J, Mueller A, Wiesinger E, et al. Comparison of different diagnostic procedures for the staging of malformations associated with Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil Steril. 2011;96(1):156–159. | ||
Fiaschetti V, Taglieri A, Gisone V, Coco I, Simonetti G. Mayer-Rokitansky-Kuster-Hauser syndrome diagnosed by magnetic resonance imaging. Role of imaging to identify and evaluate the uncommon variation in development of the female genital tract. J Radiol Case Rep. 2012;6(4):17–24. | ||
Preibsch H, Rall K, Wietek BM, et al. Clinical value of magnetic resonance imaging in patients with Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: diagnosis of associated malformations, uterine rudiments and intrauterine endometrium. Eur Radiol. 2014;24(7):1621–1627. | ||
Troiano RN, McCarthy SM. Mullerian duct anomalies: imaging and clinical issues. Radiology. 2004;233(1):19–34. | ||
Nakhal RS, Creighton SM. Management of vaginal agenesis. J Pediatr Adolesc Gynecol. 2012;25(6):352–357. | ||
Laufer MR. Congenital absence of the vagina: in search of the perfect solution. When, and by what technique, should a vagina be created? Curr Opin Obstet Gynecol. 2002;14(5):441–444. | ||
Committee on Adolescent Health Care. Committee opinion: no. 562: mullerian agenesis: diagnosis, management, and treatment. Obstet Gynecol. 2013;121(5):1134–1137. | ||
Callens N, De Cuypere G, De Sutter P, et al. An update on surgical and non-surgical treatments for vaginal hypoplasia. Hum Reprod Update. 2014;20(5):775–801. | ||
Willemsen WN, Kluivers KB. Long-term results of vaginal construction with the use of Frank dilation and a peritoneal graft (Davydov procedure) in patients with Mayer-Rokitansky-Kuster syndrome. Fertil Steril. 2015;103(1):220–227.e1. | ||
Routh JC, Laufer MR, Cannon GM Jr, Diamond DA, Gargollo PC. Management strategies for Mayer-Rokitansky-Kuster-Hauser related vaginal agenesis: a cost-effectiveness analysis. J Urol. 2010;184(5):2116–2121. | ||
McQuillan SK, Grover SR. Dilation and surgical management in vaginal agenesis: a systematic review. Int Urogynecol J. 2014;25(3):299–311. | ||
Roberts CP, Haber MJ, Rock JA. Vaginal creation for mullerian agenesis. Am J Obstet Gynecol. 2001;185(6):1349–1352. | ||
Ingram JM. The bicycle seat stool in the treatment of vaginal agenesis and stenosis: a preliminary report. Am J Obstet Gynecol. 1981;140(8):867–873. | ||
Borruto F. Mayer-Rokitansky-Kuster Syndrome: Vecchietti’s personal series. Clin Exp Obstet Gynecol. 1992;19(4):273–274. | ||
Borruto F, Camoglio FS, Zampieri N, Fedele L. The laparoscopic Vecchietti technique for vaginal agenesis. Int J Gynaecol Obstet. 2007;98(1):15–19. | ||
Creatsas G, Deligeoroglou E. Vaginal aplasia and reconstruction. Best Pract Res Clin Obstet Gynaecol. 2010;24(2):185–191. | ||
McIndoe A. The treatment of congenital absence and obliterative conditions of the vagina. Br J Plast Surg. 1950;2(4):254–267. | ||
Benedetti Panici P, Maffucci D, Ceccarelli S, et al. Autologous in vitro cultured vaginal tissue for vaginoplasty in women with Mayer-Rokitansky-Kuster-Hauser syndrome: anatomic and functional results. J Minim Invasive Gynecol. 2015;22(2):205–211. | ||
Nodale C, Vescarelli E, D’Amici S, et al. Characterization of human vaginal mucosa cells for autologous in vitro cultured vaginal tissue transplantation in patients with MRKH syndrome. Biomed Res Int. 2014;2014:201518. | ||
Cai B, Zhang JR, Xi XW, Yan Q, Wan XP. Laparoscopically assisted sigmoid colon vaginoplasty in women with Mayer-Rokitansky-Kuster-Hauser syndrome: feasibility and short-term results. BJOG. 2007;114(12):1486–1492. | ||
Mane SB, Shastri P, Dhende NP, et al. Our 10-year experience of variable Mullerian anomalies and its management. Pediatr Surg Int. 2010;26(8):795–800. | ||
Cao L, Wang Y, Li Y, Xu H. Prospective randomized comparison of laparoscopic peritoneal vaginoplasty with laparoscopic sigmoid vaginoplasty for treating congenital vaginal agenesis. Int Urogynecol J. 2013;24(7):1173–1179. | ||
Raziel A, Friedler S, Gidoni Y, Ben-ami I, Strassburger D, Ron-El R. In vitro fertilization surrogacy in rare coexisting Mayer-Rokitansky-Kuster-Hauser syndrome and triple X karyotype. Fertil Steril. 2011;95(5):1788.e11–e13. | ||
Raziel A, Friedler S, Gidoni Y, Ben Ami I, Strassburger D, Ron-El R. Surrogate in vitro fertilization outcome in typical and atypical forms of Mayer-Rokitansky-Kuster-Hauser syndrome. Hum Reprod. 2012;27(1):126–130. | ||
Esfandiari N, Claessens EA, O’Brien A, Gotlieb L, Casper RF. Gestational carrier is an optimal method for pregnancy in patients with vaginal agenesis (Rokitansky syndrome). Int J Fertil Womens Med. 2004;49(2):79–82. | ||
Brinsden PR. Gestational surrogacy. Hum Reprod Update. 2003;9(5):483–491. | ||
Ben-Rafael Z, Bar-Hava I, Levy T, Orvieto R. Simplifying ovulation induction for surrogacy in women with Mayer-Rokitansky-Kuster-Hauser syndrome. Hum Reprod. 1998;13(6):1470–1471. | ||
Cakmak H, Rosen MP. Random-start ovarian stimulation in patients with cancer. Curr Opin Obstet Gynecol. 2015;27(3):215–221. | ||
Petrozza JC, Gray MR, Davis AJ, Reindollar RH. Congenital absence of the uterus and vagina is not commonly transmitted as a dominant genetic trait: outcomes of surrogate pregnancies. Fertil Steril. 1997;67:387–389. | ||
Bean EJ, Mazur T, Robinson AD. Mayer-Rokitansky-Kuster-Hauser syndrome: sexuality, psychological effects, and quality of life. J Pediatr Adolesc Gynecol. 2009;22(6):339–346. | ||
Heller-Boersma JG, Schmidt UH, Edmonds DK. Psychological distress in women with uterovaginal agenesis (Mayer-Rokitansky-Kuster-Hauser Syndrome, MRKH). Psychosomatics. 2009;50(3):277–281. | ||
Weijenborg PT, ter Kuile MM. The effect of a group programme on women with the Mayer-Rokitansky-Kuster-Hauser syndrome. BJOG. 2000;107(3):365–368. | ||
Sanfilippo JS. Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: it’s more than the anatomy. J Pediatr Adolesc Gynecol. 2009;22(6):337–338. | ||
Poland ML, Evans TN. Psychologic aspects of vaginal agenesis. J Reprod Med. 1985;30(4):340–344. | ||
McQuillan SK, Grover SR. Systematic review of sexual function and satisfaction following the management of vaginal agenesis. Int Urogynecol J. 2014;25(10):1313–1320. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.