")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Maturity-Onset Diabetes of the Young Identified Among Algerian Probands with Early-Onset Diabetes
Authors Bouldjennet F, Gjesing AP , Azzouz M, Abderrahman SA , Guecier AE, Ali S, Oudjit B, Mennadi-Lacete F, Yargui L , Boudiba A, Chibane A, Touil-Boukoffa C, Hansen T , Raache R
Received 7 July 2020
Accepted for publication 3 November 2020
Published 8 December 2020 Volume 2020:13 Pages 4829—4837
DOI https://doi.org/10.2147/DMSO.S269251
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Faiza Bouldjennet,1,* Anette P Gjesing,2,* Malha Azzouz,3 Samir Ait Abderrahman,4 Amina El Guecier,5 Said Ali,6 Brahim Oudjit,4 Farida Mennadi-Lacete,7 Lyèce Yargui,6 Aissa Boudiba,3 Ahcène Chibane,5 Chafia Touil-Boukoffa,1 Torben Hansen,2 Rachida Raache1
1Laboratory of Cellular and Molecular Biology, Cytokine and NO Synthase Team, University of Science and Technology, Houari Boumediene (USTHB), Algiers, Algeria; 2The Novo Nordisk Foundation Center for Basic Metabolic Research, Faculty of Health Sciences, University of Copenhagen, Copenhagen, Denmark; 3Diabetology Department of Mustapha Pacha Hospital, Algiers, Algeria; 4Diabetology Department of Mohamed Seghir Nekkache Hospital, Algiers, Algeria; 5Internal Medicine Department of Djillali Bounaâma Hospital, Algiers, Algeria; 6Laboratory of Biochemistry, Mustapha Pacha, Algiers, Algeria; 7Pediatric Department of Parnet Hospital, Algiers, Algeria
*These authors contributed equally to this work
Correspondence: Anette P Gjesing; Rachida Raache Email [email protected]; [email protected]
Aim: To investigate the prevalence of variants within selected maturity-onset diabetes of the young (MODY)-genes among Algerian patients initially diagnosed with type 1 diabetes (T1D) or type 2 diabetes (T2D), yet presenting with a MODY-like phenotype.
Methods: Eight unrelated patients with early-onset diabetes (before 30 years) and six relatives with diabetes were examined by targeted re-sequencing for variants in genes known to be involved in MODY (HNF1A, GCK, HNF4A, HNF1B, INS, ABCC8, KCNJ1). Clinical data for probands were retrieved from hospital records.
Results: A total of 12 variants were identified, of which three were classified as pathogenic and one as a variant of uncertain clinical significance (VUS). Two of the pathogenic variants were found in GCK (p.Gly261Arg and p.Met210Lys, respectively) in one proband each and the remaining pathogenic variant was found in HNF1B (p.Gly76Cys) in a proband also carrying the VUS in HNF1A (p.Thr156Met).
Conclusion: Variants in known MODY-genes can be the cause of early-onset diabetes in Algerians diagnosed with T1D or T2D among patients presenting with a MODY-like phenotype; thus, genetic screening should be considered.
Keywords: MODY, type 1 diabetes, early-onset, monogenic diabetes, genes
Introduction
The monogenic form of diabetes called Maturity-Onset Diabetes of the Young (MODY) is a heterogeneous form of diabetes caused by variants in specific genes of which the majority affects insulin secretion.1 It is characterized by an autosomal-dominant mode of inheritance, onset before 25 years of age and a preservation of insulin secretion with detectable C-peptide outside the “honeymoon” period.2–4 To date, at least fourteen subtypes of MODY have been found which are caused by variants in genes implicated in pancreatic development, insulin biosynthesis and/or secretion: HNF4A (MODY1), GCK (MODY2), HNF1A (MODY3), PDX1 (MODY4), HNF1B (MODY5), NEUROD1 (MODY6), KLF11 (MODY7), CEL (MODY8), PAX4 (MODY9), INS (MODY10), BLK (MODY11), ABCC8 (MODY12), KCNJ11 (MODY13) and RFX6 (MODY14).5,6 These various MODY subtypes differ in clinical manifestation: age of onset, severity of hyperglycemia, risk of complications, response to treatment and associated extrapancreatic manifestations.7 GCK-MODY patients usually do not require any pharmacological treatment and do not develop co-morbidities8 whereas carriers of variants in the two transcription factors HNF1A and HNF4A, as well as the two genes (ABCC8 and KCNJ11) encoding a beta-cell K-ATP channel, are sensitive to sulphonylurea treatment.8–10 In contrast, patients carrying variants in the insulin gene (INS) will very likely require insulin therapy.11 Therefore, molecular genetic diagnosis is crucial as it anticipates the patient’s prognosis and can influence treatment decisions. In addition, a molecular genetic diagnosis can lead to early clinical care of asymptomatic relatives.12,13
Previous studies of the prevalence and etiology of MODY have most commonly been investigated in Caucasians and have shown that MODY represents about 1–2% of diabetes among Europeans14 with a predominance of MODY2 (GCK-MODY) and MODY3 (HNF1A-MODY).15,16 Studies investigating the prevalence of different forms of diabetes in Algeria are however limited.17 The prevalence and etiology of MODY may be different in Algeria due to differences in genetic ancestry and the long tradition of consanguine marriages17 and only one previous study has examined the presence of MODY in Algeria which reported one family with MODY caused by the p.Arg85Trp GCK variant.18 Due to the possibility to target treatment according to the genetic etiology in patients carrying variants within HNF4A, GCK, HNF1A, HNF1B, INS, ABCC8 and KCNJ11, it is important to establish if variants within these known MODY-genes are the cause of early-onset diabetes in Algerian patients.
In the present paper, we aim to assess if the presence of pathogenic variants in HNF4A, GCK, HNF1A, HNF1B, INS, ABCC and, KCNJ11 is the cause of diabetes in Algerian patients with an early onset (before 30 years of age) initially diagnosed with type 1 diabetes (T1D) or type 2 diabetes (T2D), yet, presenting with a MODY-like phenotype.
Patients and Methods
Patients
Our study included eight probands (seven males and one female) diagnosed with T1D except for one patient (P4) initially diagnosed with T2D. In addition, six family members with diabetes (two males and four females) were included (Table S1). In addition to being diagnosed with early-onset diabetes, probands were selected due to suspicion of MODY as they had at least three of the following features: 1) a family history of diabetes mimicking an autosomal-dominant inheritance (Figure 1 and Figure S1), 2) young age at diabetes diagnosis (˂ 30 years old), 3) diabetes managed with oral hypoglycemic agents (OHA) or low insulin doses (≤0.5U/Kg/J) at least three years after diagnosis, 4) BMI ˂ 30 kg/m2, 5) decreased renal threshold for glycosuria and 6) anti-body negative (when antibodies are measured) (Table 1). The patients were recruited from University Hospital Mustapha Pacha, Central Hospital of Army, Djillali Bounaâma Hospital and Parnet Hospital. Probands were from families without known consanguinity. The present study was approved by the local ethics committee “Algerian National Agency for Research in Health Sciences, ATRSS ex-ANDRS” in compliance with Helsinki declaration (Code number 43-ANDRS-2011). After obtainment of written informed consent from all participants including consent to publish familial case descriptions (from parents for participants under 18 years old), blood samples were collected for biochemical, immunological and genetic studies.
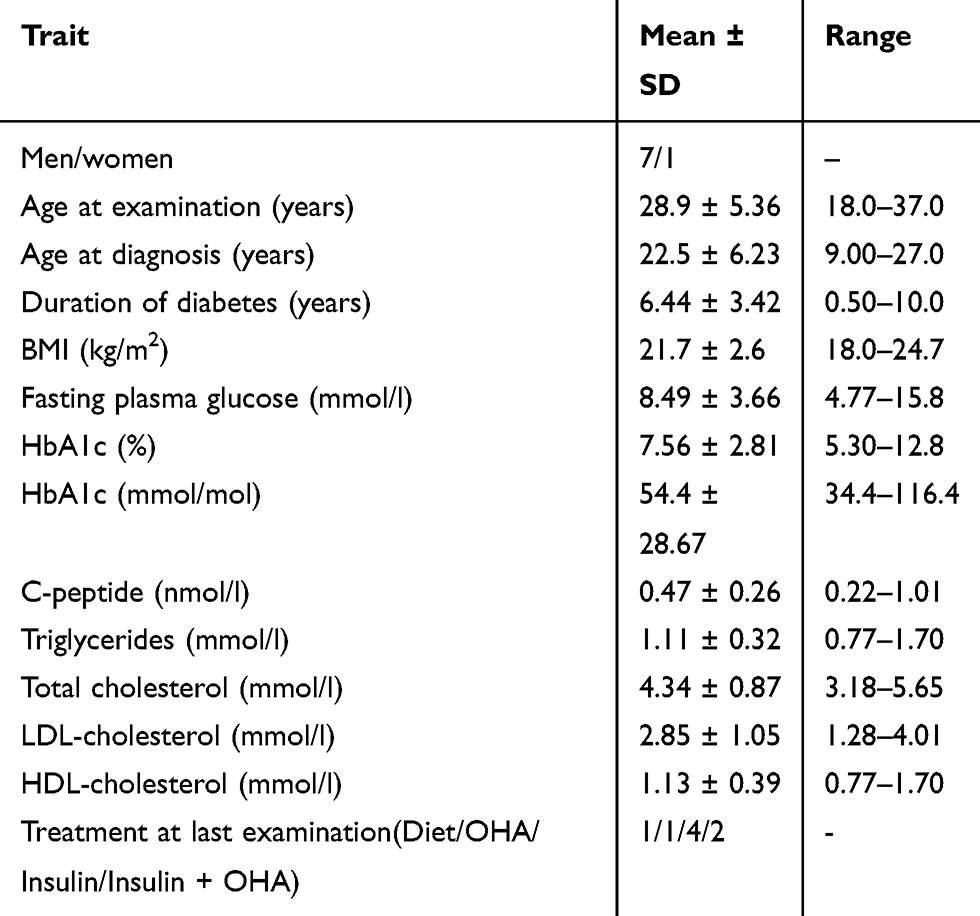 |
Table 1 Clinical and Laboratory Characteristics of the Eight Probands |
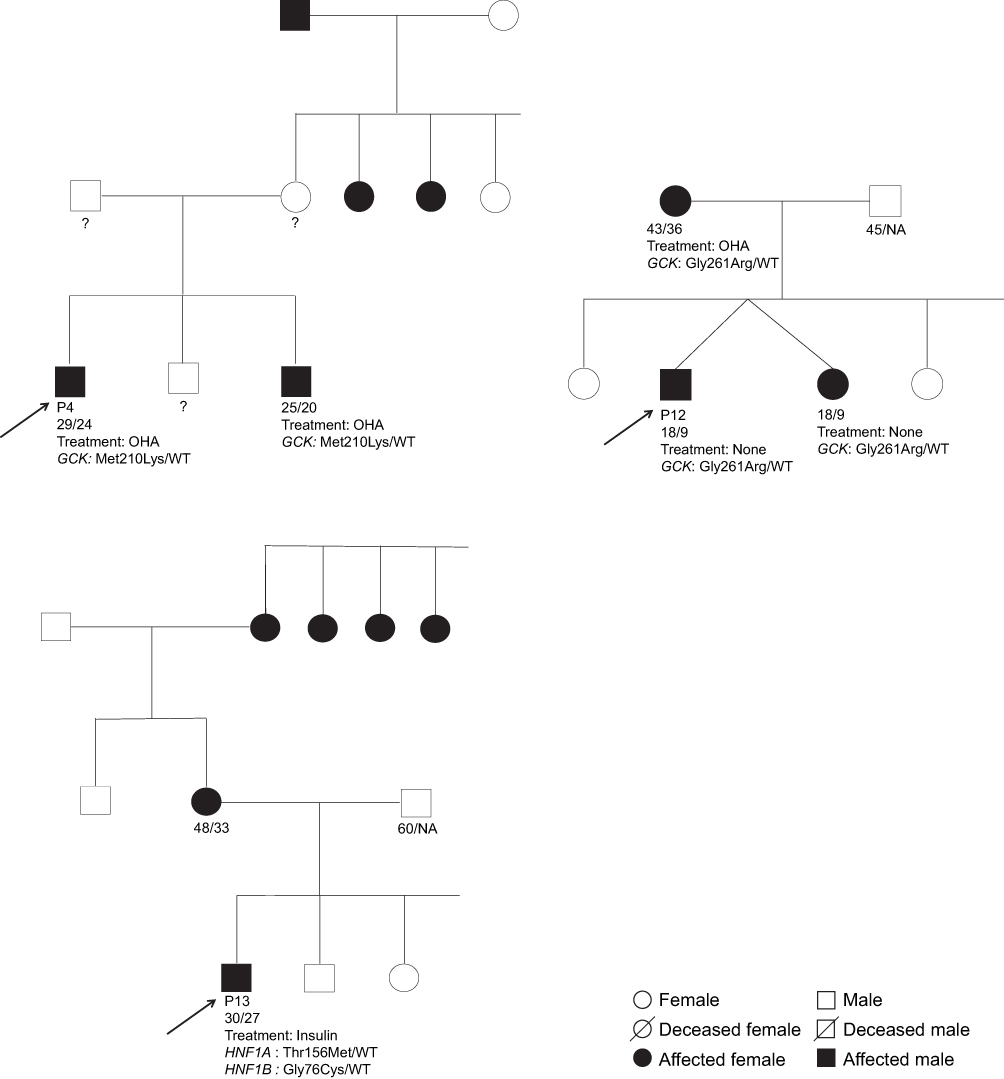 |
Figure 1 Pedigrees of the families 4, 12 and 13 where pathogenic variants were identified. Notes: The P-numbers indicate subjects undergone genetic investigation and the numbers below indicate the age at the last examination/age at diabetes onset (years). Squares represent males and circles indicate females. Black symbols indicate subjects with manifested diabetes and white symbols indicate healthy or non-investigated members. The arrow indicates the proband. Abbreviations: WT, wild-type allele; NA, not applicable. |
The mean age-of-diagnosis for probands was 22.5 ± 6.2 years old [range 9–27 years old], the diabetes duration was 6.4 ± 3.4 years [range 0.5–10 years] and the mean value of BMI was of 21.7 ± 2.6 kg/m2 [range 18–24.7 kg/m2] at the latest examination.
Clinical Data Collection, Biochemical and Immunological Measurements
The family history of diabetes, BMI, age at diabetes diagnosis, fasting plasma glucose (FPG), glycosylated hemoglobin (HbA1c) at diagnosis and treatment history were collected from hospital records. The treatment information included in Table S1 is from the latest examination.
Blood samples were drawn for DNA extraction and measurement of FPG, plasma lipids and HbA1c. Moreover, β cells autoantibodies (Tyrosine phosphatase (IA2) and/or Glutamic Acid Decarboxylase (GAD) auto-antibodies) were measured in probands. Endogenous insulin production was assessed in patients by measuring C-peptide concentrations at the last examination.
Assays
Measurement of HbA1c was performed by High-Performance Liquid Chromatography HPLC (Tosoh G8, Japan); plasma glucose, plasma triglycerides, total plasma cholesterol, high-density lipoprotein (HDL) cholesterol and low-density lipoprotein (LDL) cholesterol were evaluated on the automatic analyser (BiOLiS 24i, Japan), C-peptide by chemiluminescence Immuno-Assay (LIAISON®, DiaSorin, Germany) and IA2 and/or GAD auto-antibodies (GADA) by ELISA (Enzyme-Linked ImmunoSorbent Assay) (ELISA plate reader, PR 3100. BioRad, USA).
Screening for HNF1B Alterations
For patients having HNF1B variants, additional measurements were performed, including serum bilirubin, alanine aminotransferase (ALT), aspartate aminotransferase (AST) and γ-glutamyl transferase (GGT), serum uric acid, creatinine and potassium, measured on a Cobas 400+ Analyser.
Molecular Analysis
Genomic DNA was extracted from peripheral whole blood by standard methods.19 DNA concentration and purity were determined using a NanoDrop (MaestroNano Spectrophotometer, MAESTROGEN Technologies, TAIWAN) and adjusted to 100ng/µL. Targeted regions, including the coding regions and exon/intron boundaries of the HNF4A, GCK, HNF1A, HNF1B, INS, ABCC8 and KCNJ11 genes, were captured and sequenced using the Illumina HiSeq2000 Analyzer as described.20 Obtained results were analyzed for sequence variations using the SeqScape Software (Applied Biosystems) by comparison with published reference sequences (NM_175914, NM_000162, NM_000545.5, NM_000458.2, NM_000207.2, NM_000352 and NM_000525). Variants were classified as being either pathogenic, likely pathogenic, variants of uncertain significance (VUS) or benign, based on the standards and guidelines from American Collee of Medical Genetics and Genomics and the Association for Molecular Pathology.21 This classification is based on 1) the location and function of variant, 2) minor allele frequency (MAF) in both the Gnomad database22 and a Middle Eastern reference population,23 3) previous information of variants being involved in MODY,24– 26 4) predicted functionality based on changes in thermostability25 or CADD score (http://cadd.gs.washington.edu/info) and 5) information of the prevalence of variants in house database of approximately 6000 Danish population-based non-diabetic individuals27 in which the above genes were sequenced.
Results
Clinical Description of Probands
Clinical and biochemical characteristics of the probands are summarized in Table 1 and immunological data are presented in Table S1.
A total of 12 variants were found in GCK, HNF1A, HNF4A, HNF1B, INS, ABCC8 and KCNJ11. Eight variants were classified as benign, three as pathogenic, and one as a VUS (Table 2). The identified pathogenic variants have previously been found in MODY families.24,28
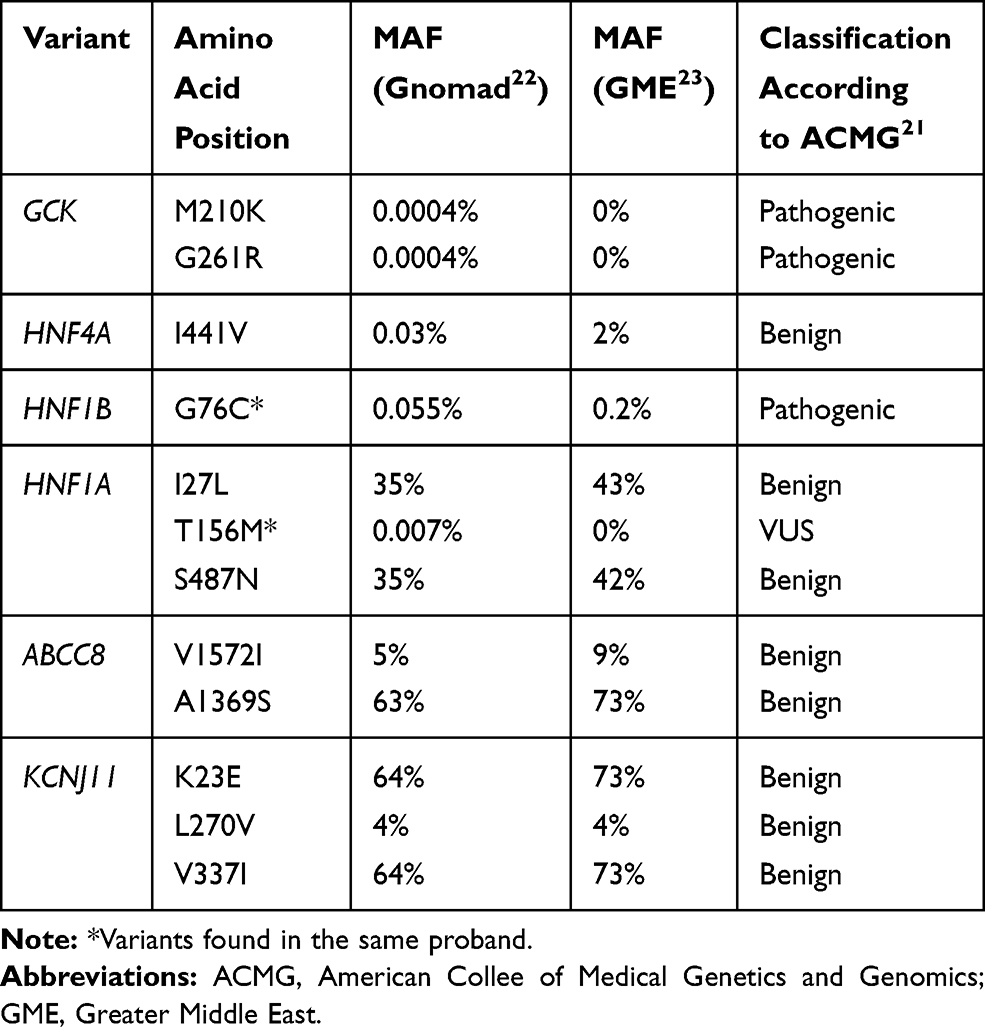 |
Table 2 Variants Identified in MODY-Genes and Their Pathogenicity Classification |
Family 4
The proband in family 4 (P4) was heterozygous for the pathogenic substitution c. 629T>A in the GCK gene, resulting in the p.Met210Lys variant. This patient was diagnosed with diabetes incidentally at age 24 years old, after pre-employment medical checkup that revealed a fasting plasma glucose of 7.11 mmol/l. Subsequent blood glucose monitoring showed that fasting plasma glucose and post-prandial glucose reached 8.89 mmol/l and 11.11 mmol/l, respectively. The patient P4 was initially treated with metformin and then with gliclazide 80 mg twice a day. However, the treatment at the latest examination was 850 mg metformin 3 times a day. HbA1c values ranged between 6.70% (49.7 mmol/mol) and 7.21% (55.3 mmol/mol). A recent oral glucose tolerance test (OGTT) (performed in 2019) showed fasting plasma glucose levels of 8.55 mmol/l and levels of 7.83 mmol/l 120 min after glucose intake.
The brother of P4 (Figure 1) was also heterozygous for this GCK variant. At diagnosis, fasting plasma glucose levels of 6.89 mmol/l were found with subsequent measures at fasting and 2-h glucose of 7.78 mmol/l and 7.72 mmol/l, respectively.
Family 12
The proband from family 12 (P12) (Figure 1) was carrying two variants: Ile441Val in HNF4A (c.1321A>G) and Gly261Arg in GCK (c.784G>A). Only the GCK Gly261Arg variant is considered pathogenic. Genetic screening of additionally two family members with diabetes from family 12 (twin sister and mother) revealed that both were carriers of the GCK Gly261Arg variant but not the HNF4A Ile441Val.
The proband of this family was born at term from non-consanguineous parents with a birth-weight of 2.95 kg. He was lethargic during infancy. In 2010 (aged 9 years old) he was admitted to the hospital for surgical intervention for hernia, and a routine laboratory test was performed which revealed impaired fasting glucose and subsequently an HbA1c value of 7.0% (53.0 mmol/mol). Insulin therapy was immediately initiated with 4x2U/day and this dosage was preserved for 8 years. The proband’s blood glucose was measured over a period of 8 years and HbA1c levels ranged between 5.4% (35.5 mmol/mol) and 7.0% (53.0 mmol/mol) and random blood glucose ranged between 4.94 and 8.89 mmol/l. During a routine visit in 2018, the patient revealed that insulin therapy had not been taken regularly for approximately one year. Yet, his fasting plasma glucose level was below 8.05 mmol/l, post-prandial values stayed below 11.11 mmol/l and fasting fingertip blood glucose values ranged between 4.17 and 7.22 mmol/l. His most recent HbA1c value was 5.5% (36.6 mmol/mol). In addition to diabetes, the patient experienced four epileptic seizures between 2017 and 2018. Three of these experienced while treated with insulin and during the most severe attack, hypoglycemia was detected with a value of 2.22 mmol/l.
The proband’s twin sister was also diagnosed with diabetes in 2010 with an HbA1c value of 7.0% (53.0 mmol/mol). Thus, insulin treatment was initiated with a dosage of 4 x 2U/day. After insulin omission, her fingertip blood glucose ranged between 5.44 and 7.94 mmol/l and her random blood glucose ranged between 6.39 and 8.33 mmol/l, except for one measure of 9.78 mmol/l during illness. Her most recent HbA1c value is 6.0% (42.1 mmol/mol). The proband’s mother was diagnosed with diabetes in 2011 and OHA treatment (metformin, glimepiride) was initiated. She has remained on this treatment regime as she has obtained a good glycemic control, with HbA1c values below 7.0% (53.0 mmol/mol). The proband’s father, elder and younger sisters all have fingertip blood glucose levels within the normal range (4.17 to 5.00 mmol/l).
Family 13
The proband in family 13 (P13) is a carrier of both a heterozygous pathogenic missense variant in HNF1B, (c. 226G>T; Gly76Cys) and the variant in HNF1A (c.467C>T; Thr156Met) with an uncertain clinical significance. The mother, maternal grandmother and maternal grandmother’s sisters are all diagnosed with diabetes (Figure 1); however, DNA and clinical information is available only for the proband.
Before diabetes onset, the proband was obese (BMI=31.7 kg/m2). He was diagnosed with diabetes at age 27, after manifesting polyuria, polydipsia, marked weight loss (~40 Kg) and ketosis. Diabetes was diagnosed based on a blood sample revealing a random blood glucose of 33.05 mmol/l. Insulin treatment was initiated. At age 29 years old (nearly 3 years after diabetes diagnosis), he was admitted to the hospital after suffering from asthenia and incoercible vomiting accompanied by ketosis. The patient’s weight was now 55 kg with a BMI of 18.4 kg/m2. Biochemical analyses revealed hypokalemia (2.5 mmol/l), increased bilirubin and liver enzymes levels (ALT, AST and GGT), negativity for GADA (Table 3), a fasting C-peptide level of 0.83 basal and 1.12 nmol/l after glucagon stimulation. In order to elucidate the etiology of the incoercible vomiting, an abdominal pelvis ultrasound and computed tomography (CT) scan was performed and the obtained imaging showed the nutcracker syndrome (Wilkie syndrome) and nephrolithiasis, without any renal, pancreatic or urogenital tracts malformations.
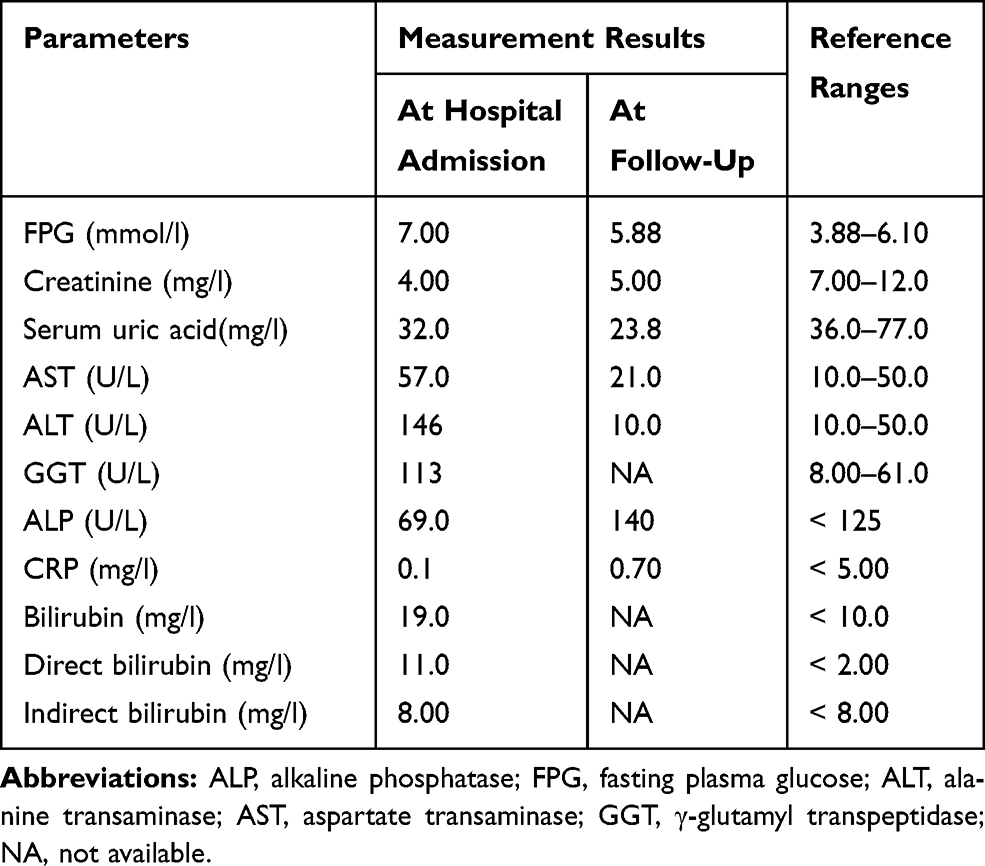 |
Table 3 Results of Biochemical Investigation of Proband P13 |
Discussion
The present study demonstrates the presence of the monogenic form of diabetes, MODY, in three out of eight (38%) Algerian North-African patients with early-onset diabetes. In our cohort, the majority of patients were initially diagnosed with T1D based on their young age at diabetes onset and a normal BMI, demonstrating the risk of MODY being misclassified as T1D and underlining the importance of genetic screening. The pathogenic variants identified have all previously been observed in MODY families24,28,29 and are therefore not specific to the Algerian population.
Two probands carrying pathogenic variants within GCK were identified and both were diagnosed incidentally and not due to the presence of diabetes symptoms. This is of little surprise, as diabetes caused by variants in GCK is characterized by only mildly elevated levels of plasma glucose.30 Several of the carriers had received insulin treatment, yet GCK-MODY patients are not responsive to oral hypoglycaemic agents such as sulphonylurea or metformin or low-dose insulin31 nor do they require treatment as GCK-MODY is not associated with progression or risk of comorbidities. Thus, cessation of treatment is recommended to the carriers of pathogenic GCK variants identified in the present study.
The GCK variant Met210Lys variant found in family P4 has previously been associated with diabetes, impaired glucose tolerance or impaired fasting glucose among related Norwegian subjects by altering glucokinase kinetic parameters.32 This is comparable to the clinical presentation in the identified family.
The second GCK variant identified in family P19, Gly261Arg, has been reported in patients with the clinical features of MODY from different populations33–41 and has previously been described in a homozygous state in patients with neonatal diabetes.42,43 Furthermore, functional studies performed by Gidh-Jain et al (1993)44 and Davis et al (1999)45 of this variant have revealed that it affects kinetic parameters. Thus, the phenotype of the twin sister and the mother of proband 12, is consistent with GCK-MODY phenotype, which is characterized by fasting plasma glucose levels ranging between 5.5 and 8.5 mmol/l, as previously established by the ISPAD consensus guidelines (International Society for Pediatric and Adolescent Diabetes).46 Discontinuation of therapy will therefore be initiated.
The Gly76Cys variant in HNF1B was present in proband P13. The clinical presentation of proband P13 is consistent with the presence of a pathogenic HNF1B-MODY variant by the presence of fluctuant elevation of ALAT, ASAT, GGT and ALP and frequent hypokalemia.47 This variant has previously been reported in a fetus with bilateral multicystic dysplasia from North African parents48 and in two probands with renal anomalies.49 This patient was also a carrier of an HNF1A-MODY variant of unknown clinical significance; thus, whether the presence of low CRP concentrations50 within this patient is a consequence of the HNF1A variant, is uncertain. As DNA was not available from the parents, it was not possible to establish whether the two variants co-segregate with diabetes, and thus causality could not be further examined. Based on the genetic diagnosis, the recommended treatment for this patient is sulphonylurea or insulin.51
Previous studies examining the prevalence of MODY, have for the majority been performed among Caucasians.16 However, it is of great importance to investigate other populations as they may identify deviating prevalences as well as clinical characteristics. Studies of HNF1A have found that 9% of South Asian MODY patients51 and 5% of Korean MODY patients are carriers of causal HNF1A variants, whereas 14% have been found in Japanese pediatric patients.52,53 For GCK, a prevalence of 2.5% were found in Korean MODY patients in contrast to 23% among Japanese pediatric patients.52–53 Thus, genetic investigation of different ethnicities is key.
The misdiagnosis observed in the present study has also previously been observed. In a study among patients stringently defined as T1D diabetes, nearly 8% had monogenic diabetes variants.55 Another study investigating the prevalence of MODY variants in antibody-negative children with diabetes found that 4.1% of these children were carrying pathogenic MODY-variants.56 These studies, as well as the present study, illustrate the importance of considering genetic testing in early-onset diabetes to initiate an appropriate intervention.
The limitation of the current study is the small number of included patients. This allows only the conclusion that common forms of MODY exist in Algerian patients with early-onset diabetes. In the present study, more than one third of the patients carried MODY variants; however, whether this prevalence is representative for a larger population cannot be determined.
Conclusion
Pathogenic variants in genes linked to common forms of MODY could explain the etiology of early-onset diabetes among Algerian patients; thus, sequencing of these genes should be considered, as the correct diagnosis can instigate accurate intervention and early diagnosis in family members.
Acknowledgments
Authors are very grateful to patients and their families for their involvement and cooperation in this study. We would also wish to thank T. H. Lorentzen for laboratory assistance and Camilla Verdich for grant management. In the memory of Pr. N. Ouadahi for his precious contribution to patient recruitment.
Author Contributions
FB, APG, TH, RR and CTB contributed to the conception and design of the study. MA, SAA, AEG, FB, BO, FL, AC and AB examined and performed the clinical evaluation of the patients in each family. FB prepared the genomic DNA samples. FB and APG performed the analysis and all authors contributed to the interpretation of data. SA and LY performed biochemical analysis and interpretation of the data. FB and APG drafted the article. Literature search was performed by FB and APG and figures were prepared by FB and APG. All authors have agreed on the journal, critically revised the manuscript, contributed to the discussion and take responsibility and are accountable for the contents of the article. The final version of the paper was read and approved by all authors.
Funding
This project received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No 667191 (authors awarded: APG, NG and TH). In addition, the study was supported by the Novo Nordisk Foundation through the Challenge Program (number 33950). The study was partially performed at The Novo Nordisk Foundation Center for Basic Metabolic Research which is an independent research center at the University of Copenhagen, partially funded by an unrestricted donation from the Novo Nordisk Foundation (www.metabol.ku.dk).
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta-cell function. Nature. 2001;414(6865):788–791. doi:10.1038/414788a
2. Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345(13):971–980. doi:10.1056/NEJMra002168
3. Owen K, Hattersley AT. Maturity-onset diabetes of the young: from clinical description to molecular genetic characterization. Best Pract Res Clin Endocrinol Metab. 2001;15(3):309–323. doi:10.1053/beem.2001.0148
4. Hattersley A, Bruining J, Shield J, Njolstad P, Donaghue KC. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2009;10(Suppl 12):33–42. doi:10.1111/j.1399-5448.2009.00571.x
5. Hattersley AT, Pearson ER. Minireview: pharmacogenetics and beyond: the interaction of therapeutic response, beta-cell physiology, and genetics in diabetes. Endocrinology. 2006;147(6):2657–2663. doi:10.1210/en.2006-0152
6. Patel KA, Kettunen J, Laakso M, et al. Heterozygous RFX6 protein truncating variants are associated with MODY with reduced penetrance. Nat Commun. 2017;8(1):888. doi:10.1038/s41467-017-00895-9
7. Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4(4):200–213. doi:10.1038/ncpendmet0778
8. Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S, Hattersley AT. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA. 2014;311(3):279–286. doi:10.1001/jama.2013.283980
9. Hansen T, Eiberg H, Rouard M, et al. Novel MODY3 mutations in the hepatocyte nuclear factor-1alpha gene: evidence for a hyperexcitability of pancreatic beta-cells to intravenous secretagogues in a glucose-tolerant carrier of a P447L mutation. Diabetes. 1997;46(4):726–730. doi:10.2337/diab.46.4.726
10. Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003;362(9392):1275–1281. doi:10.1016/S0140-6736(03)14571-0
11. Harris AG, Letourneau LR, Greeley SAW. Monogenic diabetes: the impact of making the right diagnosis. Curr Opin Pediatr. 2018;30(4):558–567. doi:10.1097/MOP.0000000000000643
12. Hattersley AT. Molecular genetics goes to the diabetes clinic. Clin Med. 2005;5(5):476–481. doi:10.7861/clinmedicine.5-5-476
13. Vaxillaire M, Froguel P. Genetic basis of maturity-onset diabetes of the young. Endocrinol Metab Clin North Am. 2006;35(2):371–384. doi:10.1016/j.ecl.2006.02.009
14. Fajans SS, Bell GI. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care. 2011;34(8):1878–1884. doi:10.2337/dc11-0035
15. Delvecchio M, Ludovico O, Menzaghi C, et al. Low prevalence of HNF1A mutations after molecular screening of multiple MODY genes in 58 Italian families recruited in the pediatric or adult diabetes clinic from a single Italian hospital. Diabetes Care. 2014;37(12):e258–60. doi:10.2337/dc14-1788
16. Pihoker C, Gilliam LK, Ellard S, et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for diabetes in youth. J Clin Endocrinol Metab. 2013;98(10):4055–4062. doi:10.1210/jc.2013-1279
17. Lamri L, Gripiotis E, Ferrario A. Diabetes in Algeria and challenges for health policy: a literature review of prevalence, cost, management and outcomes of diabetes and its complications. Global Health. 2014;10:11. doi:10.1186/1744-8603-10-11
18. Bouldjennet F, Hireche A, Kechout N, et al. Clinical characteristics of Algerian subjects with MODY p. R85W glucokinase mutation-in silico assessment of p. R85W effect on glucokinase structure and function. Meta Gene. 2019;19:268–275. doi:10.1016/j.mgene.2018.12.013
19. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:3. doi:10.1093/nar/16.3.1215
20. Gao R, Liu Y, Gjesing AP, et al. Evaluation of a target region capture sequencing platform using monogenic diabetes as a study-model. BMC Genet. 2014;15:13. doi:10.1186/1471-2156-15-13
21. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
22. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291.
23. The Greater Middle East (GME) Variome Project. 2020. Available from: http://igm.ucsd.edu/gme/index.php.
24. Osbak KK, Colclough K, Saint-Martin C, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30(11):1512–1526. doi:10.1002/humu.21110
25. Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE, Ellard S. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat. 2013;34(5):669–685.
26. De Franco E, Saint-Martin C, Brusgaard K, et al. Update of variants identified in the pancreatic beta-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum Mutat. 2020;41(5):884–905. doi:10.1002/humu.23995
27. Glumer C, Jorgensen T, Borch-Johnsen K, Inter S. Prevalences of diabetes and impaired glucose regulation in a Danish population: the Inter99 study. Diabetes Care. 2003;26(8):2335–2340. doi:10.2337/diacare.26.8.2335
28. Anik A, Catli G, Abaci A, et al. Molecular diagnosis of maturity-onset diabetes of the young (MODY) in Turkish children by using targeted next-generation sequencing. J Ped Endocrinol Metabol. 2015;28(11–12):1265–1271. doi:10.1515/jpem-2014-0430
29. Bellanne-Chantelot C, Clauin S, Chauveau D, et al. Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes. 2005;54(11):3126–3132. doi:10.2337/diabetes.54.11.3126
30. Stride A, Vaxillaire M, Tuomi T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia. 2002;45(3):427–435. doi:10.1007/s00125-001-0770-9
31. Stride A, Shields B, Gill-Carey O, et al. Cross-sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia. 2014;57(1):54–56. doi:10.1007/s00125-013-3075-x
32. Njolstad PR, Sovik O, Cuesta-Munoz A, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344(21):1588–1592. doi:10.1056/NEJM200105243442104
33. Stoffel M, Froguel P, Takeda J, et al. Human glucokinase gene: isolation, characterization, and identification of two missense mutations linked to early-onset non-insulin-dependent (type 2) diabetes mellitus. Proc Natl Acad Sci U S A. 1992;89(16):7698–7702. doi:10.1073/pnas.89.16.7698
34. Sakura H, Eto K, Kadowaki H, et al. Structure of the human glucokinase gene and identification of a missense mutation in a Japanese patient with early-onset non-insulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1992;75(6):1571–1573.
35. Velho G, Blanche H, Vaxillaire M, et al. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia. 1997;40(2):217–224. doi:10.1007/s001250050666
36. Massa O, Meschi F, Cuesta-Munoz A, et al. High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI. Diabetologia. 2001;44(7):898–905. doi:10.1007/s001250100530
37. Thomson KL, Gloyn AL, Colclough K, et al. Identification of 21 novel glucokinase (GCK) mutations in UK and European Caucasians with maturity-onset diabetes of the young (MODY). Hum Mutat. 2003;22(5):417. doi:10.1002/humu.9186
38. Cuesta-Munoz AL, Tuomi T, Cobo-Vuilleumier N, et al. Clinical heterogeneity in monogenic diabetes caused by mutations in the glucokinase gene (GCK-MODY). Diabetes Care. 2010;33(2):290–292. doi:10.2337/dc09-0681
39. Costantini S, Malerba G, Contreas G, et al. Genetic and bioinformatics analysis of four novel GCK missense variants detected in Caucasian families with GCK-MODY phenotype. Clin Genet. 2015;87(5):440–447. doi:10.1111/cge.12406
40. Carmody D, Naylor RN, Bell CD, et al. GCK-MODY in the US National Monogenic Diabetes Registry: frequently misdiagnosed and unnecessarily treated. Acta Diabetol. 2016;53(5):703–708. doi:10.1007/s00592-016-0859-8
41. Wang Z, Diao C, Liu Y, et al. Identification and functional analysis of GCK gene mutations in 12 Chinese families with hyperglycemia. J Diabetes Investig. 2019;10(4):963–971. doi:10.1111/jdi.13001
42. Bennett K, James C, Mutair A, Al-Shaikh H, Sinani A, Hussain K. Four novel cases of permanent neonatal diabetes mellitus caused by homozygous mutations in the glucokinase gene. Pediatr Diabetes. 2011;12(3 Pt 1):192–196. doi:10.1111/j.1399-5448.2010.00683.x
43. Raimondo A, Chakera AJ, Thomsen SK, et al. Phenotypic severity of homozygous GCK mutations causing neonatal or childhood-onset diabetes is primarily mediated through effects on protein stability. Hum Mol Genet. 2014;23(24):6432–6440. doi:10.1093/hmg/ddu360
44. Gidh-Jain M, Takeda J, Xu LZ, et al. Glucokinase mutations associated with non-insulin-dependent (type 2) diabetes mellitus have decreased enzymatic activity: implications for structure/function relationships. Proc Natl Acad Sci U S A. 1993;90(5):1932–1936.
45. Davis EA, Cuesta-Munoz A, Raoul M, et al. Mutants of glucokinase cause hypoglycaemia- and hyperglycaemia syndromes and their analysis illuminates fundamental quantitative concepts of glucose homeostasis. Diabetologia. 1999;42(10):1175–1186. doi:10.1007/s001250051289
46. Hattersley A, Bruining J, Shield J, Njolstad P, Donaghue K. ISPAD clinical practice consensus guidelines 2006-2007. The diagnosis and management of monogenic diabetes in children. Pediatr Diabetes. 2006;7(6):352–360. doi:10.1111/j.1399-5448.2006.00217.x
47. Chen YZ, Gao Q, Zhao XZ, et al. Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chin Med J. 2010;123(22):3326–3333.
48. Madariaga L, Moriniere V, Jeanpierre C, et al. Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin j Am Soc Nephrol. 2013;8(7):1179–1187. doi:10.2215/CJN.10221012
49. Ulinski T, Lescure S, Beaufils S, et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol. 2006;17(2):497–503. doi:10.1681/ASN.2005101040
50. Juszczak A, Pavic T, Vuckovic F, et al. Plasma Fucosylated Glycans and C-Reactive protein as biomarkers of HNF1A-MODY in young adult-onset nonautoimmune diabetes. Diabetes Care. 2019;42(1):17–26. doi:10.2337/dc18-0422
51. Urakami T. Maturity-onset diabetes of the young (MODY): current perspectives on diagnosis and treatment. Diabetes Metabol Syndr Obesity. 2019;12:1047–1056. doi:10.2147/DMSO.S179793
52. Radha V, Ek J, Anuradha S, Hansen T, Pedersen O, Mohan V. Identification of novel variants in the hepatocyte nuclear factor-1alpha gene in South Indian patients with maturity onset diabetes of young. J Clin Endocrinol Metab. 2009;94(6):1959–1965. doi:10.1210/jc.2008-2371
53. Yorifuji T, Fujimaru R, Hosokawa Y, et al. Comprehensive molecular analysis of Japanese patients with pediatric-onset MODY-type diabetes mellitus. Pediatr Diabetes. 2012;13(1):26–32. doi:10.1111/j.1399-5448.2011.00827.x
54. Hwang JS, Shin CH, Yang SW, Jung SY, Huh N. Genetic and clinical characteristics of Korean maturity-onset diabetes of the young (MODY) patients. Diabetes Res Clin Pract. 2006;74(1):75–81. doi:10.1016/j.diabres.2006.03.002
55. Yu MG, Keenan HA, Shah HS, et al. Residual beta cell function and monogenic variants in long-duration type 1 diabetes patients. J Clin Invest. 2019;129(8):3252–3263. doi:10.1172/JCI127397
56. Johansson BB, Irgens HU, Molnes J, et al. Targeted next-generation sequencing reveals MODY in up to 6.5% of antibody-negative diabetes cases listed in the Norwegian Childhood Diabetes Registry. Diabetologia. 2017;60(4):625–635. doi:10.1007/s00125-016-4167-1
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.