")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Mathematical modeling of postcoinfection with influenza A virus and Streptococcus pneumoniae, with implications for pneumonia and COPD-risk assessment
Authors Cheng YH , You SH , Lin YJ, Chen SC , Chen WY , Chou WC, Hsieh NH, Liao CM
Received 29 March 2017
Accepted for publication 5 June 2017
Published 5 July 2017 Volume 2017:12 Pages 1973—1988
DOI https://doi.org/10.2147/COPD.S138295
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Yi-Hsien Cheng,1 Shu-Han You,2 Yi-Jun Lin,3 Szu-Chieh Chen,4,5 Wei-Yu Chen,6 Wei-Chun Chou,2 Nan-Hung Hsieh,7 Chung-Min Liao3
1Institute of Computational Comparative Medicine (ICCM), Department of Anatomy and Physiology, College of Veterinary Medicine, Kansas State University, Manhattan, KS, USA; 2National Institute of Environmental Health Sciences, National Health Research Institutes, Zhunan, 3Department of Bioenvironmental Systems Engineering, National Taiwan University, Taipei, 4Department of Public Health, 5Department of Family and Community Medicine, Chung Shan Medical University Hospital, Taichung, 6Department of Biomedical Science and Environmental Biology, Kaohsiung Medical University, Kaohsiung, Taiwan; 7Department of Veterinary Integrative Biosciences, College of Veterinary Medicine and Biomedical Sciences, Texas A&M University, College Station, TX, USA
Background: The interaction between influenza and pneumococcus is important for understanding how coinfection may exacerbate pneumonia. Secondary pneumococcal pneumonia associated with influenza infection is more likely to increase respiratory morbidity and mortality. This study aimed to assess exacerbated inflammatory effects posed by secondary pneumococcal pneumonia, given prior influenza infection.
Materials and methods: A well-derived mathematical within-host dynamic model of coinfection with influenza A virus and Streptococcus pneumoniae (SP) integrated with dose–response relationships composed of previously published mouse experimental data and clinical studies was implemented to study potentially exacerbated inflammatory responses in pneumonia based on a probabilistic approach.
Results: We found that TNFα is likely to be the most sensitive biomarker reflecting inflammatory response during coinfection among three explored cytokines. We showed that the worst inflammatory effects would occur at day 7 SP coinfection, with risk probability of 50% (likely) to develop severe inflammatory responses. Our model also showed that the day of secondary SP infection had much more impact on the severity of inflammatory responses in pneumonia compared to the effects caused by initial virus titers and bacteria loads.
Conclusion: People and health care workers should be wary of secondary SP infection on day 7 post-influenza infection for prompt and proper control-measure implementation. Our quantitative risk-assessment framework can provide new insights into improvements in respiratory health especially, predominantly due to chronic obstructive pulmonary disease (COPD).
Keywords: chronic obstructive pulmonary disease, pneumonia, influenza, coinfection, modeling, risk assessment
Introduction
The upper respiratory tract is simultaneously exposed to a vast range of potential pathogenic bacteria and viruses. A well-recognized viral–bacterial coinfection in the lung is the synergy between influenza virus and Streptococcus pneumoniae (SP; or pneumococcus).1–5 Influenza can suppress the immune response to SP infection and also result in upregulation of pneumococcal adhesion molecules, leading to increased pneumococcal load and decreased survival.1,2,5,6 Therefore, secondary pneumococcal pneumonia might lead to an increase in respiratory morbidity and mortality (predominantly due to chronic obstructive pulmonary disease [COPD]) associated with influenza infections.5,7
McCullers1 indicated that although an influenza infection alone can be severe, mortality increases significantly when a bacterial superinfection occurs, as in the 1918 influenza pandemic, when millions of people died, mostly from secondary pneumococcal pneumonia. Chertow and Memoli5 indicated the clinical course and microbiology of severe influenza and bacterial coinfection. The bacterial coinfection commonly occurs within the first 6 days of influenza infection and presents similarly to influenza infection occurring alone, but with an increased risk of death.5 Chertow and Memoli5 further indicated that bacterial coinfection complicated up to 34% of 2009 pandemic influenza A virus (IAV; H1N1) infections managed in intensive care units worldwide. Globally, more than 3.1 million deaths attributable to influenza and pneumonia occur annually.8 Therefore, it is critical to quantify and understand the risk of influenza and SP coinfection and initiate potentially effective control measures and treatment strategies, as well as clinical care.
Several mechanisms have been proposed to explain the synergistic interactions observed in virus–bacteria coinfection. Influenza has been shown to increase bacterial adherence or proliferation by disruption of the epithelial barrier, production of viral neuraminidase,4 and reduction in the efficiency of the immune system, such as induced neutrophil apoptosis and dysfunction,9 as well as depressed phagocytic activity in macrophages through the expression of different cytokines (eg, IFNα/β,10 IFNγ,11 TNFα,12 and IL1013). On the other hand, the presence of bacteria might facilitate subsequent viral infection by secreting proteases that activate the infectivity of influenza14 and producing bacterial neuraminidase to enhance the release of virions.15
Shrestha et al16 developed mathematical within-host or transmission-dynamic models to characterize the nature, timing, and magnitude of coinfection interaction. In particular, they developed an “SIRS” compartmental model with influenza incidences incorporated for characterizing pneumococcal transmission where S, I, and R represent susceptible, infectious, and recovered individuals, respectively. They indicated a short-lived but strong interaction (~100-fold) of increased susceptibility to pneumococcal pneumonia postinfluenza infection. Meanwhile, they derived an immunomediated model quantifying virus–bacteria interaction and proposed advice on clinical management that antiviral treatment should be administered no later than 4 days after influenza infection.17 In addition, Smith et al15,18 established a virus–bacteria–host kinetic model to investigate interaction during coinfection based on various bacterial inoculum sizes (100 CFU versus 1,000 CFU D39) and virus strains (PR8 versus PR8-PB1-F2[1918]).
Collectively, based on the aforementioned synergistic mechanisms, well-established mathematical within-host dynamic models, and review articles regarding in vivo experiments,1,4 the modeling of secondary SP coinfection from influenza reveals three key ways where coinfection can affect transmission and disease: 1) each infection alters the ability of the immune system to mount an adequate immune response to the other infection, 2) morbidity and mortality of coinfection can be independent of pathogen burden or excessive inflammatory response, and 3) symptoms and effects of secondary pathogen coinfection may be exacerbated compared to primary pathogen infection. Therefore, understanding how coinfection affects the dynamics and control of important infectious diseases has become increasingly significant. Although changes in the course of infection of coinfected individuals have been observed, the mechanisms of how transmission is altered are poorly understood.
In this study, our modeling framework is based on the mathematical within-host dynamics of coinfection with IAV and SP developed in Smith et al.15 When and what causes the increased pneumonia-exacerbation risk from SP coinfection in influenza infection, however, is not well quantified or assessed. Therefore, explicitly modeling the interaction between influenza and SP is important for understanding how coinfection may impact the incidence, copathogenesis, and efficacy of clinical management, which relies on measures to prevent, diagnose, and treat both influenza and bacterial infection. The purpose of this study is threefold: 1) to provide an integrated probabilistic risk-assessment framework to quantify exacerbated risks in inflammatory responses for secondary pneumococcal pneumonia patients coinfected with influenza, 2) to investigate coinfection interactions between influenza and SP through identifying variations in cytokine level reflecting severity in inflammatory response of three biomarkers, and 3) to conduct the risk assessment for patients with COPD who develop the coinfection of influenza and pneumonia.
Materials and methods
Study framework
The study framework for quantifying IAV and SP coinfection-associated inflammation exacerbation-risk assessment is demonstrated in Figure 1. Briefly, we started with searching previously published mouse experimental studies related to IAV and SP coinfection and epidemiological studies regarding IAV infection-associated pneumonia to understand the potential health risks of IAV-SP coinfection (Figure 1A). This study then implemented a mathematical within-host coinfection dynamic model to quantify virus dynamically, as well as bacterial burden in mice (Figure 1B). Parameterization and uncertainty analysis of a coinfection dynamic model is carried out as well (Figure 1C). Dose–response relationships between transferred bacterial loads and normalized inflammatory effects can then be established for further implementation and implications for pneumonia and COPD-risk assessment (Figure 1D–F).
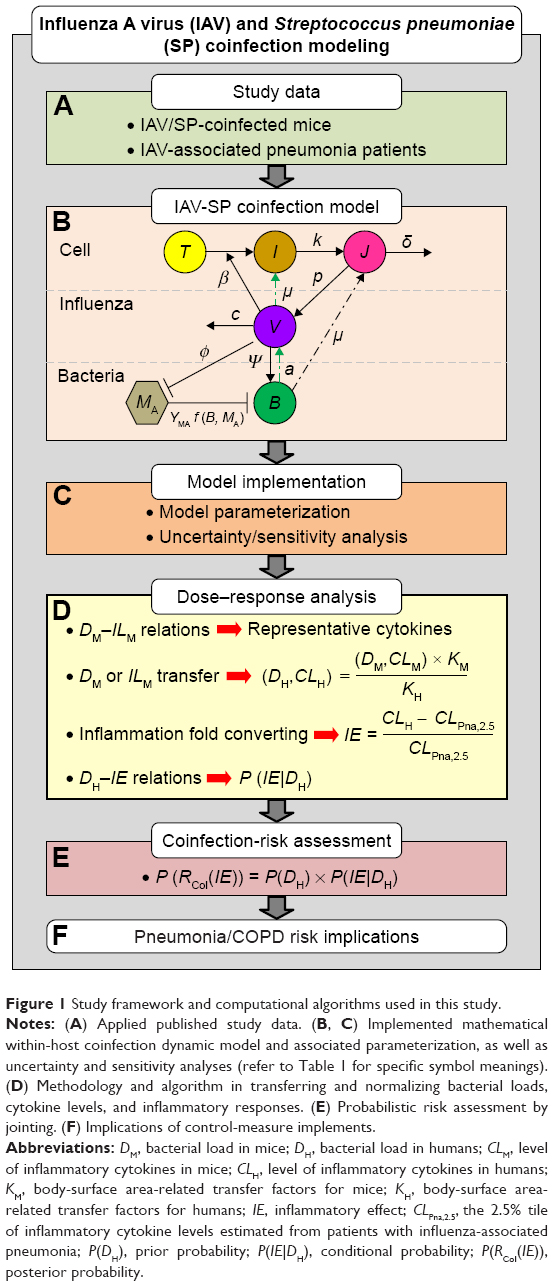 | Figure 1 Study framework and computational algorithms used in this study. |
Study data
There were sporadic human epidemiological data relating to IAV-SP coinfection. Therefore, we principally collated IAV-SP coinfection experimental data based on mouse models from previously published studies. Table S1 summarizes the data extracted from published mouse-coinfection literature covering ages and species (such as C57BL/6 and BALB/c mice), as well as inoculated strains and doses of virus and bacteria. Moreover, data in Table S1 also indicate explicitly the infection in emphysematous lung. We then extrapolated bacterial loads and corresponding cytokine levels in mice to those in humans with caution.
Based on a previously published in vivo coinfection assay, mice were infected intranasally with IAV on day 0 with doses of 1.4–1.8×105 50% tissue culture infective dose (TCID50)·mL−1 and then challenged with SP of 102–107 CFU·mL−1 at 3–14 days post-IAV infection. Following virus and bacterial inoculation, mice were anesthetized and killed at 12–72 hours post-IAV infection, and cytokine levels of IL6, IL10, keratinocyte chemoattractant (KC), TNFα, and IFNγ in supernatants extracted from lung homogenates were quantified by either enzyme-linked immunosorbent assay or such instruments as Luminex readers.
To select representative cytokines for studying bacterial load-dependent inflammation, we established preliminary linear models. As we aimed at constructing best-fit linear models, some data points potentially presenting as outliers relative to linear models were excluded; therefore, only a few studies were explored (see Figure S1 and Table S1). Additionally, both criteria, including coefficient of determination (r2>0.8) and P-value (P<0.05), were adopted simultaneously to select representative cytokines, ie, IL6, TNFα, and IFNγ (Figure S1).
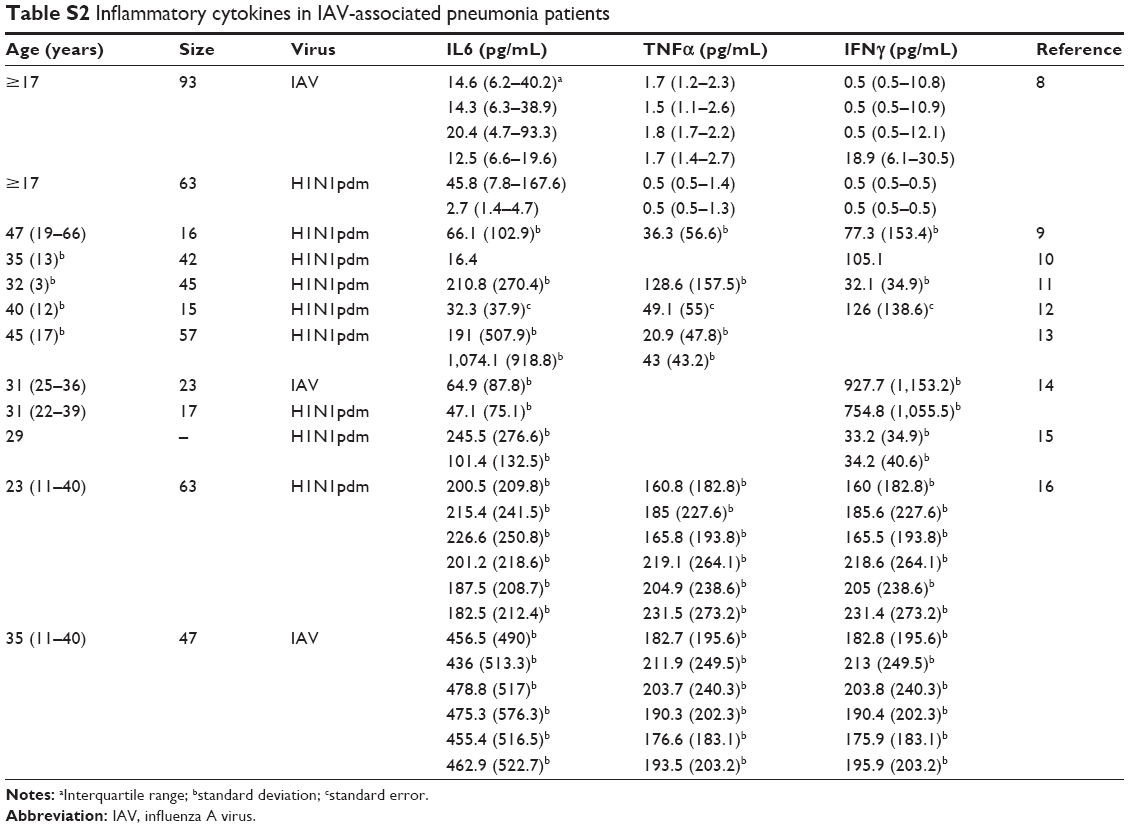
On the other hand, epidemiological data of IAV infection-associated pneumonia (Pna) patients were selected carefully for determining the severity of inflammatory effects (Table S2). Briefly, nasopharyngeal swab samples were collected from hospitalized patients for RNA isolation using a viral RNA minikit.19 Moreover, inflammatory cytokine levels were measured from serum samples collected from all Pna patients. Note that we chose three representative inflammatory cytokines existing in mice and humans – IFNγ, TNFα, and IL6 – for quantifying inflammatory effects.
Coinfection transmission-dynamic modeling
A previously well-developed IAV-SP coinfection model15 capturing three population phases – epithelial cells, virus (V), and bacteria (B) – was adopted to explore the contribution of secondary infection with SP to the interaction dynamics between influenza and pneumococcus (Figure 1B). Briefly, uninfected epithelial cells (T) would become virus-productive infected cells (J) by influenza virus (V) after 6–8 hours latency of unproductive infected cells (I). The consequences of coinfection can be quantified by four major mechanisms of virus–bacteria–immune system interactions (Figure 1B): 1) the viral infection can enhance bacterial adherence to epithelial cells, resulting in elevated bacteria-carrying capacity; 2) the carrying capacity can weaken the ability of alveolar macrophages to phagocytize bacteria; 3) with the presence of bacteria, which can synergistically promote virion release; and 4) released virions can increase epithelial cell death from bacterial adherence. The details of model equations describing the dynamics of IAV inoculation and transmission, IAV transmission postcoinfection with SP, and SP transmission given coinfection are given in Table 1. The simulation of IAV-SP coinfection-transmission dynamics was performed with Berkeley Madonna (version 8.0.1; University of California at Berkeley, Berkeley, CA, USA).
 | Table 1 Descriptions of governing equations used for influenza A virus and Streptococcus pneumoniae coinfection dynamic model15 |
To assess the performance of model predictability (ie, model validation) for mice coinfected with IAV and SP, the mean absolute percentage error (MAPE) was calculated as 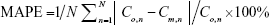 where N denotes the number of observations, Co,n the observed experimental data, and Cm,n the modeled result corresponding to data point n.
where N denotes the number of observations, Co,n the observed experimental data, and Cm,n the modeled result corresponding to data point n.
Dose–response analysis
To extrapolate the mouse doses, including bacteria load (DM) and inflammatory cytokine level (CLM), to human doses (DH and CLH) due to coinfection with IAV and SP, we applied the concept of human equivalent dose (HED) to transfer doses of mice into those of humans.20 Here, DH or CLH can be calculated accordingly as
|
|
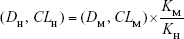
where KM and KH are the body-surface area-related transfer factors for mice and humans, respectively, of 3 and 37·kg m−2.20
Due to lack of information regarding the combinative impact of coinfection with IAV and SP on human inflammatory responses, we thus normalized Pna-associated inflammatory effect (IE) to capture the level of response in Pna patients potentially coinfected with IAV followed by SP as
|
|
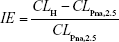
where CLH is the level of inflammatory cytokines in humans extrapolated from mice and CLPna,2.5 the percentile 2.5 of inflammatory cytokine levels (eg, IL6, TNFα, and IFNγ) estimated from patients with influenza-associated Pna as the baseline values representing coinfection, since there is no such information for cytokine values within coinfected Pna patients.
Dose–response relationships between DH and IE can then be constructed by fitting those estimated values with the most suitable equations, resulting in a conditional probability of IE given all probable DH taken into account (ie, P[IE|DH]). TableCurve 2D (version 5.01; AISN Software, Mapleton, OR, USA) was used to fit the published epidemiological data to determine the governing dose–response relationships.
Probabilistic risk assessment
Bacteria load in mouse lungs estimated from the IAV-SP coinfection dynamic-transmission model can be further transformed into bacteria load in human lungs (DH). Taking potential factors (eg, certain immune responses, underlying coinfection mechanisms, and different days of SP inoculation post-IAV infection) influencing coinfection dynamics into consideration would obtain a probability density function (ie, the prior probability (P[DH])). We then linked P(DH) with P(IE|DH) by Bayesian inference to construct inflammatory risk profiles related to coinfection-induced Pna, resulting in a joint probability function (ie, posterior probability) that can be expressed mathematically as
|
|
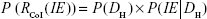
To better understand the likely excess exacerbations risks of coinfection, we used the maximum probability of 1 to subtract the cumulative risk profile, ie, 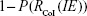 , leaving an exceedance-risk profile, indicating that there is a certain probability for inflammatory responses to exceed such a level. Moreover, to quantify extents of coinfection-exacerbated inflammation, this study defined a tertiary inflammatory response as mild (I), moderate (II), and severe (III) according to lower- and upper-tertile estimates (33rd and 67th) in probability density functions for cytokine levels of IL6, TNFα, and IFNγ from influenza-infected Pna patients. The 33rd- and 67th-percentile estimates of cytokine levels can then be transformed into inflammation folds to represent classes in inflammatory response based on our normalization methodology described.
, leaving an exceedance-risk profile, indicating that there is a certain probability for inflammatory responses to exceed such a level. Moreover, to quantify extents of coinfection-exacerbated inflammation, this study defined a tertiary inflammatory response as mild (I), moderate (II), and severe (III) according to lower- and upper-tertile estimates (33rd and 67th) in probability density functions for cytokine levels of IL6, TNFα, and IFNγ from influenza-infected Pna patients. The 33rd- and 67th-percentile estimates of cytokine levels can then be transformed into inflammation folds to represent classes in inflammatory response based on our normalization methodology described.
Uncertainty and sensitivity analysis
A Monte Carlo (MC) simulation technique was implemented to generate percentiles 2.5 and 97.5 as 95% CIs for quantifying the uncertainty of parameters and inoculation day-varied bacteria loads based on the IAV-SP coinfection dynamic model. Kolmogorov–Smirnov goodness-of-fit statistics were used to determine the optimal distributions for parameters and bacteria loads.
We also applied the MC simulation to quantify the uncertainty and its impact on the estimations of expected inflammatory risks. The MC simulation was performed with 10,000 iterations to ensure the stability of results. Crystal Ball software (version 2000.2; Decisioneering, Denver, CO, USA) was employed to implement the MC simulation. Moreover, this study employed a one-way sensitivity analysis to assess the contribution of change in each parameter used in the IAV-SP coinfection dynamic model at one time across its 95% CI to the simulation outcome.
Results
IAV-SP coinfection dynamics
We incorporated V(0) =2 TCID50·mL−1, B(0) =100 CFU·mL−1 at day 7 coinfection and the essential input-parameter values (Table 2) derived from Smith et al into the IAV-SP coinfection model15 to capture the interaction dynamics among epithelial cells, virus, and bacteria (Figure 2). Our results showed that unproductive infected cells (I) and virus-productive infected cells (J) increased significantly and peaked approximately at day 3 after IAV infection (Figure 2A). Meanwhile, peak activity was observed in virus with 6.7 log TCID50·mL−1 (Figure 2B). By contrast, as the peak of virus titer approached, uninfected epithelial cells (T) decreased dramatically by virtue of virion infection (Figure 2A).
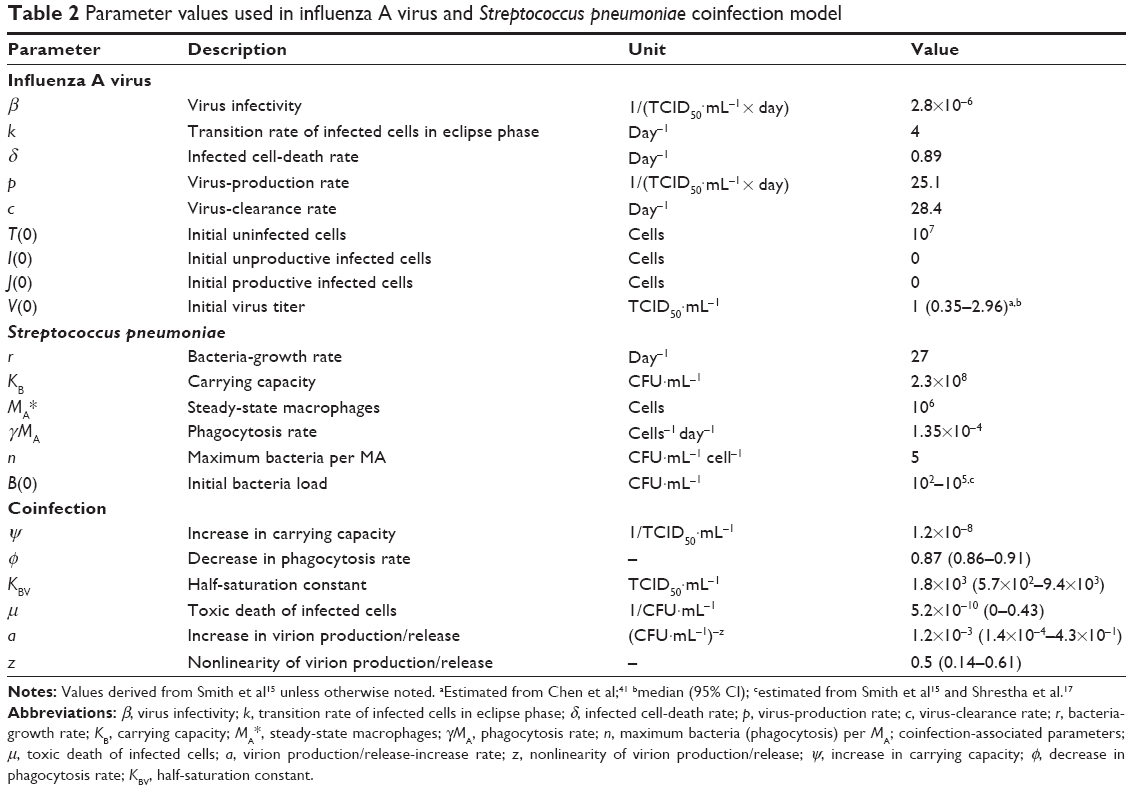 | Table 2 Parameter values used in influenza A virus and Streptococcus pneumoniae coinfection model |
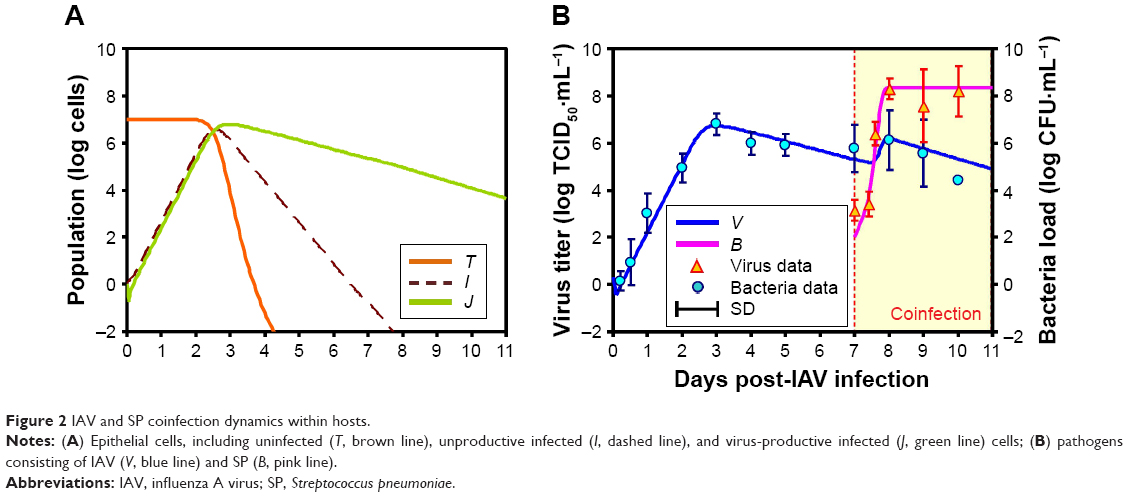 | Figure 2 IAV and SP coinfection dynamics within hosts. |
After day 7, while bacterial inoculation was occurring, a second virus titer peak was observed around day 8, with a slightly lower concentration of 6.2 log TCID50·mL−1 compared to the first peak (Figure 2B). We also found that bacteria load increased rapidly and reached the maximum value of 8.4 log CFU·mL−1, even though viral titers decreased continuously. Comparisons between predictions by the IAV-SP coinfection model and observed experimental data are demonstrated in Figure 2B, indicating that predictions were in apparent agreement with data derived from Smith et al15 for virus titer (MAPE 31.64%) and bacteria load (MAPE 14.19%), respectively.
Inflammatory effects on postcoinfection
Table 3 lists bacteria loads and inflammatory cytokine levels of IL6, TNFα, and IFNγ of mice and humans (extrapolated based on Tables S1 and S2), as well as the inflammatory responses obtained by normalizing with percentile-2.5 estimates of cytokine levels (CLPna,2.5) detected from IAV-infection Pna patients. Here, CLPna,2.5 estimates of IL6, TNFα, and IFNγ were 5.67, 0.41, and 0.23 pg·mL−1, respectively. The estimated bacteria loads in human (DHs) were highest for IL6 at 6.12±0.99, followed by TNFα at 5.74±1.44 and IFNγ at 5.71±2.73 log CFU·mL−1. On the other hand, the extrapolated cytokine levels (CLHs) for IL6, TNFα, and IFNγ were, respectively, estimated to be 2.88±0.28, 2.74±0.4, and 1.51±0.47 log pg·mL−1 (Figure 3, Table 3).
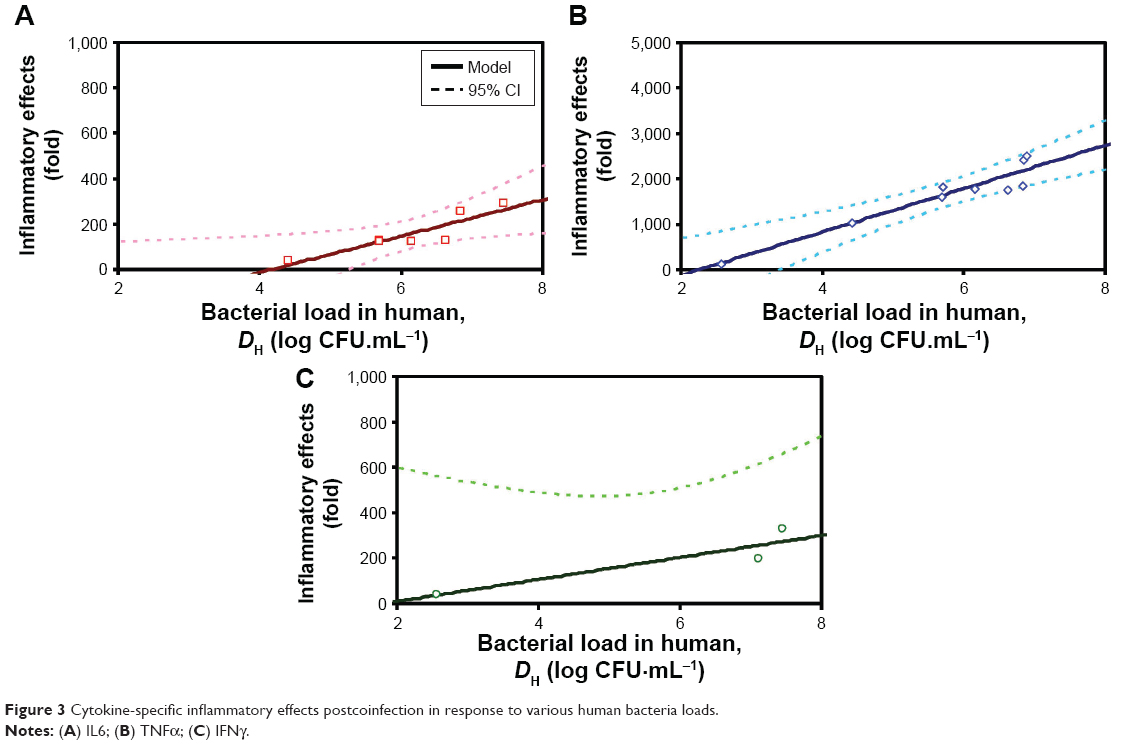 | Figure 3 Cytokine-specific inflammatory effects postcoinfection in response to various human bacteria loads. |
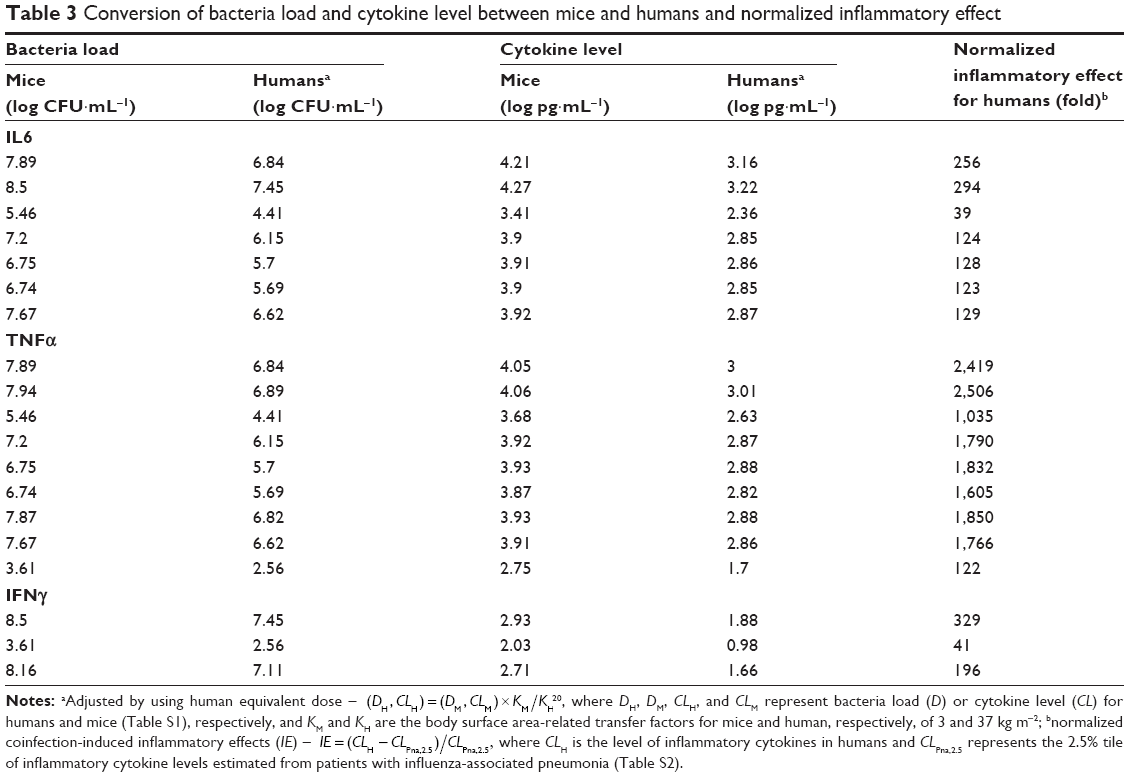 | Table 3 Conversion of bacteria load and cytokine level between mice and humans and normalized inflammatory effect |
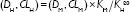 , where DH, DM, CLH, and CLM represent bacteria load (D) or cytokine level (CL) for humans and mice (
, where DH, DM, CLH, and CLM represent bacteria load (D) or cytokine level (CL) for humans and mice ( , where CLH is the level of inflammatory cytokines in humans and CLPna,2.5 represents the 2.5% tile of inflammatory cytokine levels estimated from patients with influenza-associated pneumonia (
, where CLH is the level of inflammatory cytokines in humans and CLPna,2.5 represents the 2.5% tile of inflammatory cytokine levels estimated from patients with influenza-associated pneumonia (Our results revealed that the simple linear equations were found to be adequate for capturing the relationships between log-transferred DH and IE for IAV infection Pna patients. Specifically, the simple linear equation fitted well with the DH–IE relationships of TNFα (IE =477DH −1,079, r2=0.91; P<0.001) (Figure 3B) and IL6 (IE =80DH −335, r2=0.81; P<0.01) (Figure 3A) compared to the relationship of IFNγ (IE =48DH −87, r2=0.83; P=0.09) (Figure 3C). We thus found that human bacteria loads in lungs were positively correlated with inflammatory responses. We also observed that given the same level increases in DH, TNFα displayed the significantly severer inflammatory responses compared to IL6 and IFNγ, indicating that it was likely to be the most sensitive biomarker for coinfection-exacerbated inflammation among the three cytokines explored in this study.
Pneumonia exacerbation-risk estimates
Results of sensitivity analyses demonstrated that changes in each coinfection parameter across its median and 95% CI contributed approximately the same to the mouse-coinfection dynamics at day 7 postcoinfection, resulting in an average bacteria load of 1.78×108 CFU·mL−1 with 95% CI of 1.02–36.35×107 CFU·mL−1 (Figure 4). On the other hand, changes in different levels of IAV and SP inoculation did not impact much on bacteria load, given the same day of SP inoculation post-IAV infection considered, except for initial virus titer (V0) of 0.35 TCID50·mL−1 and initial bacteria load (B0) of 102 CFU·mL−1 with SP inoculating at days 1 and 9, respectively (Figure 5A and B). Moreover, we recognized that days of secondary SP inoculation (ie, days 1, 7, and 9) impacted more on coinfection than that by considering various initial IAV (0.35–1.45 TCID50·mL−1) and SP doses (102–105 CFU·mL−1) (Figure 5C). We also found SP inoculation at day 7 post-IAV infection would lead to the highest bacteria loads of 21.98±4.42×107 CFU·mL−1 (ie, susceptibility to SP inoculation) compared to SP inoculation at day 1 or 9 (Figure 5C).
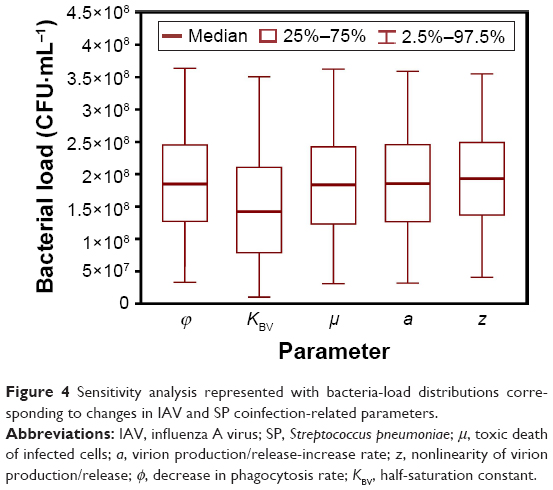 | Figure 4 Sensitivity analysis represented with bacteria-load distributions corresponding to changes in IAV and SP coinfection-related parameters. |
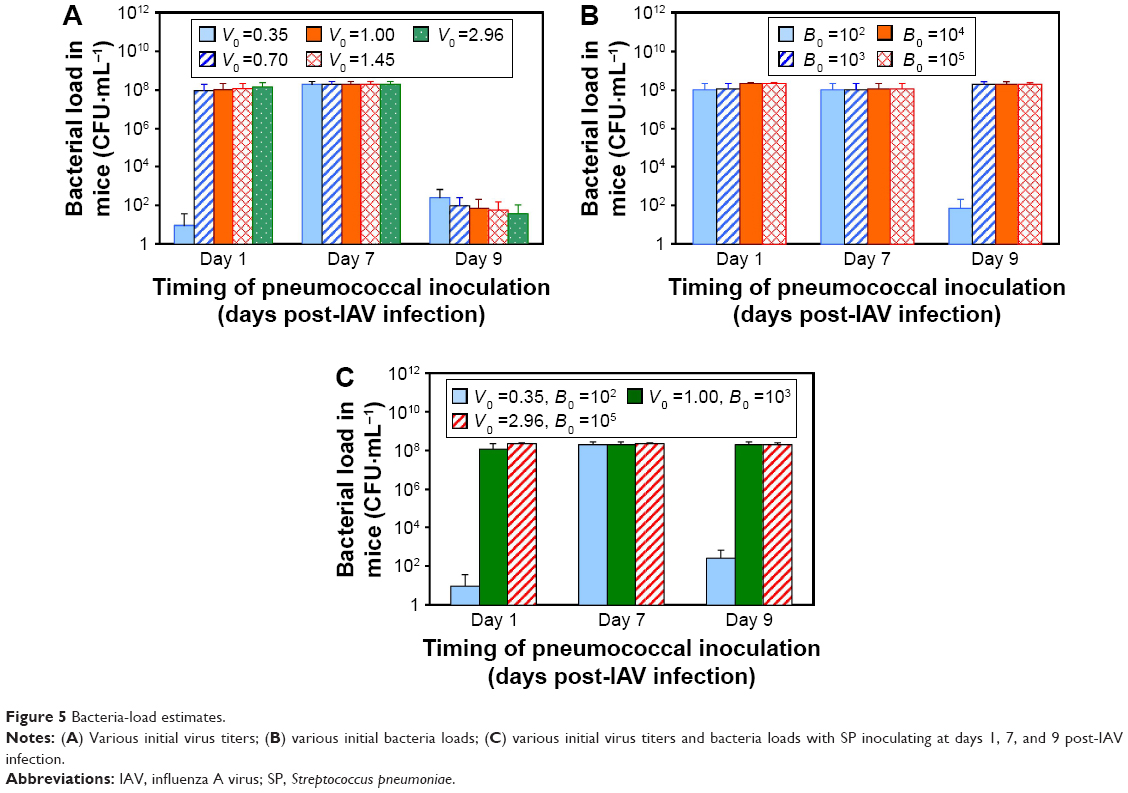 | Figure 5 Bacteria-load estimates. |
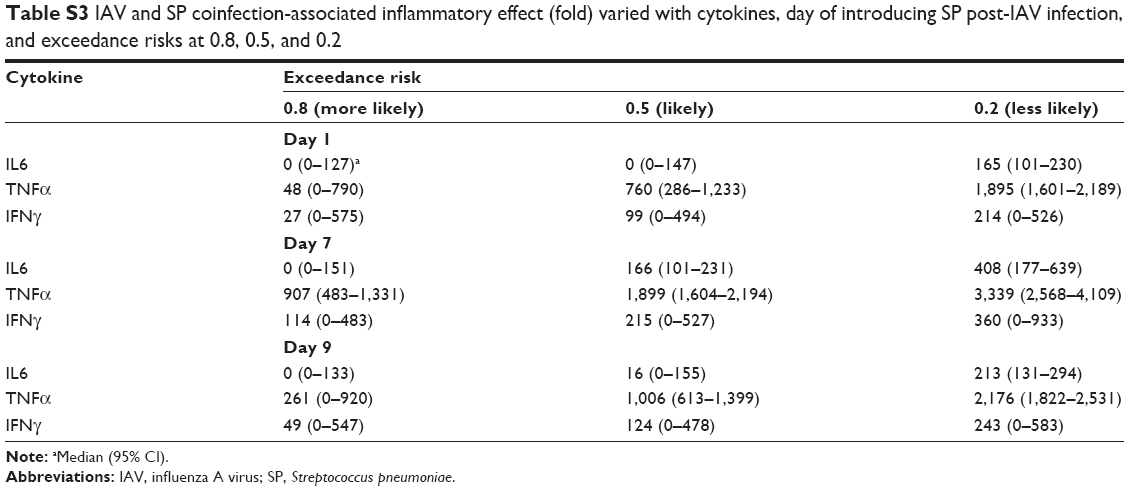
This study hence exclusively considered different days of SP coinfection post-IAV infection varied with high, medium, and low initial IAV and SP loads and incorporated them into the coinfection dynamics to better assess the IAV-SP Pna-exacerbation risks. Figure 6A–C demonstrates the probability density profiles of DH, which can be well described by lognormal distributions with estimated geometric means of 3.88 (95% CI 1.26–10.98), 6.25 (95% CI 2.5–14.6), and 4.43 (95% CI 1.6–11.81) log CFU·mL−1 for days 1, 7, and 9 post-IAV infection. This study thus inferred the worst inflammatory effects would occur with SP introduction at day 7 post-IAV infection, which had 50% risk probability for inflammatory responses of IL6, TNFα, and IFNγ to exceed 166 (95% CI 101–231), 1,899 (95% CI 1,604–2,194), and 215 (95% CI 0–527)-fold, respectively (Figure 6D–I, Table S3).
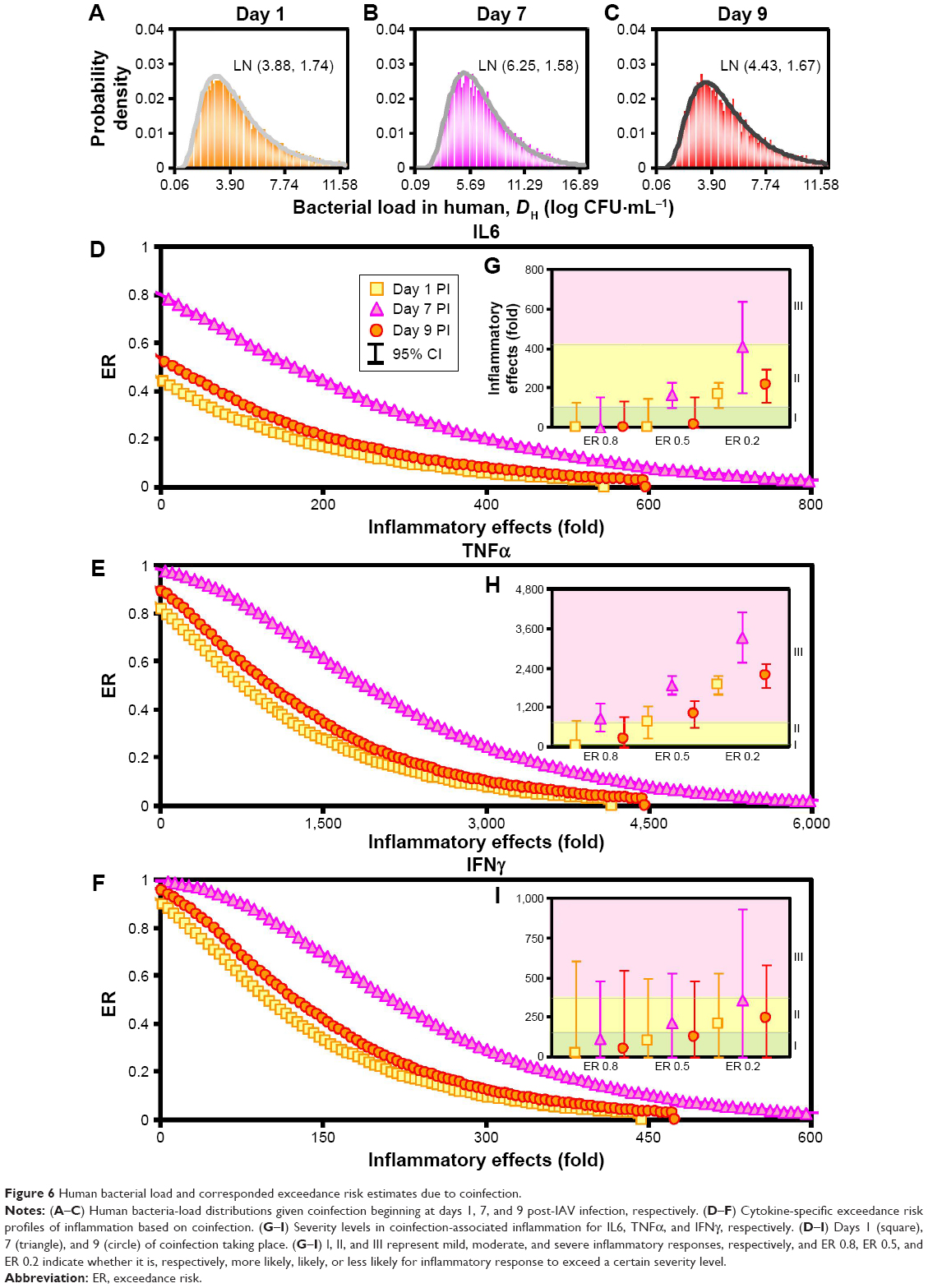 | Figure 6 Human bacterial load and corresponded exceedance risk estimates due to coinfection. |
Furthermore, we classified inflammatory responses of IL6, TNFα, and IFNγ accordingly as I<101<II<414<III, I<63<II<708<III, and I<155<II<369<III, where I, II, and III represent categorized regions of mild, moderate, and severe inflammatory responses, respectively. However, this study found that only while considering the worst Pna-induced inflammatory effects, eg, 20% risk probability (less likely to occur), would there be inflammatory effects of IL6 and IFNγ fall at regions II and III for day 1, 7, and 9 coinfection scenario (Figure 6G and I). On the other hand, for TNFα, all scenarios (ie, various risk probabilities) in IAV-SP coinfection (ie, on days 1, 7, and 9 coinfection) would unexceptionally develop moderate and mainly severe inflammatory responses, implying that TNFα was a potentially sensitive indicator for coinfection-exacerbated inflammation among the three cytokines studied (Figure 6H).
Discussion
Coinfection mathematical modeling
Mathematical models have long been recognized as useful tools in exploring complicated relationships with underlying infectious disease-transmission processes.21 The accuracy of the predictions obtained from mathematical modeling studies depends on the accuracy of the estimates for parameters governing the model dynamics. Good parameter estimates are needed to better understand and model the potential spread of influenza and SP coinfection.15,17 Therefore, interpretation of available data from experimental studies provides a platform to link mathematical models, such as infection dynamics, corresponding responses, and efficacy of different control measures for influenza and secondary bacterial coinfection.
Smith et al15,18 developed a kinetic model that can quantify and describe the mechanisms of influenza coinfection with SP, making it helpful to develop potentially effective therapeutic strategies. It can also help in understanding interactions of copathogenesis between influenza and bacterial pathogens, eg, influenza infection enhances bacterial adherence to epithelial cells, as well as alveolar macrophage dysfunction.1,2 On the other hand, increased bacterial adherence enhances viral release from infected cells and elevates the death rate of infected cells. Our simulated results of pathogen dynamics were in good agreement with Smith et al,15 with MAPE of nearly 30% and 14%, respectively, for influenza titer and bacteria load. To the best of our knowledge, this is the first study estimating simultaneously bacteria loads of SP and quantifying the probable inflammation risks as well as the corresponsive severity regimes of inflammatory responses in influenza-infected Pna patients coinfected with SP at different days (ie, days 1, 7, and 9).
Cytokine-specific inflammatory effects
Cytokines can cause both specific and aspecific effects, such as immune damage and viral replication in the lungs. In animal models, cytokines can lead to irreparable tissue destruction as proinflammatory damage in the alveoli.22 Proinflammatory cytokines, such as IL6, IFNγ, and TNFα, are required for immunoprotection, yet releasing large proinflammatory cytokines can become toxic. Onishi et al23 suggested that rapid bacterial growth may be caused by excess proinflammatory cytokine response due to rapid viral replication in a vulnerable lung environment. On the other hand, the present study has confirmed that the antiviral effects occurred during coinfection with the presence of proinflammatory cytokines. Li et al24 suggested that SP infection was linked to high concentration of type I IFN in animal model.
Studies have demonstrated that cytokine induction after infection with proinflammatory stimuli, or even with SP, in vitro and in vivo.13,25 They suggested that proinflammatory cytokines, such as IFNγ, TNFα, and IL6, were more likely to be key mediators of acute influenza-induced symptoms. One human experimental study has indicated that an intranasal influenza inoculation was followed by increased levels of TNFα and IL6 in nasal lavage fluid.26 Similarly, significant induction of cytokines, including TNFα and IL6, was found in human lung tissue, followed by pneumococcal infection. These studies imply that the relationships among bacteria loads/virus titers and cytokine levels could be treated in a dose–response manner for reflecting the severity of symptoms followed by coinfection with influenza and pneumococcus.
Our study constructed a Pna-associated inflammatory risk profile using Bayesian inference to predict the risk of inflammatory response postcoinfection with IAV and SP. Specifically, IL6 and TNFα are well-known mediators on the effects of attraction and activation of inflammatory cells, increased microvascular permeability, bronchoconstriction, bronchial hyperreactivity, and changes in mucus and surfactant production.27 On the other hand, IFNγ is a known cytokine critical in innate and adaptive immunity against viral, some bacterial, and protozoal infections.
This study selected five common cytokines (IL6, IL10, KC, TNFα, and IFNγ) initially to characterize coinfection-exacerbated inflammation. Due to limited experimental studies and our aim to construct best-fit linear dose–response relationships, only three cytokines (IL6, TNFα, and IFNγ) and few studies were adopted in quantifying exacerbated inflammatory responses, though several studies supported our idea to adopt these cytokines.25–29 This study further indicated TNFα might be the most sensitive biomarker and set as an early warning signal among the three explored cytokines for reflecting inflammatory response during coinfection. However, we recognize and suggest that robust TNFα responses are observed in a myriad of infections, and are in no way specific to influenza and SP coinfection.
There have been sporadic clinical studies simultaneously quantifying bacterial loads and cytokine levels in IAV-SP-coinfected Pna patients, and this study could thus only extrapolate bacterial loads and cytokine levels from mice to humans prudently based on an adjusted factors relating to body weight and body-surface area.20 Still, this study may have failed to consider various influences on pathogenicity posed by ages, strains, and doses. More animal experimental data and clinical studies regarding IAV-SP coinfection might be helpful to minimize uncertainties and assess health risks of coinfection more adequately and accurately in future study. In addition, Lee et al indicated that ideally local (lung) cytokine production should be studied, yet lower-respiratory secretion and tissue samples are difficult to obtain.30 Moreover, there are currently no standardized assay methods. Given the lungs are highly vascular, studying circulating cytokines may still provide a reasonably good approximation on their response patterns.30
Limitations and implications
Inflammatory cytokines could be a protective mechanism, and cytokine level could be correlated with systemic symptom dynamics. Canini and Carrat31 incorporated systemic symptom dynamics and cytokine production into an influenza kinetic model such that the cytokine compartment included all cytokines, such as IL6 and IFNγ. Based on IL6- and IFNγ-induced mechanisms, including immune response and symptom development, IL6 is the acute-phase response to viral infection, and IFNγ is the important immunoresponse that could inhibit viral replication directly. IL6 and IFNγ levels from nasal wash could thus be linked to body temperature, mucus production, viral titer, and symptom scores.26,32 Cytokine production is related to activated macrophages or infected cells.33 However, there are limited studies linking IAV-SP coinfection-induced cytokines and systemic symptom scores. Our study hence assessed cytokine responses alone as the end point of health risk.
Secondary bacterial Pna leads to increased disease severity, leading to a significant percentage of deaths during influenza pandemics.34 Pneumococcal diseases and influenza have caused substantial clinical and economic burden in Taiwan, particularly in young children and people aged ≥65 years.35 Moreover, the Taiwan Centers for Disease Control has incorporated severely complicated influenza and invasive pneumococcus into their list of categories III and IV notifiable communicable diseases. Continuous surveillance of respiratory illnesses and influenza antigen-detection tests are necessary to identify disease outbreaks so that infection-control measures can be promptly initiated in the inpatient and outpatient settings.
To date, the 13-valent pneumococcal conjugate vaccine (Pcv13) has been available on the private market in Taiwan to protect infants and young children against disease caused by the pneumococcus. On the other hand, Katsura et al36 indicated that the hemagglutinin segment encoding the pneumococcal surface protein is a promising bivalent vaccine candidate that simultaneously confers protective immunity against both SP and influenza virus.
Under the assumptions and limitations of the variables used in the IAV-SP coinfection model, we demonstrated that 7 days’ lag in bacterial coinfection resulted in the most serious susceptibility. Several studies have investigated the time course of susceptibility to SP infection after IAV infection and estimated that on average these individuals developed coinfection within 6.2 days (1.3–11.1 days) after IAV infection.5,9,13,15,17 This indicates that secondary SP infection may occur concurrently with or shortly after influenza infection.5 Several mechanisms have been demonstrated to affect the later stages of secondary pneumococcal infection (at 24 hours or later), rather than the initial clearance of bacterial populations.11,15 The increase of susceptibility to secondary pneumococcal infection due to the suppression of phagocytosis by alveolar macrophages is mainly attributed to IFNγ abundance instead of the viral burden.11
Shrestha et al17 suggested that the timing of administering treatment to coinfected individuals was best within 4 days post-IAV infection to prevent enhanced susceptibility to secondary pneumococcal infection. Therefore, based on our studied results of lag timing in coinfection, we recommend that people and health care workers should be wary of secondary SP infection on day 7 postinfluenza infection for prompt and proper control-measure implementations.
In view of our findings, we conclude that SP may be a risk factor of COPD exacerbations, a sudden flare-up of COPD symptoms. A higher prevalence of bacterial colonization was found in the airway of patients with stable COPD.37,38 The interaction between SP and COPD can cause serious complications. Moreover, both viral and bacterial infections increase the exacerbation severity of COPD, and the patients should be hospitalized.39 Viral respiratory infections, particularly those caused by influenza, increase the incidence of secondary bacterial infection, such as Pna.40 There is considerable potential for significant virus–bacteria interaction in COPD.40 As such, coinfection of influenza and pneumococcal strains should be acknowledged to enhance control effectiveness for influenza and pneumococcus of coinfected patients with COPD. Taken together, patients with COPD have a higher risk of getting influenza or developing Pna. People with COPD who develop Pna are highly likely to represent a mortality risk from influenza infection. Understanding the link between COPD and influenza/Pna coinfection could ensure effective and prompt treatment. Influenza and pneumococcal vaccine could reduce the risk of COPD exacerbations.40
Conclusion
Our study provides an integrated risk-assessment framework for quantifying pneumococcus-exacerbation risks associated with inflammatory response based on a well-built mathematical IAV-SP coinfection dynamic model, as well as mechanistically mathematical models representing relationships between bacteria load and severity of inflammatory response. We believe that TNFα might be the most sensitive biomarker among the three explored cytokines for reflecting inflammatory response during coinfection with IAV and SP. Our research highlights that SP inoculation at day 7 post-IAV infection impacts much more on Pna-associated inflammatory responses. People and health care workers should be wary of secondary SP infection on day 7 postinfluenza infection. We hope that our quantitative risk-assessment framework can provide new insights into improvements in respiratory health, especially predominantly due to COPD.
Acknowledgment
The authors acknowledge the financial support of the Ministry of Science and Technology, Republic of China, under grant MOST 104-2221-E-002-030-MY3.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev. 2006;19:571–582. | ||
McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol. 2014;12:252–262. | ||
Blyth CC, Webb SA, Kok J, et al. The impact of bacterial and viral co-infection in severe influenza. Influenza Other Respir Viruses. 2013;7:168–176. | ||
Bosch AA, Biesbroek G, Trzcinski K, Sanders EA, Bogaert D. Viral and bacterial interactions in the upper respiratory tract. PLoS Pathog. 2013;9:e1003057. | ||
Chertow DS, Memoli MJ. Bacterial coinfection in influenza: a grand rounds review. JAMA. 2013;309:275–282. | ||
Kosai K, Seki M, Tanaka A, et al. Increase of apoptosis in a murine model for severe pneumococcal pneumonia during influenza A virus infection. Jpn J Infect Dis. 2011;64:451–457. | ||
Weinberger DM, Simonsen L, Jordan R, Steiner C, Miller M, Viboud C. Impact of the 2009 influenza pandemic on pneumococcal pneumonia hospitalizations in the United States. J Infect Dis. 2012;205:458–465. | ||
World Health Organization. The top 10 causes of death. 2017. Available from: http://www.who.int/mediacentre/factsheets/fs310/en. Accessed June 12, 2017. | ||
McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun. 2006;74:6707–6721. | ||
Shahangian A, Chow EK, Tian X, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest. 2009;119:1910–1920. | ||
Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-γ during recovery from influenza infection. Nat Med. 2008;14:558–564. | ||
Raza SL, Cornelius LA. Matrix metalloproteinases: pro-and anti-angiogenic activities. J Investig Dermatol Symp Proc. 2000;5:47–54. | ||
van der Sluijs KF, van Elden LJ, Nijhuis M, et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. 2004;172:7603–7609. | ||
Tashiro M, Ciborowski P, Klenk HD, Pulverer G, Rott R. Role of Staphylococcus protease in the development of influenza pneumonia. Nature. 1987;325:536–537. | ||
Smith AM, Adler FR, Ribeiro RM, et al. Kinetics of coinfection with influenza A virus and Streptococcus pneumoniae. PLoS Pathog. 2013;9:e1003238. | ||
Shrestha S, Foxman B, Weinberger DM, Steiner C, Viboud C, Rohani P. Identifying the interaction between influenza and pneumococcal pneumonia using incidence data. Sci Transl Med. 2013;5:191ra84. | ||
Shrestha S, Foxman B, Dawid S, et al. Time and dose-dependent risk of pneumococcal pneumonia following influenza: a model for within-host interaction between influenza and Streptococcus pneumoniae. J R Soc Interface. 2013;10:20130233. | ||
Smith AM, McCullers JA, Adler FR. Mathematical model of a three-stage innate immune response to a pneumococcal lung infection. J Theor Biol. 2011;276:106–116. | ||
Bautista-Márquez A, Richardson V, Ortiz-Orozco O, et al. Prevalence of pneumococcal disease, serotype distribution, and antimicrobial susceptibility in Mexican children younger than 5 years of age. Arch Med Res. 2013;44:142–150. | ||
Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. | ||
Keeling MJ, Rohani P. Modeling Infectious Diseases in Humans and Animals. Princeton, NJ: Princeton University Press; 2008. | ||
Cauley LS, Vella AT. Why is coinfection with influenza virus and bacteria so difficult to control? Discov Med. 2015;19:33–40. | ||
Onishi M, Kitano M, Taniguchi K, et al. Intravenous peramivir inhibits viral replication, and leads to bacterial clearance and prevention of mortality during murine bacterial co-infection caused by influenza A(H1N1)pdm09 virus and Streptococcus pneumoniae. Antiviral Res. 2015;117:52–59. | ||
Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of γδ T cells. J Virol. 2012;86:12304–12312. | ||
Endeman H, Meijvis SC, Rijkers GT, et al. Systemic cytokine response in patients with community-acquired pneumonia. Eur Respir J. 2011;37:1431–1438. | ||
Hayden FG, Fritz R, Lobo MC, Alvord W, Strober W, Straus SE. Local and systemic cytokine responses during experimental human influenza A virus infection: relation to symptom formation and host defense. J Clin Invest. 1998;101:643–649. | ||
Kragsbjerg P, Holmberg H, Vikerfors T. Dynamics of blood cytokine concentrations in patients with bacteremic infections. Scand J Infect Dis. 1996;28:391–398. | ||
Mehta D, Petes C, Gee K, Basta S. The role of virus infection in deregulating the cytokine response to secondary bacterial infection. J Interferon Cytokine Res. 2015;35:925–934. | ||
Shi X, Zhou W, Huang H, et al. Inhibition of the inflammatory cytokine tumor necrosis factor-alpha with etanercept provides protection against lethal H1N1 influenza infection in mice. Crit Care. 2013;17:R301. | ||
Lee N, Wong CK, Chan PK, et al. Cytokine response patterns in severe pandemic 2009 H1N1 and seasonal influenza among hospitalized adults. PLoS One. 2011;6:e26050. | ||
Canini L, Carrat F. Population modeling of influenza A/H1N1 virus kinetics and symptom dynamics. J Virol. 2011;85:2764–2770. | ||
Kaiser L, Fritz RS, Straus SE, Gubareva L, Hayden FG. Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J Med Virol. 2001;64:262–268. | ||
Pichlmair A, Sousa CR. Innate recognition of viruses. Immunity. 2007;27:370–383. | ||
Kash JC, Walters KA, Davis AS, et al. Lethal synergism of 2009 pandemic H1N1 influenza virus and Streptococcus pneumoniae coinfection is associated with loss of murine lung repair responses. MBio. 2011;2:e00172. | ||
Wu DB, Chong HY, Lee KK. Cost-effectiveness analysis of joint vaccination with 13-valent pneumococcal conjugate vaccine (Pcv13) and influenza vaccine in Taiwan during seasonal influenza. Value Health. 2014;17:A273. | ||
Katsura H, Piao Z, Iwatsuki-Horimoto K, et al. A bivalent vaccine based on a replication-incompetent influenza virus protects against Streptococcus pneumoniae and influenza virus inflection. J Virol. 2014;88:13410–13417. | ||
Albrich WC, Monnet DL, Harbarth S. Antibiotic selection pressure and resistance in Streptococcus pneumoniae and Streptococcus pyogenes. Emerg Infect Dis. 2004;10:514–517. | ||
Sethi S, Sethi R, Eschberger K, et al. Airway bacterial concentrations and exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;176:356–361. | ||
Papi A, Bellattato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173:1114–1121. | ||
Mallia P, Johnston SL. Influenza infection and COPD. Int J Chron Obstruct Pulmon Dis. 2007;2:55–64. | ||
Chen SC, You SH, Liu CY, Chio CP, Liao CM. Using the experimental human influenza infections to validate a viral dynamic model and the implications for prediction. Epidemiol Infect. 2012;140:1557–1568. |
Supplementary materials
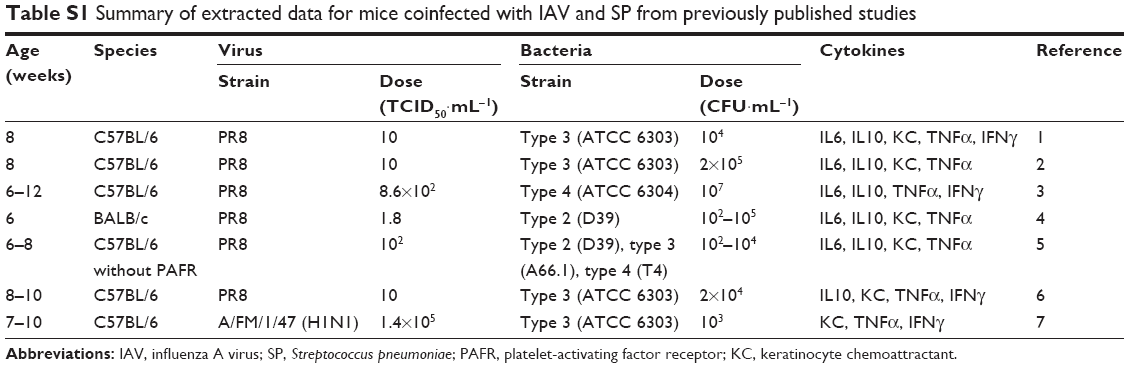 | Table S1 Summary of extracted data for mice coinfected with IAV and SP from previously published studies |
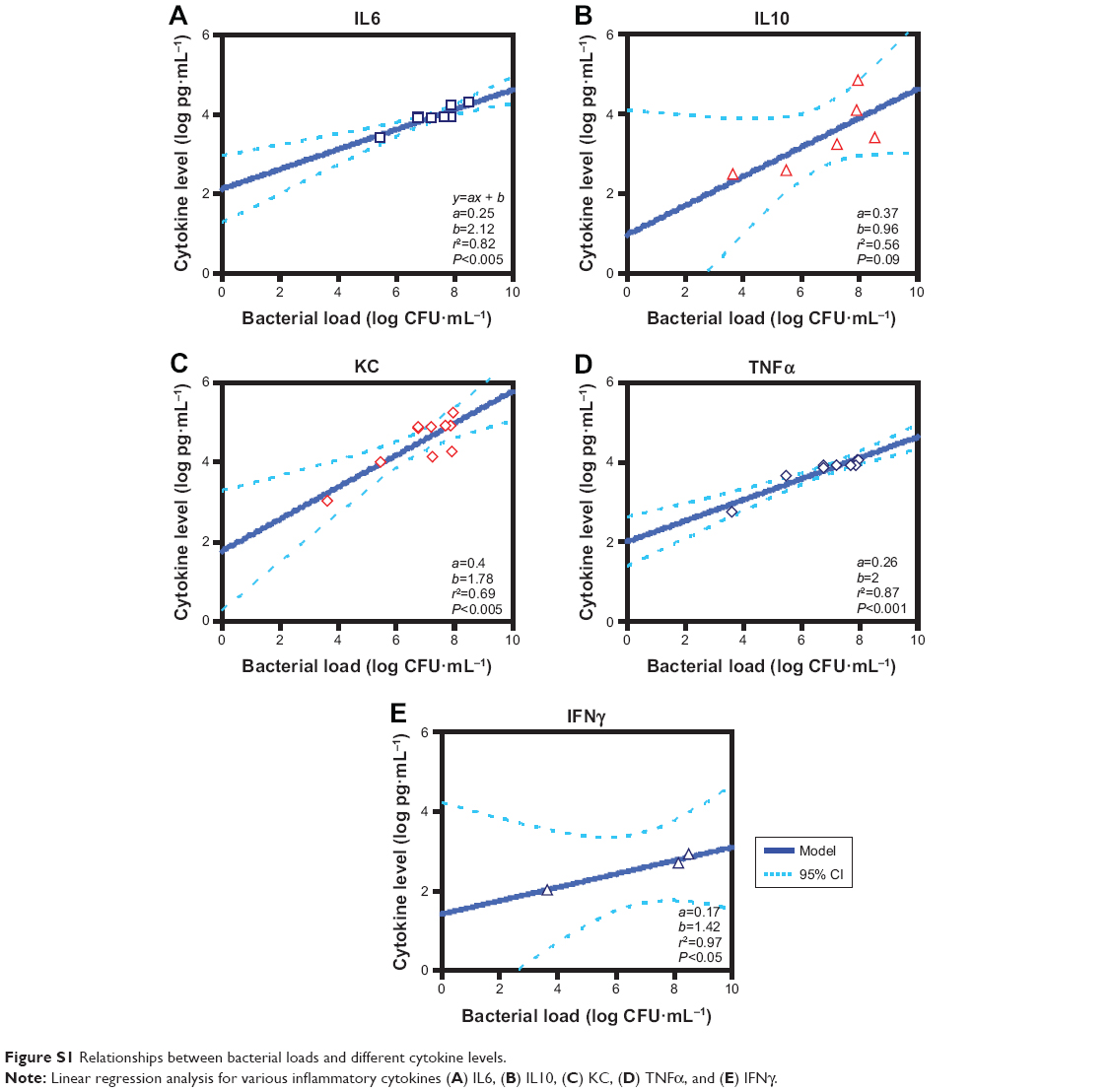 | Figure S1 Relationships between bacterial loads and different cytokine levels. |
 | Table S2 Inflammatory cytokines in IAV-associated pneumonia patients |
 | Table S3 IAV and SP coinfection-associated inflammatory effect (fold) varied with cytokines, day of introducing SP post-IAV infection, and exceedance risks at 0.8, 0.5, and 0.2 |
References
van der Sluijs KF, van Elden LJ, Nijhuis M, et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. 2004;172:7603–7609. | ||
van der Sluijs KF, van Elden LJ, Nijhuis M, et al. Involvement of the platelet-activating factor receptor in host defense against Streptococcus pneumoniae during postinfluenza pneumonia. Am J Physiol Lung Cell Mol Physiol. 2006;290:L194–L199. | ||
McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun. 2006;74:6707–6721. | ||
Chockalingam AK, Hickman D, Pena L, et al. Deletions in the neuraminidase stalk region of H2N2 and H9N2 avian influenza virus subtypes do not affect postinfluenza secondary bacterial pneumonia. J Virol. 2012;86:3564–3573. | ||
McCullers JA, Iverson AR, McKeon R, Murray PJ. The platelet activating factor receptor is not required for exacerbation of bacterial pneumonia following influenza. Scand J Infect Dis. 2008;40:11–17. | ||
Dessing MC, van der Sluijs KF, Florquin S, Akira S, van der Poll T. Toll-like receptor 2 does not contribute to host response during postinfluenza pneumococcal pneumonia. Am J Respir Cell Mol Biol. 2007;36:609–614. | ||
Damjanovic D, Lai R, Jeyanathan M, Hogaboam CM, Xing Z. Marked improvement of severe lung immunopathology by influenza-associated pneumococcal superinfection requires the control of both bacterial replication and host immune responses. Am J Pathol. 2013;183:868–880. | ||
Lee N, Wong CK, Chan PK, et al. Cytokine response patterns in severe pandemic 2009 H1N1 and seasonal influenza among hospitalized adults. PLoS One. 2011;6:e26050. | ||
Wen Y, Deng BC, Zhou Y, et al. Immunological features in patients with pneumonitis due to influenza A H1N1 infection. J Investig Allergol Clin Immunol. 2011;21:44–50. | ||
Zúñiga J, Torres M, Romo J, et al. Inflammatory profiles in severe pneumonia associated with the pandemic influenza A/H1N1 virus isolated in Mexico City. Autoimmunity. 2011;44:562–570. | ||
Liu Y, Chen H, Sun Y, Chen F. Antiviral role of Toll-like receptors and cytokines against the new 2009 H1N1 virus infection. Mol Biol Rep. 2012;39:1163–1172. | ||
Bautista E, Arcos M, Jimenez-Alvarez L, et al. Angiogenic and inflammatory markers in acute respiratory distress syndrome and renal injury associated to A/H1N1 virus infection. Exp Mol Pathol. 2013;94:486–492. | ||
Gürbüz Y, Tütüncü EE, Öztürk DB, Solay AH, Sencan I. TNF-α, IL-1β and IL-6 levels in pandemic influenza A (H1N1) 2009 patients and effect on mortality. Mediterr J Infect Microb Antimicrob. 2013;2:2. | ||
Haran JP, Buglione-Corbett R, Lu S. Cytokine markers as predictors of type of respiratory infection in patients during the influenza season. Am J Emerg Med. 2013;31:816–821. | ||
Rodriguez A, Falcon A, Cuevas MT, et al. Characterization in vitro and in vivo of a pandemic H1N1 influenza virus from a fatal case. PLoS One. 2013;8:e53515. | ||
Tiwari N, Kapoor P, Dhole TN. Antibody and inflammatory response-mediated severity of pandemic 2009 (pH1N1) influenza virus. J Med Virol. 2014;86:1034–1040. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.