")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Maresin 1 mitigates renal ischemia/reperfusion injury in mice via inhibition of the TLR4/MAPK/ NF-κB pathways and activation of the Nrf2 pathway
Authors Qiu Y, Wu Y, Zhao H, Sun H, Gao S
Received 25 September 2018
Accepted for publication 29 January 2019
Published 20 February 2019 Volume 2019:13 Pages 739—745
DOI https://doi.org/10.2147/DDDT.S188654
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Yun Qiu, Yichen Wu, Hongmei Zhao, Hong Sun, Sumin Gao
Department of Emergency Medicine, The Affiliated Huaian No 1 People’s Hospital of Nanjing Medical University, Huai’an, Jiangsu Province, China
Background: Inflammation and oxidative stress play a crucial role in the pathogenesis of renal ischemia/reperfusion injury (IRI). Maresin 1 (MaR1), which has shown strong anti-inflammatory and antioxidant effects, was recently reported to have protective properties in several different animal models.
Aim: The objectives of our study were to determine whether MaR1 alleviates renal IRI and to identify the underlying mechanisms.
Materials and methods: The mouse model in this study was induced by ischemia of the left kidney for 45 minutes and by nephrectomy of the right kidney. All mice were intravenously injected with a vehicle or MaR1. Renal histopathologic changes, function, proinflammatory cytokines, and oxidative stress were assessed. The expression of proteins was measured by Western blot.
Results: The results indicated that MaR1 markedly protected against renal IRI. The protective effects were accompanied by the reduction of histologic changes and reduction of renal dysfunction. Meanwhile, MaR1 remarkably mitigated renal IRI-induced inflammation and oxidative stress. In addition, our results showed that MaR1 significantly inhibited the expression of TLR4 and the expression of phosphorylated Erk, JNK, and P38. Furthermore, MaR1 decreased the nuclear translocation of NF-κB and increased the nuclear translocation of Nrf2.
Conclusion: MaR1 protects against renal IRI by inhibiting the TLR4/MAPK/NF-κB pathways, which mediate anti-inflammation, and by activating the Nrf2 pathway, which mediates antioxidation.
Keywords: renal ischemia/reperfusion injury, Maresin 1, TLR4, MAPK, NF-κB, Nrf2
Introduction
Acute kidney injury (AKI) is a major clinical problem that can result from renal ischemia/reperfusion injury (IRI), leading to acute kidney failure with increased morbidity and mortality in critically ill adults.1 Renal IRI is present in various types of surgeries including renal transplantation, vascular surgery, and cardiac surgery.2–4 It is recognized that inflammation and oxidative stress are perhaps the most crucial pathophysiological processes involved in the propagation of renal IRI.5 Effective measures to attenuate renal IRI may, therefore, improve patients’ postoperative survival.
The inflammatory response mediated by neutrophils and macrophages plays a main role in the pathogenesis of renal injury following IRI.6 Several studies have shown that a self-limited inflammatory response at the early phase could be a possible way to prevent renal IRI.7,8 Pro-resolving lipid mediators such as maresins and resolvins, which are derived from polyunsaturated fatty acids, play an important role in controlling inflammation and oxidative stress.9,10 Maresin 1 (MaR1) is derived from docosahexaenoic acid.11 A recent investigation has also shown that MaR1 can promote the resolution of acute inflammation in sepsis12 and enhance the activation of the antioxidant pathway in lung IRI.13 However, whether MaR1 has protective effects in renal IRI has not been reported.
In this context, we investigated the impact of MaR1 on renal IRI and explored the possible mechanisms involved in this process. The objective of the present study was threefold: 1) to determine whether MaR1 alleviates kidney damage after IRI; 2) to determine whether renoprotection induced by MaR1 is associated with anti-inflammation; and 3) to determine whether renoprotection induced by MaR1 is associated with antioxidation.
Materials and methods
Animals
Male C57BL/6 mice (7–8 weeks old; weight 23–25 g) were purchased from Beijing HFK Bioscience Co., Ltd. All animals received humane care in compliance with the Animal Care and Use Committee of Nanjing Medical University. All animals were housed in temperature-controlled cages with free access to food and water.
Experimental protocol
The mice were randomly divided into three groups: a sham-operated group (Sham), an ischemia/reperfusion group (IR), and an IR plus MaR1 group (MaR). All mice were subjected to renal IRI as previously described.14 The right kidney was exposed and removed. Then, the left renal pedicle was clamped for 45 minutes by a nontraumatic microvascular clip to induce ischemia. After renal ischemia, the vascular clamp was removed to allow reperfusion for 24 hours. All animal procedures were approved by the Animal Care and Use Committee of Nanjing Medical University.
MaR1 was purchased from Cayman Chemical (Ann Arbor, MI, USA). MaR1 (1.0 ng) was dissolved in 0.1 mL normal saline, which was injected through the tail vein at the start of the reperfusion.13 All groups except the MaR group were intravenously given the same volume of the vehicle at the reperfusion period. Mice in different groups received different treatments, and the kidneys were collected 24 hours after reperfusion and they were subjected to analysis.
Measurement of renal function
Renal function was monitored by measuring the levels of blood urea nitrogen (BUN) and creatinine (Cr) using commercial kits (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions.
Histologic evaluation
The kidney tissues were stained with H&E as previously described.14 Ten high-magnification (×400) fields of the cortex and the outer stripe of the outer medulla were randomly selected. Histologic assessment of renal damage was determined by a semi-quantitative analysis of tubular injury including the presence of tubular cell necrosis, loss of the brush border, vacuolization, tubule dilation, or cast formation. These factors were scored as follows: no injury (0); mild: <25% (1); moderate: <50% (2); severe: <75% (3); and very severe: >75% (4).15
Measurement of tissue tumor necrosis factor (TNF)-α, IL-6, and IL-10
Renal tissue homogenates were collected for the measurement of TNF-α, IL-6, and IL-10 by the ELISA method using commercially available assay kits (Neobioscience, Inc., Beijing, China) according to the manufacturer’s instructions.
Measurement of 15-F2t-isoprostane and malondialdehyde (MDA) levels
MDA was assessed by the thiobarbituric acid method using an assay kit (Jiancheng Bioengineering Institute, Nanjing, China), and 15-F2t-isoprostane was measured using an enzyme immunoassay kit (Cayman Chemical) according to the manufacturer’s protocol.
Western blot analysis
Cytoplasmic and nuclear proteins were extracted from the kidney tissue using the Protein Extraction Kit (KeyGen Biotech Co. Ltd., Nanjing, China) containing protease inhibitor and phosphatase inhibitor cocktails (Sigma Aldrich, Darmstadt, Germany) according to the manufacturer’s protocols. Samples (40 μg per lane) were loaded and then separated on SDS-PAGE (10%) gels and electrotransferred to polyvinylidene difluoride membranes. The membranes were incubated with the following primary antibodies: TLR4, p-Erk, NF-κB p65, Nrf2, HO-1, and NQO-1 (Abcam, Cambridge, UK) at 1:1,000; p-P38 and p-JNK (Cell Signaling Technology, Danvers, MA, USA) at 1:1,000; and SOD, GAPDH, or Histone3 (ABclonal, Wuhan, China) at 1:500 at 4°C overnight. Following extensive washing, the primary antibody bindings were detected with a secondary antibody (Cell Signaling Technology) at 1:5,000. The immunoreactive bands were visualized and photographed using an EC3 Imaging System (UVP, Inc., Upland, CA, USA).
Statistical analysis
All values were expressed as the mean ± SD. Comparisons across more than two groups involved the use of one-way ANOVA and Student–Newman–Keuls tests. Animal survival differences between the groups were determined using the log-rank test. P≤0.05 was set as the significance level.
Results
MaR1 alleviated renal injury
The renal morphological change was assessed by H&E staining (Figure 1A). Histologic kidney injury, including necrotic tubules, loss of the brush border, vacuolar degeneration, and cast formation, was observed after renal IRI. MaR1 attenuated histologic kidney injury as shown in Figure 1A. Compared with the Sham group, the injury score in the IR group was markedly increased (P<0.05; Figure 1B). Then, compared with the IR group, the injury score in the MaR group was significantly lowered (P<0.05). The concentrations of plasma BUN and Cr, which are indicative of kidney function, were measured by commercial kits (Figure 1C and D). Compared with the IR group, MaR1 reduced the increase of BUN and Cr levels after renal IRI (P<0.05).
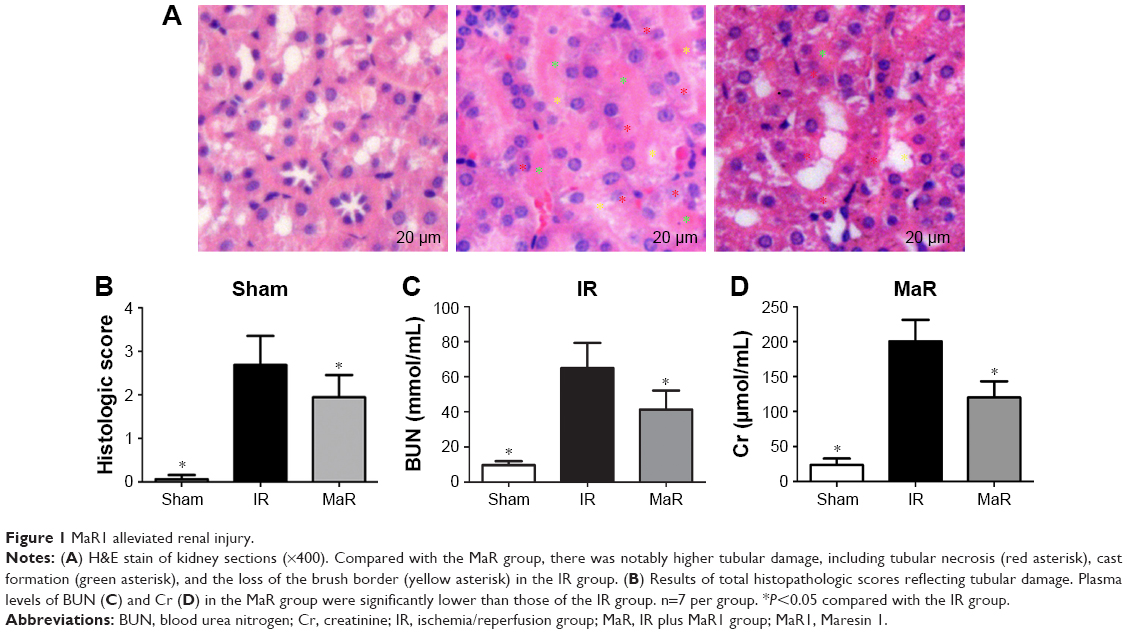 | Figure 1 MaR1 alleviated renal injury. |
MaR1 decreased inflammation in the kidney after IRI
As shown in Figure 2A–C, compared with the Sham group, ELISA detected an increase in the levels of inflammatory cytokines, including TNF-α and IL-6 in the IR group (P<0.05). Compared with the levels in the IR group, MaR1 significantly decreased the levels of TNF-α and IL-6, as well as further increased the level of IL-10 (P<0.05).
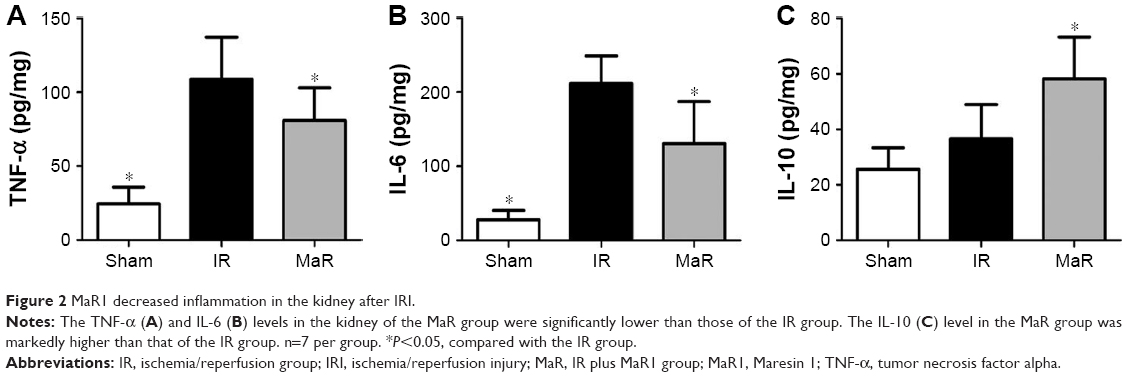 | Figure 2 MaR1 decreased inflammation in the kidney after IRI. |
MaR1-mediated renoprotection against IRI was associated with the TLR4/MAPK/NF-κB pathways in mice
The TLR4 signaling pathway plays a critical role in regulating inflammation cytokine production. To investigate the anti-inflammatory mechanism of MaR1, TLR4/MAPK/NF-κB pathways were analyzed by Western blot. As shown in Figure 3, compared with the Sham group, the expression of TLR4 in the IR group was significantly increased, while administration of MaR1 reduced the TLR4 expression (P<0.05).
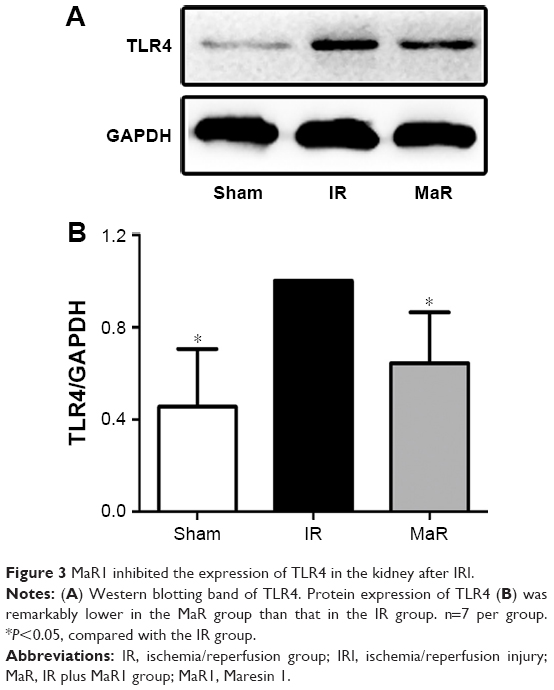 | Figure 3 MaR1 inhibited the expression of TLR4 in the kidney after IRI. |
Compared with the Sham group, as shown in Figure 4A–D, renal ischemia/reperfusion resulted in significantly increased phosphorylation of Erk, P38, and JNK. However, treatment with MaR1 remarkably inhibited phosphorylation of Erk, P38, and JNK after renal IRI (P<0.05).
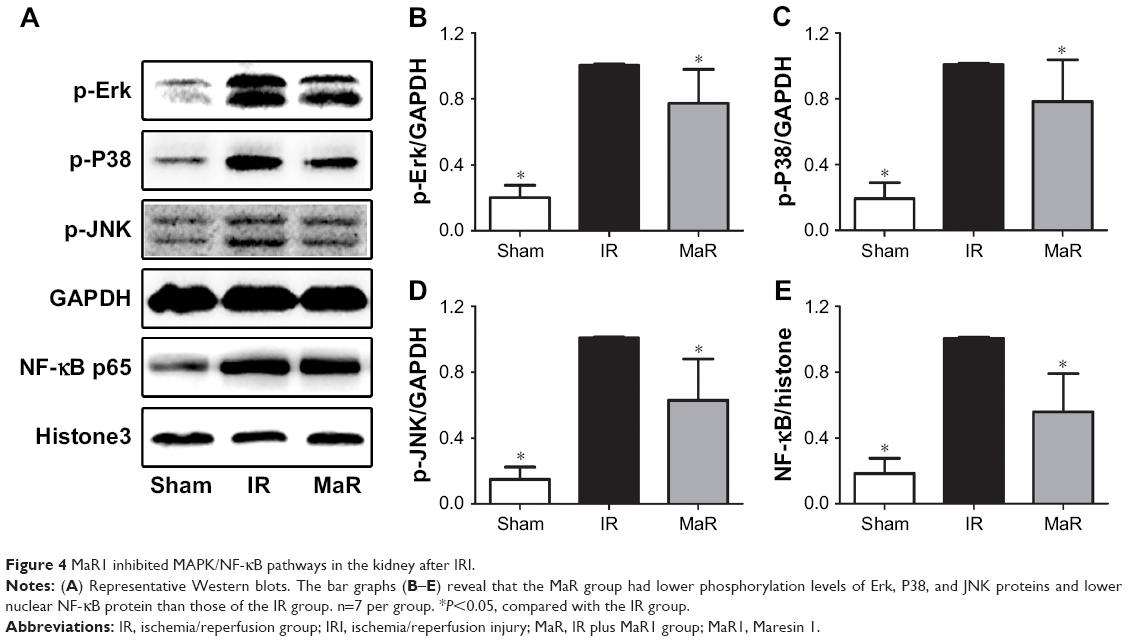 | Figure 4 MaR1 inhibited MAPK/NF-κB pathways in the kidney after IRI. |
As shown in Figure 4A and E, compared with the IR group, the nuclear translocation of NF-κB p65 was increased in the kidney after the renal IRI, while MaR1 significantly suppressed the nuclear translocation of NF-κB p65 (P<0.05).
MaR1 suppressed oxidative stress in the kidney after IRI
Compared with the Sham group, the MDA and 15-F2t-isoprostane levels were significantly elevated in the IR group (P<0.05; Figure 5A and B). However, the levels of these indicators were much lower in the MaR group than in the IR group (P<0.05).
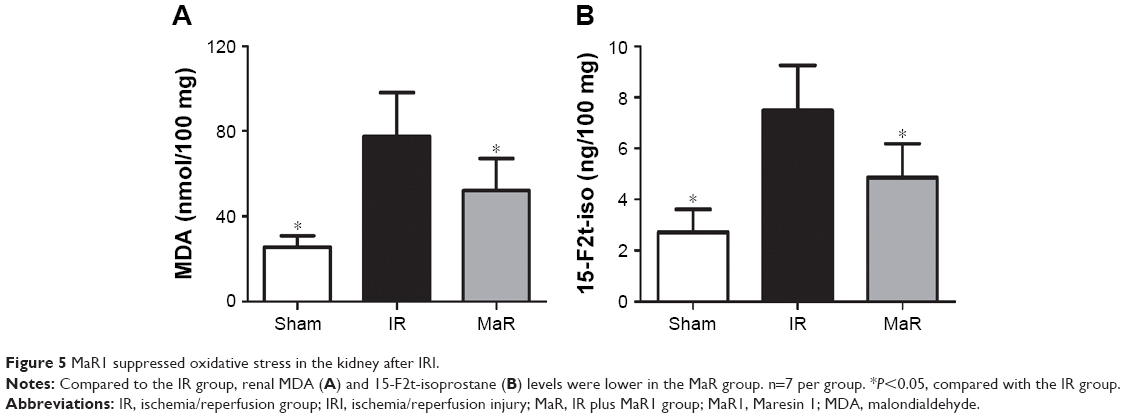 | Figure 5 MaR1 suppressed oxidative stress in the kidney after IRI. |
MaR1-mediated renoprotection against IRI was associated with the Nrf2 pathway in mice
As shown in Figure 6A and B, the nuclear translocation of Nrf2 was significantly induced by renal ischemia/reperfusion, and it was further increased by MaR1 (P<0.05). Furthermore, compared with the IR group, the levels HO-1, SOD, and NQO-1 also remained elevated in the kidney of mice treated with MaR1 (P<0.05; Figure 6C–E).
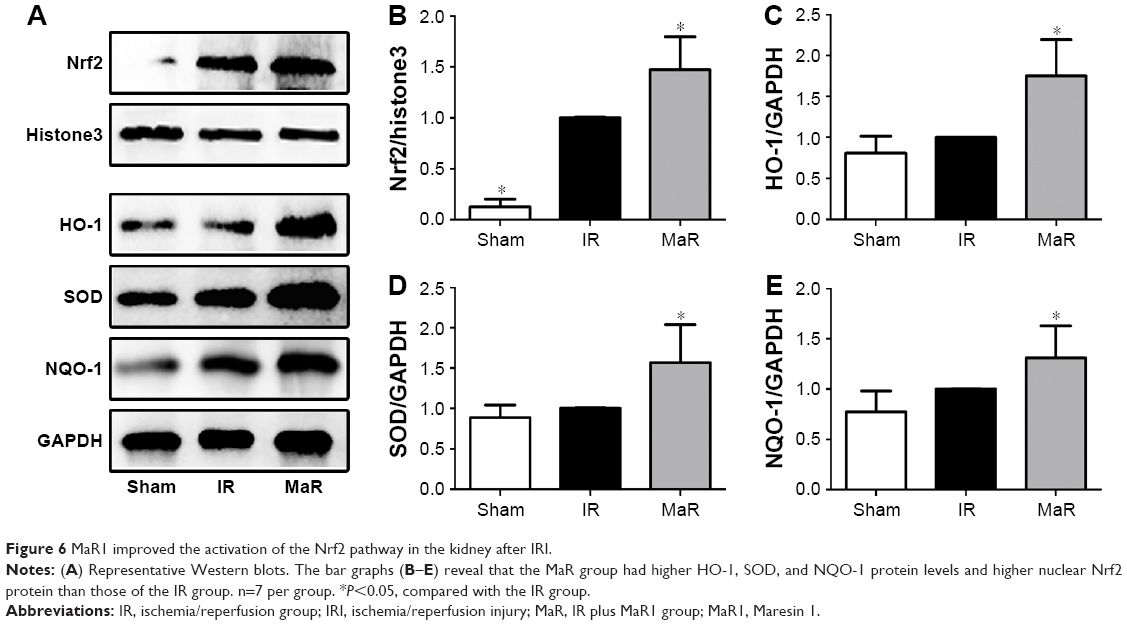 | Figure 6 MaR1 improved the activation of the Nrf2 pathway in the kidney after IRI. |
Discussion
Renal IRI is a common cause of AKI and can lead to irreversible kidney damage and high mortality. Therefore, interventions after renal IRI would be beneficial, such as anti-inflammation16 and antioxidation.17 In this study, we found that MaR1 blocked the deterioration of renal function in the IRI model. The renoprotection involved anti-inflammatory and antioxidative capabilities because MaR1 markedly suppressed the production of proinflammatory factors via inhibition of the TLR4 pathway and elevated the antioxidative effects via activation of the Nrf2 pathway.
MaR1 has been widely applied to ameliorate acute lung injury,18 lung IRI,13 and carbon tetrachloride-induced liver injury.19 However, whether MaR1 could mitigate renal IRI in mice has not been reported. In our study, the results indicated that MaR1 ameliorated renal function, as reflected by the reduced elevation of BUN and Cr after IRI. MaR1 remarkably alleviated tubular cell necrosis and attenuated the loss of cast formation and brush border. Therefore, our study indicated that MaR1 could attenuate kidney damage after IRI.
The mechanisms by which MaR1 protected against renal IRI remain unclear. The inflammatory cascade is a major component in the pathogenesis of renal IRI, causing kidney tissue damage by releasing several mediators.20 Many studies have indicated that kidney injury leads to the production of proinflammatory cytokines, such as TNF-α and IL-6, followed by the release of anti-inflammatory cytokine IL-10 during the ischemia/reperfusion period.21,22 Early and effective regulation of the inflammatory response is vital for the prevention and treatment of renal injury.21,22 Therefore, we speculated that the anti-inflammatory effect could act as a pivotal part in the renoprotection induced by MaR1. As demonstrated by ELISA, MaR1 treatment reduced IRI-induced production of proinflammatory cytokines, such as TNF-α and IL-6, and it increased the anti-inflammatory cytokine IL-10. The TLR4/MAPK/NF-κB pathways mediated inflammation in the kidney.23,24 The activation of TLR4 propagated the initial injury by inducing inflammatory cytokine production.25 In our study, we found that renal IRI led to the evident expression of TLR4 and its downstream factors. Importantly, MaR1 treatment effectively decreased the level of TLR4. MAPKs, including the Erks, the JNK, and P38, are a family of structurally related serine/threonine kinases. The MAPK pathway is a critical component in regulating renal inflammation and cell death.26 Treatment with MaR1 significantly inhibited phosphorylation of Erk, JNK, and P38 in the kidney. Activation of NF-κB resulted in its nuclear translocation to modulate the expression of numerous genes that have a pivotal role in the inflammatory response.27 In our study, MaR1 suppressed the nuclear translocation of NF-κB p65 and attenuated renal IRI. Therefore, our results were consistent with the previous hypothesis that MaR1 might be associated with suppression of the inflammatory response and inactivation of TLR4/MAPK/NF-κB signaling pathways.
Free radicals have a pivotal role in the pathogenesis of renal IRI, resulting in facilitated inflammatory cell infiltration and neutrophil activation.28 The elimination of reactive free radicals is primarily achieved by a series of antioxidant enzymes. The transcription factor Nrf2 critically regulates the expression of antioxidant enzymes, including HO-1, SOD, and NQO-1.29 The levels of MDA and 15-F2t-isoprostane, which are the products of lipid peroxidation, were significantly increased in the kidney after IRI. In our experiments, treatment with MaR1 markedly attenuated the levels of MDA and 15-F2t-isoprostane in the kidney. Moreover, the nuclear translocation of Nrf2 and the expression of HO-1, SOD, and NQO-1 were elevated by MaR1. Therefore, our results indicated that MaR1 attenuated oxidative stress against renal IRI by increasing the nuclear localization of Nrf2 and by elevating antioxidant enzymes.
Limitations
There are some limitations to this study. We found that MaR1 treatment can protect against renal IR injury at the early stage, but it is still unknown whether it could induce renoprotective effects at the chronic stage. In addition, the receptors for MaR1 action are still unclear. Our study indicated that MaR1 can attenuate the expression of TLR4 in the kidney after IRI. Previous studies indicated that TLR4 pathway could be found in the vascular endothelial cells, tubular epithelial cells, and podocytes.30–32 However, there was evidence that NF-κB activity in podocytes did not contribute to the pathogenesis of renal IRI.33 Therefore, we speculated that the vascular endothelial and renal tubular cells could be the target of MaR1-conferred renoprotection. Further research is needed for a better understanding of the specific mechanism of renoprotection induced by MaR1.
Conclusion
Our findings showed that MaR1 protects against renal IRI via inhibition of the TLR4/MAPK/NF-κB pathways, which mediate anti-inflammation, and via activation of the Nrf2 pathway, which mediates antioxidation. Taken together, these data suggest that MaR1 represents a potentially new therapeutic approach for the treatment of renal IRI.
Acknowledgments
This study was supported by grants from Nanjing Medical University (2016NJMUZD084) and Six One Project (LGY2017052).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Doyle JF, Forni LG. Acute kidney injury: short-term and long-term effects. Crit Care. 2016;20(1):188. | ||
Perico N, Cattaneo D, Sayegh MH, Remuzzi G. Delayed graft function in kidney transplantation. Lancet. 2004;364(9447):1814–1827. | ||
Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med. 2016;67(1):293–307. | ||
di Lullo L, Bellasi A, Russo D, Cozzolino M, Ronco C. Cardiorenal acute kidney injury: epidemiology, presentation, causes, pathophysiology and treatment. Int J Cardiol. 2017;227:143–150. | ||
Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7(4):189–200. | ||
Jo SK, Sung SA, Cho WY, Go KJ, Kim HK. Macrophages contribute to the initiation of ischaemic acute renal failure in rats. Nephrol Dial Transplant. 2006;21(5):1231–1239. | ||
Furuichi K, Wada T, Iwata Y, et al. Administration of FR167653, a new anti-inflammatory compound, prevents renal ischaemia/reperfusion injury in mice. Nephrol Dial Transplant. 2002;17(3):399–407. | ||
Gigliotti JC, Huang L, Ye H, et al. Ultrasound prevents renal ischemia–reperfusion injury by stimulating the splenic cholinergic anti-inflammatory pathway. J Am Soc Nephrol. 2013;24(9):1451–1460. | ||
Francos-Quijorna I, Santos-Nogueira E, Gronert K, et al. Maresin 1 promotes inflammatory resolution, neuroprotection, and functional neurological recovery after spinal cord injury. J Neurosci. 2017;37(48):11731–11743. | ||
Gao Y, Zhang H, Luo L, et al. Resolvin D1 improves the resolution of inflammation via activating NF-κB p50/p50–mediated cyclooxygenase-2 expression in acute respiratory distress syndrome. J Immunol. 2017;199(6):2043–2054. | ||
Serhan CN, Dalli J, Karamnov S, et al. Macrophage proresolving mediator Maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012;26(4):1755–1765. | ||
Li R, Wang Y, Ma Z, et al. Maresin 1 mitigates inflammatory response and protects mice from sepsis. Mediators Inflamm. 2016;2016(2):1–9. | ||
Sun Q, Wu Y, Zhao F, Wang J. Maresin 1 ameliorates lung ischemia/reperfusion injury by suppressing oxidative stress via activation of the Nrf-2-Mediated HO-1 signaling pathway. Oxid Med Cell Longev. 2017;2017(7):1–12. | ||
Gao S, Zhu Y, Li H, et al. Remote ischemic postconditioning protects against renal ischemia/reperfusion injury by activation of T-LAK-cell-originated protein kinase (TOPK)/PTEN/Akt signaling pathway mediated anti-oxidation and anti-inflammation. Int Immunopharmacol. 2016;38:395–401. | ||
Jablonski P, Howden BO, Rae DA, et al. An experimental model for assessment of renal recovery from warm ischemia. Transplantation. 1983;35(3):198–204. | ||
Wu SH, Chen XQ, Lu J, Wang MJ. BML-111 attenuates renal ischemia/reperfusion injury via peroxisome proliferator-activated receptor-α-regulated heme oxygenase-1. Inflammation. 2016;39(2):611–624. | ||
Leonard MO, Kieran NE, Howell K, et al. Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia–reperfusion injury. FASEB J. 2006;20(14):2624–2626. | ||
Gong J, Wu ZY, Qi H, et al. Maresin 1 mitigates LPS-induced acute lung injury in mice. Br J Pharmacol. 2014;171(14):3539–3550. | ||
Li R, Wang Y, Zhao E, et al. Maresin 1, a proresolving lipid mediator, mitigates carbon tetrachloride-induced liver injury in mice. Oxid Med Cell Longev. 2016;2016(11):1–13. | ||
Okusa MD. The inflammatory cascade in acute ischemic renal failure. Nephron. 2002;90(2):133–138. | ||
Bai M, Zhang L, Fu B, et al. IL-17A improves the efficacy of mesenchymal stem cells in ischemic–reperfusion renal injury by increasing Treg percentages by the COX-2/PGE2 pathway. Kidney Int. 2018;93(4):814–825. | ||
Nishikawa H, Taniguchi Y, Matsumoto T, et al. Knockout of the interleukin-36 receptor protects against renal ischemia–reperfusion injury by reduction of proinflammatory cytokines. Kidney Int. 2018;93(3):599–614. | ||
Zhang B, Ramesh G, Uematsu S, Akira S, Reeves WB. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J Am Soc Nephrol. 2008;19(5):923–932. | ||
Ye HY, Jin J, Jin LW, et al. Chlorogenic acid attenuates lipopolysaccharide-induced acute kidney injury by inhibiting TLR4/NF-κB signal pathway. Inflammation. 2017;40(2):523–529. | ||
Arslan F, Keogh B, Mcguirk P, Parker AE. TLR2 and TLR4 in ischemia reperfusion injury. Mediators Inflamm. 2010;2010(4):1–8. | ||
Basu A, Krishnamurthy S. Cellular responses to cisplatin-induced DNA damage. J Nucleic Acids. 2010;2010(10):1–16. | ||
Sanz AB, Sanchez-Niño MD, Ramos AM, et al. NF-kappaB in renal inflammation. J Am Soc Nephrol. 2010;21(8):1254–1262. | ||
Annuk M, Zilmer M, Lind L, Linde T, Fellström B. Oxidative stress and endothelial function in chronic renal failure. J Am Soc Nephrol. 2001;12(12):2747–2752. | ||
Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47(1):89–116. | ||
Chen J, John R, Richardson JA, et al. Toll-like receptor 4 regulates early endothelial activation during ischemic acute kidney injury. Kidney Int. 2011;79(3):288–299. | ||
El-Achkar TM, Wu XR, Rauchman M, et al. Tamm–Horsfall protein protects the kidney from ischemic injury by decreasing inflammation and altering TLR4 expression. Am J Physiol Renal Physiol. 2008;295(2):F534–F544. | ||
Banas MC, Banas B, Hudkins KL, et al. TLR4 links podocytes with the innate immune system to mediate glomerular injury. J Am Soc Nephrol. 2008;19(4):704–713. | ||
Yamashita M, Yoshida T, Hayashi M. Podocyte NF-κB is dispensable for the pathogenesis of renal ischemia–reperfusion injury. Physiol Rep. 2016;4(16):e12912. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.