")
Back to Journals » Journal of Inflammation Research » Volume 16
MAPK Signaling Pathways in Hepatic Ischemia/Reperfusion Injury
Authors Yu B, Zhang Y, Wang T, Guo J, Kong C, Chen Z, Ma X, Qiu T
Received 22 November 2022
Accepted for publication 16 March 2023
Published 27 March 2023 Volume 2023:16 Pages 1405—1418
DOI https://doi.org/10.2147/JIR.S396604
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam D Bachstetter
Bo Yu,* Yalong Zhang,* Tianyu Wang, Jiayu Guo, Chenyang Kong, Zhongbao Chen, Xiaoxiong Ma, Tao Qiu
Department of Organ Transplantation, Renmin Hospital of Wuhan University, Wuhan, Hubei Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Tao Qiu, Department of Organ Transplantation, Renmin Hospital of Wuhan University, Wuhan, Hubei Province, People’s Republic of China, Tel +86-13995632367, Email [email protected]
Abstract: The mitogen-activated protein kinase signaling pathway can be activated by a variety of growth factors, cytokines, and hormones, and mediates numerous intracellular signals related to cellular activities, including cell proliferation, motility, and differentiation. It has been widely studied in the occurrence and development of inflammation and tumor. Hepatic ischemia-reperfusion injury (HIRI) is a common pathophysiological phenomenon that occurs in surgical procedures such as lobectomy and liver transplantation, which is characterized by severe inflammatory reaction after ischemia and reperfusion. In this review, we mainly discuss the role of p38, ERK1/2, JNK in MAPK family and TAK1 and ASK1 in MAPKKK family in HIRI, and try to find an effective treatment for HIRI.
Keywords: ischemia/reperfusion injury, mitogen-activated protein kinases, liver
Introduction
Signal transduction by mitogen activated protein kinase (MAPK) is carried out in a three-level cascade. That is, the kinase of MAPKK kinase (MAPKKK) phosphorylates and activates MAPK kinase (MAPKK), then phosphorylates MAPK to activate it, and then translocates to the nucleus to regulate the basic physiological processes of cells.1
MAPKs are a group of highly conserved serine/threonine protein kinases that control cellular activity via signal transduction, substitution, amplification, and integration.2 Dozens of members of the MAPKs family have been identified so far, and the role of apoptosis signal-regulating kinase 1(ASK1) and transforming growth factor β-activated kinase 1 (TAK1) of the MAP3K family in hepatic ischemia-reperfusion injury is increasingly studied in HIR.3–7 MAPKKK acts on MAPKK to phosphorylate and activate it. Activated MAPKK then acts on MAPK family signaling molecules to phosphorylate and activate it. Activated MAPK enters the nucleus to regulate transcriptional regulation. We pay more attention to the function of extracellular signal regulated kinase 1 / 2 (ERK1 / 2), c-Jun N-terminal kinase (JNK) and p38MAPK in HIRI.
Hepatic ischemia/reperfusion injury (HIRI) includes “cold” IRI and “warm” IRI (which is very common in liver transplantation).8–10 HIRI mainly involves hepatocytes, Kupffer cells (KCs) and immune cells including t cells and monocytes. The pathophysiological process is biphasic: initial ischemic liver injury and reperfusion injury after restoration of the blood supply.11–14 During initial ischemic liver injury, blood flow into the liver is blocked, which results in local ischemia. The latter leads to hepatic hypoxia, consumption of adenosine triphosphate and glycogen, mitochondrial dysfunction and, eventually, hepatocyte necrosis.15,16 Large numbers of inflammatory cells such as neutrophils, KCs, dendritic cells, circulating lymphocytes and monocytes infiltrate and accumulate in the damaged area. This action leads to the activation and secretion of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and other proinflammatory factors, which results in inflammatory reactions and direct damage to hepatocytes.17,18 After restoration of the blood supply, activated KCs release many proinflammatory factors, which activate neutrophils and T cells further, and aggravate tissue damage.19,20 Monocytes in the blood circulation are activated by proinflammatory factors and differentiate into macrophages with similar function to KCs, and are then recruited to the liver to maintain the immune response and amplify the inflammatory response further, which aggravates liver injury. IRI-mediated hepatocyte injury in a donor liver leads to poor liver function and acute/chronic rejection after transplantation, which limits clinical application of the donor liver.19,21
The mechanism of HIRI stated above involves cytokines and mediators, which are related to several cellular pathways (Figure 1).13,22–24 If HIRI occurs, then p38, JNK and ERK are activated by specific cells to elicit a series of physiological and pathological reactions.2,7,25 Therefore, these MAPK-related molecules are considered to be potential therapeutic targets for HIRI.
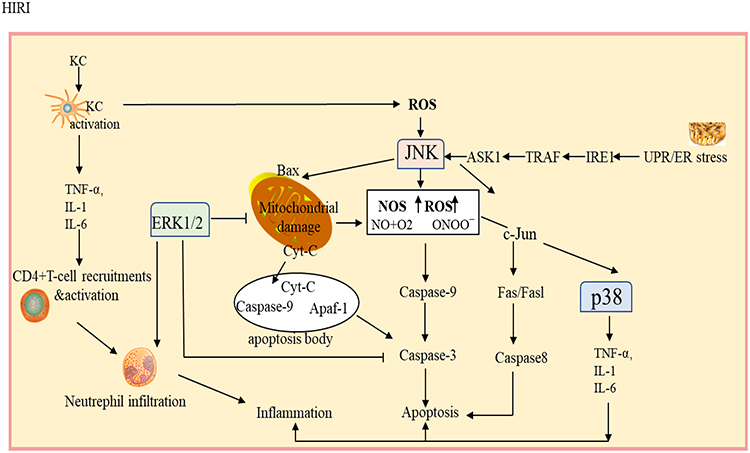 |
Figure 1 MAPK-related signaling pathways involved in hepatic ischemia-reperfusion injury. p38, Jnk and ERK participate in the regulation of liver ischemia-reperfusion injury through death receptor pathway, mitochondrial apoptosis pathway, endoplasmic reticulum reactive apoptosis pathway and oxidative stress pathway, etc. Abbreviations: Cyt-C, cytochrome C; NOS, reactive oxygen species; NOS, nitric oxide synthase; ER, endoplasmic reticulum; TRAF, Tumor necrosis factor receptor-associated factor; IRE1, inositol requires enzyme 1; UPR, unfolded protein response; Apaf-1, apoptotic protease activating factor-1.3-kinase; RBP4, retinol binding protein 4; ROS, reactive oxygen species; SLPI, secretory leukocyte protease inhibitor; SOD, superoxide dismutase; TNF-α, tumor necrosis factor-alpha; VCAM, vascular cell adhesion molecule; X/XOD, xanthine/xanthine oxidase. |
Role and Therapeutic Importance of p38 in HIRI
During HIRI, p38 is activated and participates in the inflammatory response of the liver.26–28 In general, it is believed that p38 has a different role at different times and in different types of liver disease. In a non-steatotic liver, phosphorylation and activation of p38 are involved in the necrosis and apoptosis of hepatocytes.29 In a steatotic liver, p38 is involved in liver inflammation because it induces reactive oxygen species (ROS) production indirectly.9,30 Inhibiting the production or activation of p38 has a positive role in HIRI treatment.31 Table 1 shows the current regulatory factors and therapeutic potential of p38 in HIRI.
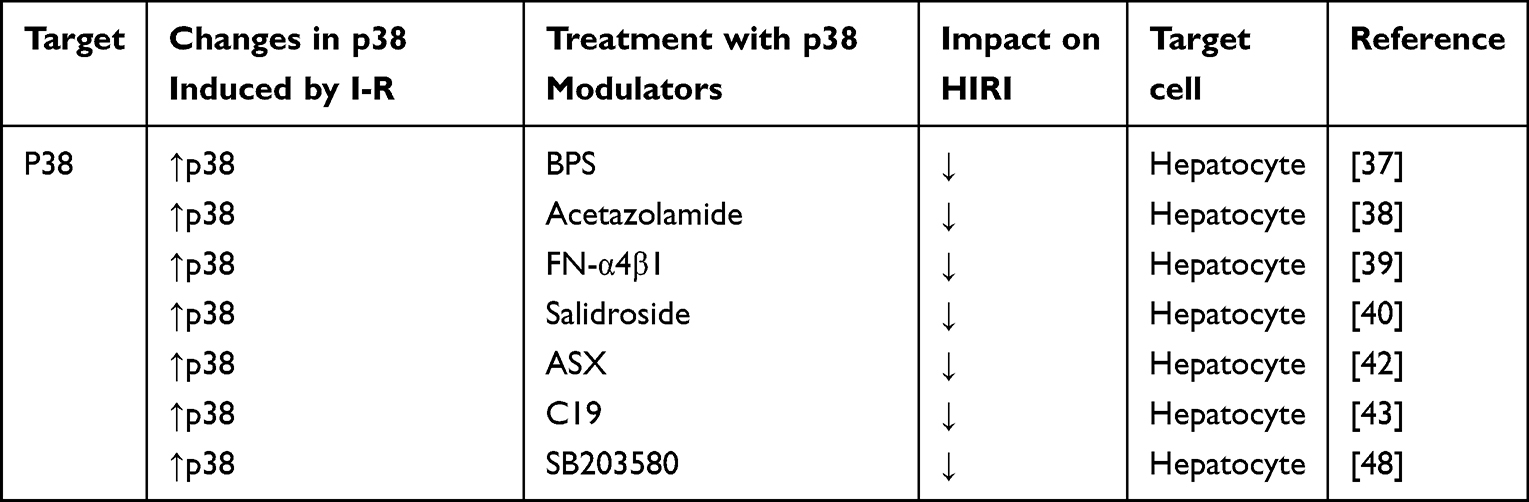 |
Table 1 Effect of Strategies That Regulate p38 in Experimental Models of Hepatic Ischemia- Reperfusion Using Livers |
Autophagy is a highly conserved program in eukaryotic cells. Autophagy plays an important part in counteracting adverse external stimuli such as IR.32,33 An “inflammatory cascade” involving activated p38 and JNK is closely related to the aggravation of inflammation and autophagic cell death observed during HIRI.34–36 Some scholars have reported that activation of p38 or JNK can be blunted by inhibiting expression of proinflammatory mediators such as TNF-α and IL-1β to reduce such inflammatory cascades. Apoptosis can be triggered by either external or internal factors, which is an important feature of HIRI pathogenesis. Inhibition of phosphorylation of p38 or JNK appears to protect against apoptosis, which can reduce HIRI or other types of acute liver injury.37
Acetazolamide has been used as a diuretic. However, Bejaoui et al showed that acetazolamide can improve HIRI by inhibiting activation of p38, ERK, and JNK.38
In vitro studies have shown that beraprost sodium (BPS) can inhibit TNF expression in human monocytes by inhibiting the phosphorylation of p38, JNK, and ERK-α to reduce lipopolysaccharide-induced inflammation. In the mesangial cells of rats, BPS has been shown to reduce ROS production and increase antioxidant activity, potentially preventing diabetic nephropathy. In vivo studies have revealed that BPS activity is related to its anti-inflammatory or antioxidant properties, and that the liver is an important target for BPS to intervene in acute or chronic progressive liver disease. As a result, BPS may be a promising HIRI treatment.37
CS-1 peptide-promoted blockade of interactions between fibronectin-a4b1 and integrins inhibits phosphorylation of p38MAPK (which is considered to be an attractive drug-intervention target), which suggests the regulatory role of fibronectin on activation of p38MAPK in HIRI. In addition, there is evidence that the p38MAPK kinase pathway controls fibronectin-mediated induction of matrix metallopeptidase (MMP)-9 and membrane-type matrix metalloproteins(MT1-MMP/MMP-14)by macrophages.39
One study on salidroside using mouse livers showed that it could protect hepatocytes from IRI by reducing the serum level of liver enzymes.40 Those findings were confirmed by histopathology. Therefore, salidroside was postulated to inhibit activation of MAPK signaling (including phosphorylation of p38, JNK, and ERK) and protect hepatocytes from IRI. Salidroside ameliorated inflammatory infiltration, apoptosis and autophagy in damaged liver of mice.
Two studies focused on use of astaxanthin. After creation of a HIRI model, astaxanthin was administered: ROS production decreased. Also, expression of phosphorylated (p)-p38MAPK, p-ERK, and p-JNK in liver tissue decreased. That observation indicated that astaxanthin inhibited ROS production and phosphorylation activation of key proteins related to the MAPK family, which may be an important strategy to reduce apoptosis and autophagy.41,42
During HIR, the mRNA and protein expression of chemokine-like factor (CKLF)1 is upregulated. Immunohistochemical staining has shown neutrophil infiltration to be increased in ischemic livers, and myeloperoxidase activity to be increased significantly, after reperfusion. Levels of TNF-α and IL-1β are increased during HIRI. Administration of an antagonist of CKLF1, C19, was shown to significantly reduce levels of alanine aminotransferase(ALT) and aspartate (AST) aminotransferase and the necrotic area of liver tissue. Further studies showed that C19 treatment suppressed phosphorylation of p38 and JNK.43
It has been reported that thioredoxin interacting protein (TXNIP) expression is upregulated in a rat model of HIRI.44–47 In one study, TXNIP overexpression was found to upregulate phosphorylation of p38 and JNK significantly, and this effect was inhibited significantly by TXNIP knockout.48 In addition, the p38-specific inhibitor SB203580 eliminated the effect of TXNIP overexpression on oxygen glucose deprivation/reoxygenation (OGD/R)-induced cell injury. Taken together, those results suggested that TXNIP knockout reduced hepatocyte IRI by preventing activation of the p38/JNK pathway. Therefore, TXNIP may be a new therapeutic target for HIRI treatment.
Role and Therapeutic Importance of JNK in HIRI
JNK, like p38, is activated during stress and inflammation, and participates in HIRI.7,31,49–53 If IR occurs, JNK is activated by the ROS produced by mitochondria, which is an important factor in HIRI.49,50 Previous studies have shown that inhibition of JNK activation attenuates HIRI.31,51,54,55 However, some studies have shown that JNK activation can inhibit liver regeneration after liver transplantation, which may be related to the occurrence or severity of IRI after liver surgery. However, JNK plays an important role in HIRI. Studying the mechanism of action of JNK in HIRI treatment is very important.31,56 Table 2 shows the current regulatory factors and therapeutic potential of JNK in HIRI.
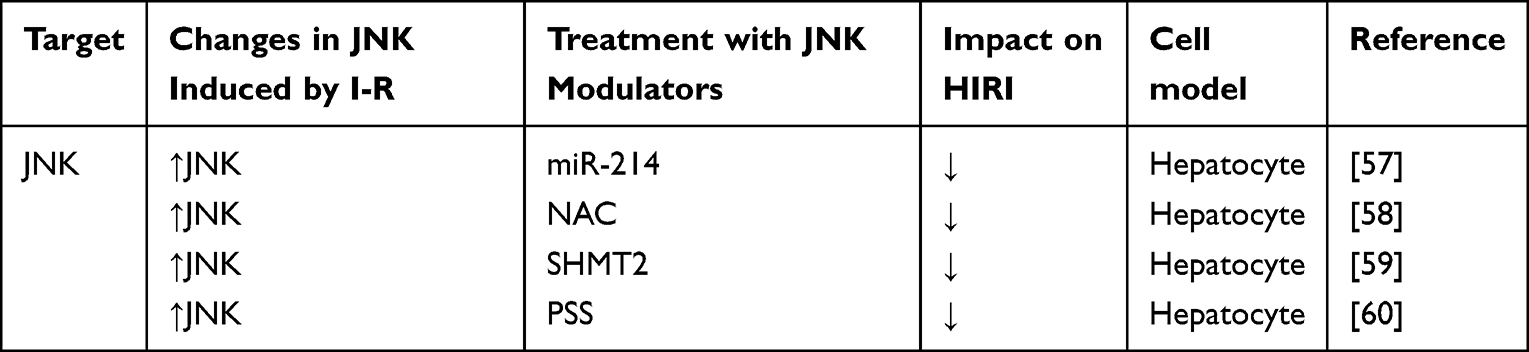 |
Table 2 Effect of Strategies That Regulate JNK in Experimental Models of Hepatic Ischemia- Reperfusion Using Livers |
Autophagy is a self-degrading and ubiquitous process in the body for recycling cellular contents, and protects organisms from various diseases (including HIRI). Activated JNK can induce B-cell lymphoma (Bcl)-2 phosphorylation, lead to separation of beclin-1 and Bcl-2, and lead to the death of autophagic cells. Therefore, the inflammation, apoptosis, and autophagy involved in HIRI are (at least in part) mediated by p38 and JNK signaling.37
Huang et al showed that simulation with microRNA (miR)-214 could reduce expression of TRAF1 (TNF receptor associated factor 1), p-ASK1 (Apoptosis signal-regulating kinase 1), and p-JNK in HIRI and inhibit the ASK1/JNK pathway. In their elegant study, miR-214 expression was downregulated in hepatocytes that underwent IRI and H/R, and miR-214 attenuates I/R-induced hepatocyte apoptosis by inhibiting the TRAF1/ASK1/JNK pathway, which expected to be a potential target for the prevention and treatment of HIRI.57 N-acetylcysteine (NAC) inhibits I/R-mediated apoptosis and autophagy by affecting the JNK signaling pathway. The mechanism of action of NAC may involve weakening of expression of JNK and p-JNK by ROS-scavenging, an indirect increase in the Bcl-2 level and, finally, a change in the balance of expression between beclin 1 and Bcl-2. Phosphorylation of Bcl-2 can lead to separation of Bcl-2 from beclin 1, which reduces the inhibitory effect on beclin 1. Therefore, JNK activation can involve a change in the balance of expression between Bcl-2 and beclin 1, and may be the way that JNK regulates autophagy. Those results suggest potential clinical treatment of HIRI by NAC.58
Studies have demonstrated that JNK can affect mitochondria directly and cause apoptosis. JNK is activated during warm HIRI and cold HIRI induced by liver transplantation. Some studies have revealed expression of serine hydroxymethyltransferase (shmt)2 in a mouse model of IR. Wu and collaborators showed that impaired expression of shmt2 could induce JNK activation, promote apoptosis, and aggravate HIRI.59
Xu et al investigated the protective function and mechanism of action of propylene glycol alginate sodium sulfate (PSS) in a mouse model of HIRI. PSS pretreatment could significantly reduce histologic injury, release of transaminases, and production of proinflammatory cytokines. Compared with the control (IR model) group, PSS protected the liver from IRI by inhibiting MAPK signal transduction and downregulating inflammation, apoptosis.60
Role and Therapeutic Importance of ERK1/2 in HIRI
ERK1/2 is expressed widely in the human body. It is activated mainly by cell proliferation-related products, such as cell-growth factors, cell division-stimulating factors, and hormones.61 It can also be activated by the extracellular matrix and ROS.26,61,62 ERK1/2 is associated with proliferation, but some studies have shown that activation and expression of ERK1/2 can aggravate IRI,63–66 which may be related to neutrophil infiltration and cell death.67–70 While some studies also have shown that activation of ERK1/2 may protect against IRI,71 which seems to be related to the time and severity of IR.69,72–75 Table 3 shows the current regulatory factors and therapeutic potential of ERK1/2 in HIRI.
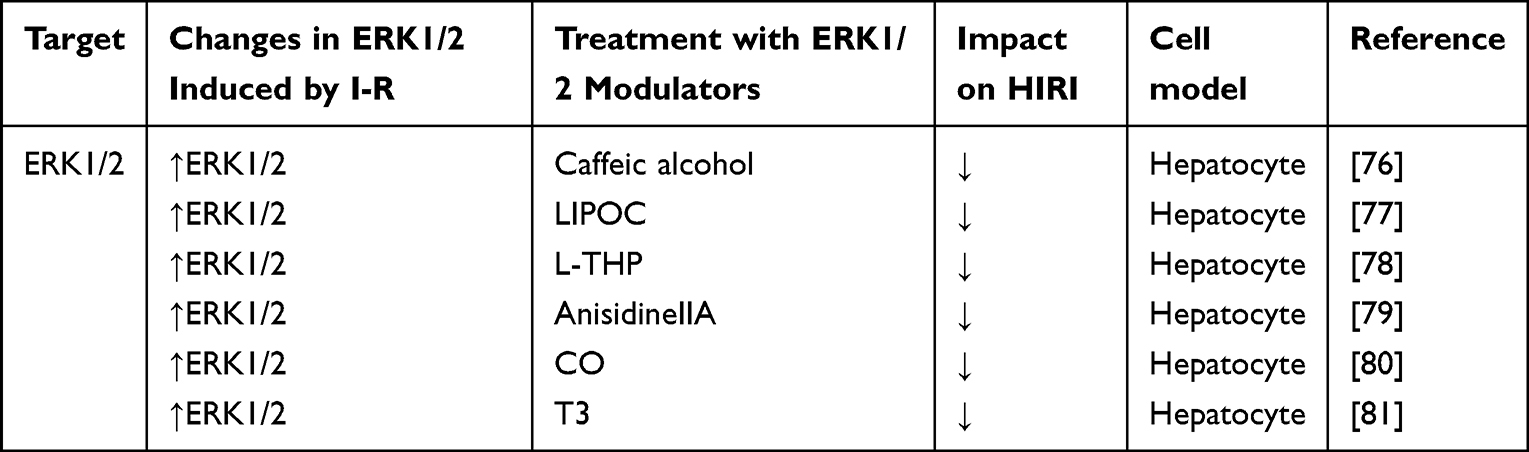 |
Table 3 Effect of Strategies That Regulate ERK1/2 in Experimental Models of Hepatic Ischemia- Reperfusion Using Livers |
Many scholars have reported the ERK pathway to be closely related to the inflammatory response, apoptosis, and autophagy. Inhibition of the ERK pathway has been found to be protective during HIRI. Studies have shown that peroxisome proliferator activated receptor (PPAR)γ expression is closely related to the proliferation, differentiation, and apoptosis of cells. In addition, several studies have shown that p-ERK can activate PPARγ. Caffeic alcohol inhibits inflammation, apoptosis, and autophagy through the ERK/PPARγ pathway, thereby protecting the liver (at least in part) from damage during IRI. Further experimental verification of that postulated mechanism is needed.76
Limb ischemic postconditioning (lipoc) is a method employed to reduce IRI. In a study on the protective effect of lipoc on HIRI and its mechanism of action, lipoc helped to protect against HIRI in rats, but this protective effect could be eliminated by a blocker of ERK1/2: PD98059. Hence, Liposomes mediate protection against IRI through activation of the ERK1/2 signaling pathway. However, how ERK1/2 may have a protective role after being activated by lipoc needs to be studied further.77
Studies have shown that L-tetrahydropalmatine (L-THP) pretreatment can reduce IR-induced hepatocyte injury and secretion of proinflammatory cytokines such as IL-6 and TNF-α. In addition, L-THP can inhibit the ERK/nuclear factor-kappa B (NF-κB) signaling pathway, thereby inhibiting the apoptosis and autophagy of hepatocytes. The protective effect of L-THP has been shown to be positively correlated with its dose.78
Wang et al used a HIRI model in mice to investigate the role of T lymphocytes in regulating autophagy and their subsequent protective effect on hepatocytes. Pretreatment with anisidineIIA before HIR could enhance the autophagy of hepatocytes significantly through the MEK/ERK/mammalian target of rapamycin (mTOR) pathway. Enhanced autophagy could reduce ROS production by scavenging damaged mitochondria so as to protect against HIR. This protective effect was manifested in a reduced serum level of enzymes, liver-tissue injury, infiltration of inflammatory cells, proinflammatory cytokines, and hepatocyte apoptosis.79
Carbon monoxide (CO) is a product of heme-oxygenase degradation. CO has been shown to provide cellular protection in various models of tissue injury. Some studies have investigated the effect of exogenous inhalation of CO on cold IR after liver transplantation. Cold HIRI was found to be related to the rapid phosphorylation of MAPK in liver grafts 1 h after orthotopic liver transplantation, and to significantly inhibit phosphorylation of ERK1/2, MAPK, an upstream transcription factor (MEK1/2) and downstream transcription factor (c-myc). Those results showed that exogenous CO could: (i) inhibit expression of early proinflammatory and stress-response genes, and improve HIRI; (ii) downregulate the MEK/ERK1/2 signaling pathway.80
Yang and collaborators studied triiodothyronine (T3) treatment in male C57BL/6 mice that underwent HIRI. Mice pretreated with T3 before IR induction showed increased autophagy mediated by the MEK/ERK/mTORC1 pathway. Pretreatment with T3 could protect against HIRI by enhancing autophagy. Therefore, T3 preconditioning may be a potential method for HIR treatment.81
Role and Therapeutic Importance of ASK1 in HIRI
The initiation of MAPK cascade can be initiated by the pressure of cell membrane receptors, intracellular inflammatory response bodies, or organelles such as mitochondria and endoplasmic reticulum. However, the sustained activation of MAPK requires the involvement of ASK1. ASK1 is usually inactivated by binding with various inhibitors such as thioredoxin or glutathione transferase.82 These inhibitors are redox sensitive. Therefore, ROS generated in the process of hepatic ischemia-reperfusion injury can activate ASK1 from the inhibited state and aggravate the inflammatory injury through the downstream JNK signaling pathway. Thus, JNK and ASK1 remove irreparably damaged cells through mechanisms that regulate endoplasmic reticulum stress and mitophagy.83 Table 4 shows the current regulatory factors and therapeutic potential of ASK1 in HIRI.
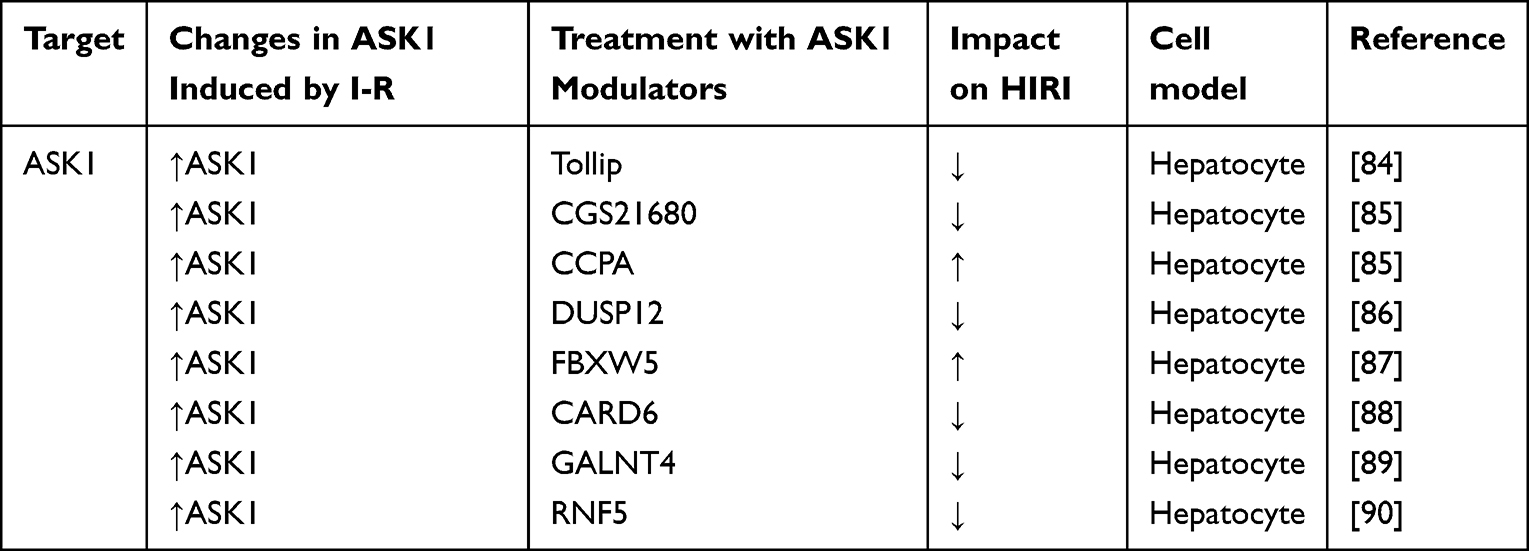 |
Table 4 Effect of Strategies That Regulate ASK1 in Experimental Models of Hepatic Ischemia- Reperfusion Using Livers |
Some studies have found that the expression of toll interacting protein (Tollip) is closely related to the process of HIRI. RNA sequencing analysis and phenotypic detection showed that Tollip deficiency could significantly reduce HIRI in vivo and in hepatocytes. Tollip is a regulator of HIRI by promoting ASK1 N-terminal dimerization and the resulting activation of JNK/p38 signal. Inhibiting Tollip or its interaction with ASK1 may be an effective strategy to treat HIRI.84
Adenosine receptors (ARs) are potentially effective therapeutic targets for liver diseases. The effects of drugs on A2AR and A1R activation were studied. CGS21680, an A2AR agonist, protected primary adipose mouse hepatocytes from IR-induced ASK1 and JNK activation. This effect was attributed to the inhibition of ASK1 by PI3K/Akt. In contrast, the A1R agonist CCPA increased IRI, intracellular lipoatrophy, and oxidative species (OS) production, which increased lipid/OS-dependent ASK1-JNK stimulation. These findings point to a new mechanism for CGS21680-mediated hepatoprotection: PI3K/Akt-dependent inhibition of ASK1. CGS2168 and CCPA may reduce or increase IRI in fatty liver by inhibiting or activating the cytotoxic ASK1/JNK axis, respectively.85
DUSP12 belongs to the DUSP (dual specific phosphatase) family and some DUSPs have been identified as being involved in HIRI regulation. Researchers found a significant decrease in DUSP12 expression in the mouse model of HIRI in vivo and the hypoxia / reoxygenation (H/R) model in vitro in one study. The study discovered that DUSP12 attenuated I/R-induced liver injury in DUSP12 transgenic mice. Furthermore, both in vitro and in vivo, DUSP12 inhibited hepatic inflammatory responses and attenuated apoptosis. ASK1 is required for DUSP12 to play its role in HIRI, and inhibiting ASK1 prevented inflammation and apoptosis in DUSP12 deficient mice and hepatocytes. The ASK1-JNK/p38 signaling pathway is primarily responsible for DUSP12’s regulatory role.86
F-box/WD repeat-containing protein 5 (FBXW5) is a key regulator of stress signaling and has recently been reported to be involved in liver immunity. In one study, inhibition of FBXW5 was found to significantly reduce MAPK and nuclear factors κB kinase (IKK) inhibitor pathway, leading to cytokine release, liver pathological damage and apoptosis. Overexpression of FBXW5 achieved the opposite effect. Studies on its mechanism have shown that FBXW5 exacerbates liver inflammation by promoting the phosphorylation of ASK1, and blocking TRAF6 can eliminate this process. Indicating that FBXW5 may become a key accelerator of HIRI by enhancing the activation of ASK1 in a TRAF6 dependent manner.87
Caspase recruitment domain family member 6 (CARD6) was initially shown to be expressed in NF- κB plays an important role in activation. Some researchers found that the expression levels of CARD6 were significantly downregulated in transplanted livers after exposure to IRI in liver transplant patients and mouse liver transplant models, and explored the molecular mechanism of CARD6 function in vivo and in vitro. CARD6 was found to be a new protective factor against HIRI, which inhibits inflammation and hepatocyte death by inhibiting ASK1 signaling pathway.88
Glycosyltransferases have been identified as potential targets for the treatment of HIRI, but their role in HIRI remains to be studied. We investigated the exact function and molecular mechanism of the glycosyltransferase N-acetylgalactosaminytransferase-4 (GALNT4) in HIRI. It was found that there was a close correlation between GALNT4 expression and HIRI related molecular events in mouse models. In patients receiving liver transplantation and mouse models, GALNT4 expression levels were significantly upregulated after reperfusion surgery. We found that GALNT4 overexpression significantly attenuated the extent of I/ R-induced liver parenchymal injury, inflammatory cell infiltration and hepatocyte death, whereas GALNT4 knockdown resulted in the opposite phenotype. In contrast, mechanistic studies suggest that GALNT4 directly binds ASK1 and inhibits its n-terminal dimerization and subsequent phosphorylation, leading to robust inactivation of downstream JNK/p38 and NF-κB signaling. Interestingly, the inhibitory ability of Galnt4 on ASK1 activation was independent of its glycosyltransferase activity.89
RNF5 plays an important role as an E3 ubiquitin ligase in the regulation of cell differentiation, growth and transformation. It has been found that RNF5 is significantly downregulated during HIR in mice and hepatocytes. Subsequently, RNF5 knockdown and cell line overexpression were stimulated by hypoxia reoxygenation. The results showed that inflammatory infiltration and hepatocyte apoptosis were significantly increased in the liver of RNF5 knockout mice, while RNF5 overexpression attenuated IR-induced liver injury. Mechanistically, RNF5 interacts with phosphoglycerate mutase family member 5 (PGAM5) and mediates the degradation of PGAM5 k48-linked ubiquitination, thereby inhibiting the activation of the ASK1/JNK/p38 pathway. This ultimately suppressed the inflammatory response and apoptosis in HIRI.90
Role and Therapeutic Importance of TAK1 in HIRI
TAK1 is a NF- κB and intracellular protein kinase upstream of JNK, which takes an important part in HIRI. The autophosphorylation of TAK1 is dependent on the ubiquitination regulation of TAK1 itself.91 Ubiquitination and deubiquitination are essential for regulating TAK1 mediated activation and have been confirmed in numerous disease model studies.92 Table 5 shows the current regulatory factors and therapeutic potential of TAK1 in HIRI.
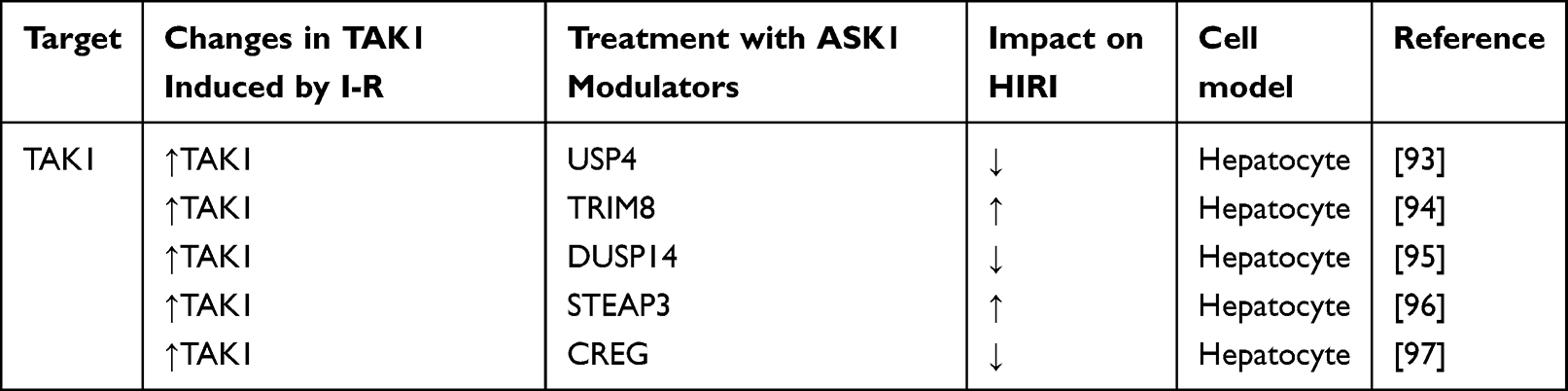 |
Table 5 Effect of Strategies That Regulate TAK1 in Experimental Models of Hepatic Ischemia- Reperfusion Using Livers |
Ubiquitin specific peptidase 4 (USP4) protein is a deubiquitinated enzyme related to many important biological processes. A study has explored the role of USP4 in HIRI. The results showed that USP4 was significantly upregulated in the liver of mice suffering from HIRI. USP4 knockout mice showed aggravated HIRI. In addition, Overexpression of USP4 reduced hepatic inflammatory infiltration and apoptosis upon HIR stimulation. USP4 deficiency has adverse effects on HIRI by inducing the activation of TAK1 / JNK signaling pathway. Studies have shown that USP4 deficiency plays a harmful role in HIRI by promoting the activation of TAK1 / JNK signaling pathway.93
Ternary motif (TRIM)8 is an E3 ubiquitin ligase that interacts with and ubiquitinates with a variety of substrates and is closely related to innate immunity. A study has explored the role of TRIM8 in HIRI. The results showed that TRIM8 was significantly upregulated in the liver of mice suffering from HIRI. TRIM8 knockdown can alleviate the injury of hepatocytes caused by I/R. In vitro and in vivo, inhibition of TRIM8 expression alleviates hepatic inflammatory response and inhibits apoptosis. Mechanistically, studies have shown that TRIM8 deficiency may alleviate IRI by inhibiting the activation of TAK1 and its downstream JNK/P38 pathway. TAK1 requires TRIM8 function in HIRI, because TAK1 activation abrogates TRIM8 function in vitro. In addition, TRIM27 was found to be a key regulator of HIRI by mediating the degradation of TAK1 binding protein(TAB)2/3 and the inhibition of downstream TAK1-JNK/p38 signal. TRIM27 may be a promising method to protect the liver of transplant recipients from I / R-mediated hepatocyte injury.94
The investigators found that DUSP14 was significantly down regulated in liver tissues of liver transplant patients and mice undergoing liver I/R surgery. DUSP14 knockout and DUSP14 transgenic mouse models showed that DUSP14 reduced cell death, improved inflammation, and promoted hepatocyte proliferation/regeneration. Dusp14 also inhibits MAPK and NF-κB signaling through physical interaction with TAK1. The results show that DUSP14 is a protective factor of HIRI, and DUSP14 plays an important role in ameliorating HIRI by inhibiting the TAK1-JNK1/2 pathway. Targeting regulation of this axis may be a novel strategy to prevent or intervene in this pathological process.95
Six-transmembrane epithelial antigen of the prostate (STEAP3) are key regulators of iron uptake and participate in the immune and apoptotic processes of various cell types. In one study, researchers found that STEAP3 expression was significantly upregulated in the liver tissues of mice undergoing liver I/R surgery and primary hepatocytes subjected to hypoxia/reoxygenation injury. Subsequently, the molecular mechanism of STEAP3 function was explored in vivo and in vitro. The results showed that STEAP3 deficiency could inhibit TAK1 activation and downstream JNK and p38 signaling. STEAP3 is a mediator of HIRI and regulates inflammatory response and apoptosis through TAK1 dependent activation of JNK/p38 pathway.96
The cellular repressor of E1A-stimulated genes (CREG) is a key regulator of cell proliferation, plays a protective role in cardiovascular disease, and is involved in steatosis and inflammatory responses in the liver. However, other effects of CREG on the liver and on IRI are unknown. However, the role of CREG in HIRI remains largely unknown. One study used genetic engineering technology to explore the role of CREG in HIRI and found that CREG inhibits MAPK signaling by inhibiting TAK1 phosphorylation. The results showed that CREG could prevent HIRI. CREG plays a protective role in HIRI by directly interacting with TAK1 and inhibiting the phosphorylation of TAK1 and activation of the downstream JNK/p38 pathway, thereby attenuating the inflammatory response and apoptosis.97
MAPK Pathway in KCs
KCs participate directly in the inflammatory process during HIRI. Therefore, exploring the role of MAPK pathways in KCs is very important when studying HIRI. Recent studies on KCs in HIRI are as follows. (Tables 6).
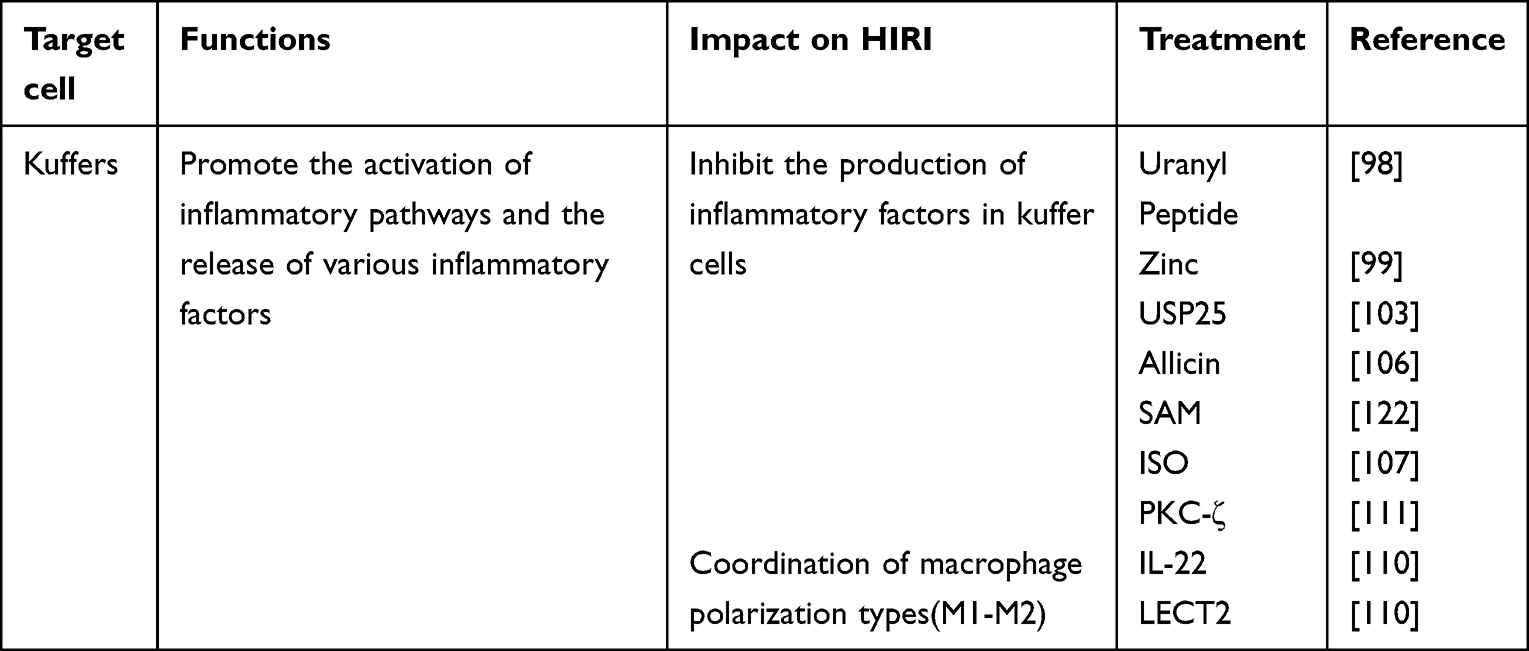 |
Table 6 Effects of Strategies to Regulate MAPK on KCs |
Liu et al showed that KCs increased levels of urotensin II (UII), urotensin II receptor (UIIR), TNF-α, and IL-1β significantly after stimulation with lipopolysaccharide. Pretreatment with uranyl peptide (UIIR antagonist) could significantly inhibit the effects of lipopolysaccharide stimulation of KCs in terms of expression of UII/UIIR, TNF-α, and IL-1β. LPS stimulation induced phosphorylation of nuclear p38MAPK and NF-κB, expression of the p65 subunit, and enhancement of NF-κB binding activity to DNA molecules, whereas pretreatment with uranyl peptide significantly inhibited the nuclear expression and activity of these molecules stimulated by lipopolysaccharide in KCs. Therefore, the UII/UIIR system may mediate the production and release of lipopolysaccharide-stimulated proinflammatory cytokines through KCs. This mediating effect may be dependent (at least in part) on p38MAPK and NF-κB.98
Adequate zinc levels are essential for reducing inflammatory processes in the body, and studies have shown that zinc supplementation can improve the prognosis of patients with chronic liver disease.99–101 Some studies have discussed the clinical application of serum zinc in patients with chronic liver disease and the anti-infection mechanism of zinc supplementation.99 Zinc pretreatment has been found to increase the level of lactate dehydrogenase in KCs and HSCs, and reduce gene expression of myeloid differentiation primary response (MyD)88, MAPK, and NF-κB. Those data indicate that zinc supplementation acts on KCs through a MyD88/MAPK-related pathway to reduce the inflammatory effect on the liver.
Inhibition of tumor necrosis factor receptor related factor (TRAF)3 degradation induces endotoxin tolerance in macrophages.102 However, the mechanism leading to TRAF3 inhibition is largely unknown. Ubiquitin-specific peptidase 25 (USP25) is a deubiquitinase. USP25 interacts with TRAF3 and stabilizes endotoxin tolerance in KCs.103 The mechanism suggests that USP25 induces endotoxin tolerance in KCs by removing the TRAF3 K48-linked ubiquitin chain thereby inhibiting its degradation and reducing the phosphorylation level of downstream JNK/P38, promoting the secretion of the inflammatory factor IL-10.
Acrylamide in heated processed foods has attracted extensive attention because of its hepatotoxicity.104 Allicin is a plant-derived antioxidant which has a significant protective effect against acrylamide-induced hepatotoxicity, but its mechanism of action is not known.105 Steib et al studied the mechanism of action of allicin in KCs.106 Allicin was found to inhibit activation of MAPK and NF-κB pathways and downregulate phosphorylation of JNK, ERK, p38, p65, and nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκBα). Allicin pretreatment seemed to inhibit acrylamide-induced inflammation.
The volatile anesthetic isoflurane has an immunomodulatory effect, whereas the fungal component fermentase induces inflammation through TLR2 or dectin-1 signaling pathways.107,108 Wang et al studied the molecular effect of isoflurane (0.7%) on inflammatory activation of mouse KCs induced by fermentase.109 In mouse KCs, isoflurane treatment significantly reduced fermentase-induced upregulation of cyclooxygenase-2 expression, prostaglandin E2 release, and the production of proinflammatory cytokines and chemokines. Wang et al showed (for the first time) that isoflurane treatment partially improved the fermentase-induced inflammatory response of KCs in vivo and in vitro by reducing ROS production, and that p38MAPK was regulated by ROS.
Su et al found that exogenous IL-22 significantly inhibited the fibrogenic factors and macrophage phenotype-changing factors secreted by M1 KCs while increasing the number of M2 KCs during the progression of carbon tetrachloride-induced liver fibrosis in mice.110 A high M2/M1 KC ratio inhibited collagen production and HSC activation in a coculture of HSCs and KCs from mice treated with IL-22. These findings suggested that IL-22 could boost the ratio of M2 KCs to M1 KCs, slowing the progression of liver fibrosis. IL-22 promoted the polarization of lipopolysaccharide-treated macrophages from the M1 to the M2 phenotype, according to in vitro mechanistic studies. Cytokines achieved these effects by activating the signal transducer and activator of transcription (STAT)3 pathway while inhibiting the ERK1/2 and protein kinase C pathways.
Peng and collaborators transfected protein kinase C (PKC)-ζ small interfering RNA into a mouse KC line (mkcl3-2). Then, they treated the cells with elastase alone or an ERK inhibitor (U0126). The PKC-ζ protein/activity of cell extracts was determined, as was TLR-4 expression, NF-κB nuclear translocation, phosphorylation of ERK1/2, activated caspase-3, and DNA fragmentation. KC activation led to upregulated PKC-ζ activity, increased apoptosis, and induction of NF-κB nuclear translocation through an ERK1/2-dependent pathway. Inhibition of PKC-ζ activity in KCs significantly decreased apoptosis as well as activation of NF-κB and ERK1/2.111
Leukocyte-derived chemokine (LECT)2 is a hepatocyte factor that “senses” fat in the liver and whose expression is upregulated before weight gain.112–114 LECT2 has been shown to selectively enhance lipopolysaccharide-induced JNK phosphorylation, but not to enhance phosphorylation of ERK or p38 in a mouse line of KCs (kup5). Consistently, LECT2 enhanced lipopolysaccharide-induced phosphorylation of two upstream activators of JNK: MKK4 (Mitogen-activated protein kinase kinase 4) and Table 2. The increase in LECT2 expression transformed the residual macrophages in the liver into the M1-like phenotype and contributed to the development of liver inflammation.115 Those findings revealed that the hepatocyte factor LECT2 could be used as a target for treatment of inflammatory diseases of the liver.
Conclusions and Perspectives
For various end-stage liver diseases, liver transplantation is the last option to save life. IRI is the first complication of liver transplantation. IRI involves complex mechanisms and has a huge impact on grafts. Avoiding or reducing IRI occupies the thoughts of all surgeons. MAPKs have great potential for the prevention and treatment of IR. p38, JNK, ERK1/2, ASK1 and TAK1 have been shown to have important roles in the occurrence and development of IR. We summarized some researches (Tables 1–6).However, some of the mechanisms are not clear, and some proteins have dual roles. For example, in most studies, inhibiting JNK helps reduce HIRI, but other studies have found that JNK activation inhibits liver regeneration after liver transplantation.These factors may be related to the type of liver, cold ischemia time, and other factors. Our goal is to discover how these different factors affect the liver and MAPKs, and whether study designs can be improved. After solving these problems, MAPKs could be important targets for HIRI treatment.
Autophagy is a highly conserved cellular program in the body. Through the regulation of autophagy related genes, cells can selectively transfer some specific organelles or metabolites to lysosomes for dissolution and transformation. Under stress, autophagy is considered to be a mechanism to protect cells. In the case of HIRI, autophagy can inhibit the production of ROS by mediating the phagocytosis of damaged mitochondria, and the production of ROS is the key link of liver IRI. Therefore, it is very important to find autophagy related regulators. In previous studies, it was found that inhibiting multiple members of MAPK family can enhance autophagy, indicating that HIRI can be effectively reduced by regulating related members of MAPK family.
LIPOC is a method to improve IRI, which has been studied more in recent years. Previous studies have found that LIPOC has a good protective effect on IRI of heart and kidney. Recently, it was found that LIPOC can protect the liver by activating ERK1 / 2 signaling pathway in HIRI. Compared with traditional ischemic preconditioning, LIPOC has stronger operability and less side effects. It is a good method to reduce HIRI, however, the specific mechanism needs to be investigated further. In addition, some other pre-treatment methods may also improve HIRI.For example, Fisetin can mediate GSK3β/AMPK/NLRP3 inflammasome pathway can fight inflammatory reaction and protect HIRI.116
In addition, we also pay attention to the polarization of intrahepatic macrophages. Due to their plasticity and activation mode, intrahepatic macrophages are mainly divided into classically activated macrophages (M1 type) and selectively activated macrophages (M2 type), while M1 type macrophages are mainly involved in initiating and maintaining inflammatory responses and M2 type macrophages are mainly involved in inflammation regression. If we can interfere with the polarization direction of macrophages, with M2 macrophages as the main differentiation direction, it will well protect hepatocytes. It is reported that miR-450b-5p suppressed alpha B-crystallin (CRYAB), CRYAB activates M2 polarization through Akt1/ mTOR target, thus alleviating HIRI.117
There are other ways and factors involved in the occurrence and development of HIRI. For example, miR-24-3p can improve the inflammatory response and cell apoptosis in HIRI process by targeting STING.118 Liver selective MMP inhibition can prevent the hydrolytic cleavage of liver VEGF protein, thus enhancing the recruitment and implantation of bone marrow after liver injury.119 This can improve the injury and accelerate liver regeneration. miR-125b protects liver from HIRI via inhibiting TRAF6 and NF-κB signal pathway.120 FAM49B, constrained by miR-22, alleviates HIRI by inhibiting the TRAF6/IKK signal pathway in a Rac1-dependent manner.121
Taken together, we primarily concentrate on the related roles of p38, JNK and ERK1 / 2 in MAPK family in IR. It is worth noting that different researchers use different methods and time to create animal models, which may require more in-depth horizontal and vertical studies to compare these differences caused by different operation methods. At the same time, we also pay attention to some promising research, and discussed it in the previous article, hoping to provide some ideas for researchers.
Disclosure
The authors report no conflict of interest in this study.
References
1. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–252. doi:10.1016/S0092-8674(00)00116-1
2. Kobayashi M, Takeyoshi I, Yoshinari D, et al. The role of mitogen-activated protein kinases and the participation of intestinal congestion in total hepatic ischemia-reperfusion injury. Hepatogastroenterology. 2006;53(68):243–248.
3. Mccubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8(9–10):1775–1789. doi:10.1089/ars.2006.8.1775
4. Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19(4):2435–2444. doi:10.1128/MCB.19.4.2435
5. Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2(9):717–726. doi:10.1038/nrd1177
6. Schnabl B, Bradham CA, Bennett BL, et al. TAK1/JNK and p38 have opposite effects on rat hepatic stellate cells. Hepatology. 2001;34(5):953–963. doi:10.1053/jhep.2001.28790
7. Toledo-Pereyra LH, Toledo AH, Walsh J, et al. Molecular signaling pathways in ischemia/reperfusion. Exp Clin Transplant. 2004;2(1):174–177.
8. Casillas-Ramírez A, Mosbah IB, Ramalho F, et al. Past and future approaches to ischemia-reperfusion lesion associated with liver transplantation. Life Sci. 2006;79(20):1881–1894. doi:10.1016/j.lfs.2006.06.024
9. Saidi RF, Chang J, Brooks S, et al. Ischemic preconditioning and intermittent clamping increase the tolerance of fatty liver to hepatic ischemia-reperfusion injury in the rat. Transplant Proc. 2007;39(10):3010–3014. doi:10.1016/j.transproceed.2007.09.044
10. Clavien PA, Harvey PR, Strasberg SM. Preservation and reperfusion injuries in liver allografts. An Over Syn Curr Stu Transpl. 1992;53(5):957–978.
11. Selzner N, Rudiger H, Graf R, et al. Protective strategies against ischemic injury of the liver. Gastroenterology. 2003;125(3):917–936. doi:10.1016/S0016-5085(03)01048-5
12. Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, et al. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res. 2008;147(1):153–159. doi:10.1016/j.jss.2007.06.015
13. Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284(1):G15–26. doi:10.1152/ajpgi.00342.2002
14. Gracia-Sancho J, Villarreal G, Zhang Y, et al. Flow cessation triggers endothelial dysfunction during organ cold storage conditions: strategies for pharmacologic intervention. Transplantation. 2010;90(2):142–149. doi:10.1097/TP.0b013e3181e228db
15. Gasbarrini A, Borle AB, Farghali H, et al. Effect of anoxia on intracellular ATP, Na+i, Ca2+i, Mg2+i, and cytotoxicity in rat hepatocytes. J Biol Chem. 1992;267(10):6654–6663. doi:10.1016/S0021-9258(19)50477-X
16. Peralta C, Bartrons R, Riera L, et al. Hepatic preconditioning preserves energy metabolism during sustained ischemia. Am J Physiol Gastrointest Liver Physiol. 2000;279(1):G163–71. doi:10.1152/ajpgi.2000.279.1.G163
17. Kalogeris T, Baines CP, Krenz M, et al. Ischemia/Reperfusion. Compr Physiol. 2016;7(1):113–170. doi:10.1002/cphy.c160006
18. Brown DA, Hale SL, Baines CP, et al. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J Cardiovasc Pharmacol Ther. 2014;19(1):121–132. doi:10.1177/1074248413508003
19. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17(5):306–321. doi:10.1038/nri.2017.11
20. Shapouri-Moghaddam A, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–6440. doi:10.1002/jcp.26429
21. Lu TF, Yang TH, Zhong CP, et al. Dual effect of hepatic macrophages on liver ischemia and reperfusion injury during liver transplantation. Immune Netw. 2018;18(3):e24. doi:10.4110/in.2018.18.e24
22. Banga NR, Homer-Vanniasinkam S, Graham A, et al. Ischaemic preconditioning in transplantation and major resection of the liver. Br J Surg. 2005;92(5):528–538. doi:10.1002/bjs.5004
23. Fan C, Zwacka RM, Engelhardt JF. Therapeutic approaches for ischemia/reperfusion injury in the liver. J Mol Med. 1999;77(8):577–592. doi:10.1007/s001099900029
24. Lentsch AB, Kato A, Yoshidome H, et al. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32(2):169–173. doi:10.1053/jhep.2000.9323
25. Bradham CA, Stachlewitz RF, Gao W, et al. Reperfusion after liver transplantation in rats differentially activates the mitogen-activated protein kinases. Hepatology. 1997;25(5):1128–1135. doi:10.1002/hep.510250514
26. Widmann C, Gibson S, Jarpe MB, et al. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79(1):143–180. doi:10.1152/physrev.1999.79.1.143
27. Han J, Lee JD, Bibbs L, et al. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265(5173):808–811. doi:10.1126/science.7914033
28. Lee JC, Laydon JT, Mcdonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372(6508):739–746. doi:10.1038/372739a0
29. Massip-Salcedo M, Zaouali MA, Padrissa-Altés S, et al. Activation of peroxisome proliferator-activated receptor-alpha inhibits the injurious effects of adiponectin in rat steatotic liver undergoing ischemia-reperfusion. Hepatology. 2008;47(2):461–472. doi:10.1002/hep.21935
30. Fernández L, Carrasco-Chaumel E, Serafín A, et al. Is ischemic preconditioning a useful strategy in steatotic liver transplantation? Am J Transplant. 2004;4(6):888–899. doi:10.1111/j.1600-6143.2004.00447.x
31. Massip-Salcedo M, Casillas-Ramirez A, Franco-Gou R, et al. Heat shock proteins and mitogen-activated protein kinases in steatotic livers undergoing ischemia-reperfusion: some answers. Am J Pathol. 2006;168(5):1474–1485. doi:10.2353/ajpath.2006.050645
32. Nikoletopoulou V, Markaki M, Palikaras K, et al. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833(12):3448–3459. doi:10.1016/j.bbamcr.2013.06.001
33. Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I Ultrastructural and cytochemical changes. Am J Pathol. 1980;98(2):425–444.
34. Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi:10.1056/NEJMra1205406
35. Sakoda H, Ogihara T, Anai M, et al. Activation of AMPK is essential for AICAR-induced glucose uptake by skeletal muscle but not adipocytes. Am J Physiol Endocrinol Metab. 2002;282(6):E1239–44. doi:10.1152/ajpendo.00455.2001
36. Matsui Y, Takagi H, Qu X, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914–922. doi:10.1161/01.RES.0000261924.76669.36
37. Deng J, Feng J, Liu T, et al. Beraprost sodium preconditioning prevents inflammation, apoptosis, and autophagy during hepatic ischemia-reperfusion injury in mice via the P38 and JNK pathways. Drug Des Devel Ther. 2018;12:4067–4082.
38. Bejaoui M, Pantazi E, De Luca V, et al. Acetazolamide protects steatotic liver grafts against cold ischemia reperfusion injury. J Pharmacol Exp Ther. 2015;355(2):191–198. doi:10.1124/jpet.115.225177
39. Duarte S, Shen XD, Fondevila C, et al. Fibronectin-α4β1 interactions in hepatic cold ischemia and reperfusion injury: regulation of MMP-9 and MT1-MMP via the p38 MAPK pathway. Am J Transplant. 2012;12(10):2689–2699. doi:10.1111/j.1600-6143.2012.04161.x
40. Feng J, Zhang Q, Mo W, et al. Salidroside pretreatment attenuates apoptosis and autophagy during hepatic ischemia-reperfusion injury by inhibiting the mitogen-activated protein kinase pathway in mice. Drug Des Devel Ther. 2017;11:1989–2006.
41. Li J, Wang F, Xia Y, et al. Astaxanthin pretreatment attenuates hepatic ischemia reperfusion-induced apoptosis and autophagy via the ROS/MAPK pathway in mice. Mar Drugs. 2015;13(6):3368–3387. doi:10.3390/md13063368
42. Li S, Takahara T, Fujino M, et al. Astaxanthin prevents ischemia-reperfusion injury of the steatotic liver in mice. PLoS One. 2017;12(11):e0187810. doi:10.1371/journal.pone.0187810
43. Li FF, Zhou X, Chu SF, et al. Inhibition of CKLF1 ameliorates hepatic ischemia-reperfusion injury via MAPK pathway. Cytokine. 2021;141(155429):155429. doi:10.1016/j.cyto.2021.155429
44. Nishiyama A, Matsui M, Iwata S, et al. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 1999;274(31):21645–21650. doi:10.1074/jbc.274.31.21645
45. Szpigel A, Hainault I, Carlier A, et al. Lipid environment induces ER stress, TXNIP expression and inflammation in immune cells of individuals with type 2 diabetes. Diabetologia. 2018;61(2):399–412. doi:10.1007/s00125-017-4462-5
46. Zhou R, Tardivel A, Thorens B, et al. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–140. doi:10.1038/ni.1831
47. Anthony TG, Wek RC. TXNIP switches tracks toward a terminal UPR. Cell Metab. 2012;16(2):135–137. doi:10.1016/j.cmet.2012.07.012
48. Zhang Y, Miao LS, Cai YM, et al. TXNIP knockdown alleviates hepatocyte ischemia reperfusion injury through preventing p38/JNK pathway activation. Biochem Biophys Res Commun. 2018;502(3):409–414. doi:10.1016/j.bbrc.2018.05.185
49. Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290(4):G583–9. doi:10.1152/ajpgi.00422.2005
50. Bendinelli P, Piccoletti R, Maroni P, et al. The MAP kinase cascades are activated during post-ischemic liver reperfusion. FEBS Lett. 1996;398(2–3):193–197. doi:10.1016/S0014-5793(96)01228-8
51. Liang R, Nickkholgh A, Hoffmann K, et al. Melatonin protects from hepatic reperfusion injury through inhibition of IKK and JNK pathways and modification of cell proliferation. J Pineal Res. 2009;46(1):8–14. doi:10.1111/j.1600-079X.2008.00596.x
52. Cursio R, Filippa N, Miele C, et al. Involvement of protein kinase B and mitogen-activated protein kinases in experimental normothermic liver ischaemia-reperfusion injury. Br J Surg. 2006;93(6):752–761. doi:10.1002/bjs.5329
53. Teoh N, Dela Pena A, Farrell G. Hepatic ischemic preconditioning in mice is associated with activation of NF-kappaB, p38 kinase, and cell cycle entry. Hepatology. 2002;36(1):94–102. doi:10.1053/jhep.2002.33134
54. Uehara T, Xi Peng X, Bennett B, et al. c-Jun N-terminal kinase mediates hepatic injury after rat liver transplantation. Transplantation. 2004;78(3):324–332. doi:10.1097/01.TP.0000128859.42696.28
55. Uehara T, Bennett B, Sakata ST, et al. JNK mediates hepatic ischemia reperfusion injury. J Hepatol. 2005;42(6):850–859. doi:10.1016/j.jhep.2005.01.030
56. Lee KH, Kim SE, Lee YS. SP600125, a selective JNK inhibitor, aggravates hepatic ischemia-reperfusion injury. Exp Mol Med. 2006;38(4):408–416. doi:10.1038/emm.2006.48
57. Huang X, Gao Y, Qin J, et al. miR-214 down-regulation promoted hypoxia/reoxygenation-induced hepatocyte apoptosis through TRAF1/ASK1/JNK pathway. Dig Dis Sci. 2019;64(5):1217–1225. doi:10.1007/s10620-018-5405-9
58. Wang C, Chen K, Xia Y, et al. N-acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PLoS One. 2014;9(9):e108855. doi:10.1371/journal.pone.0108855
59. Wu H, Bai H, Duan S, et al. Downregulating serine hydroxymethyltransferase 2 deteriorates hepatic ischemia-reperfusion injury through ROS/JNK/P53 signaling in mice. Biomed Res Int. 2019;2019:2712185.
60. Xu S, Niu P, Chen K, et al. The liver protection of propylene glycol alginate sodium sulfate preconditioning against ischemia reperfusion injury: focusing MAPK pathway activity. Sci Rep. 2017;7(1):15175. doi:10.1038/s41598-017-15521-3
61. Rosseland CM, Wierød L, Oksvold MP, et al. Cytoplasmic retention of peroxide-activated ERK provides survival in primary cultures of rat hepatocytes. Hepatology. 2005;42(1):200–207. doi:10.1002/hep.20762
62. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298(5600):1911–1912. doi:10.1126/science.1072682
63. Zhai Y, Shen XD, Gao F, et al. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology. 2008;47(1):207–214. doi:10.1002/hep.21986
64. Kang JW, Koh EJ, Lee SM. Melatonin protects liver against ischemia and reperfusion injury through inhibition of toll-like receptor signaling pathway. J Pineal Res. 2011;50(4):403–411. doi:10.1111/j.1600-079X.2011.00858.x
65. Pratap A, Panakanti R, Yang N, et al. Inhibition of endogenous hedgehog signaling protects against acute liver injury after ischemia reperfusion. Pharm Res. 2010;27(11):2492–2504. doi:10.1007/s11095-010-0246-z
66. Zeng S, Dun H, Ippagunta N, et al. Receptor for advanced glycation end product (RAGE)-dependent modulation of early growth response-1 in hepatic ischemia/reperfusion injury. J Hepatol. 2009;50(5):929–936. doi:10.1016/j.jhep.2008.11.022
67. Pratap A, Panakanti R, Yang N, et al. Cyclopamine attenuates acute warm ischemia reperfusion injury in cholestatic rat liver: hope for marginal livers. Mol Pharm. 2011;8(3):958–968. doi:10.1021/mp200115v
68. Ben Mosbah I, Alfany-Fernández I, Martel C, et al. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010;1(7):e52. doi:10.1038/cddis.2010.29
69. Alfany-Fernandez I, Casillas-Ramirez A, Bintanel-Morcillo M, et al. Therapeutic targets in liver transplantation: angiotensin II in nonsteatotic grafts and angiotensin-(1-7) in steatotic grafts. Am J Transplant. 2009;9(3):439–451. doi:10.1111/j.1600-6143.2008.02521.x
70. Iannelli A, De Sousa G, Zucchini N, et al. Anti-apoptotic pro-survival effect of clotrimazole in a normothermic ischemia reperfusion injury animal model. J Surg Res. 2011;171(1):101–107. doi:10.1016/j.jss.2010.03.035
71. Park SW, Kim M, Chen SW, et al. Sphinganine-1-phosphate protects kidney and liver after hepatic ischemia and reperfusion in mice through S1P1 receptor activation. Lab Invest. 2010;90(8):1209–1224. doi:10.1038/labinvest.2010.102
72. Song J, Zhang YW, Yao AH, et al. Adenoviral cardiotrophin-1 transfer improves survival and early graft function after ischemia and reperfusion in rat small-for-size liver transplantation model. Transpl Int. 2008;21(4):372–383. doi:10.1111/j.1432-2277.2007.00616.x
73. Kamo N, Shen XD, Ke B, et al. Sotrastaurin, a protein kinase C inhibitor, ameliorates ischemia and reperfusion injury in rat orthotopic liver transplantation. Am J Transplant. 2011;11(11):2499–2507. doi:10.1111/j.1600-6143.2011.03700.x
74. EipelC C, Hübschmann U, Abshagen K, et al. Erythropoietin as additive of HTK preservation solution in cold ischemia/reperfusion injury of steatotic livers. J Surg Res. 2012;173(1):171–179. doi:10.1016/j.jss.2010.09.008
75. Moore C, Shen XD, Fondevila C, et al. Fibronectin-alpha4beta1 integrin interactions modulate p42/44 MAPK phosphorylation in steatotic liver cold ischemia-reperfusion injury. Transplant Proc. 2005;37(1):432–434. doi:10.1016/j.transproceed.2004.12.206
76. Ji J, Wu L, Feng J, et al. Cafestol preconditioning attenuates apoptosis and autophagy during hepatic ischemia-reperfusion injury by inhibiting ERK/PPARγ pathway. Int Immunopharmacol. 2020;84:106529.
77. Gao Y, Zhou S, Wang F, et al. Hepatoprotective effects of limb ischemic post-conditioning in hepatic ischemic rat model and liver cancer patients via PI3K/ERK pathways. Int J Biol Sci. 2018;14(14):2037–2050. doi:10.7150/ijbs.28435
78. Yu Q, Wu L, Liu T, et al. Protective effects of levo-tetrahydropalmatine on hepatic ischemia/reperfusion injury are mediated by inhibition of the ERK/NF-κB pathway. Int Immunopharmacol. 2019;70:435–445.
79. Wang Y, Ni Q, Ye Q, et al. Tanshinone IIA activates autophagy to reduce liver ischemia-reperfusion injury by MEK/ERK/mTOR pathway. Pharmazie. 2018;73(7):396–401. doi:10.1691/ph.2018.7509
80. Kaizu T, Ikeda A, Nakao A, et al. Protection of transplant-induced hepatic ischemia/reperfusion injury with carbon monoxide via MEK/ERK1/2 pathway downregulation. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G236–44. doi:10.1152/ajpgi.00144.2007
81. Yang J, Wang Y, Sui M, et al. Tri-iodothyronine preconditioning protects against liver ischemia reperfusion injury through the regulation of autophagy by the MEK/ERK/mTORC1 axis. Biochem Biophys Res Commun. 2015;467(4):704–710. doi:10.1016/j.bbrc.2015.10.080
82. Weijman JF, Kumar A, Jamieson SA, et al. Structural basis of autoregulatory scaffolding by apoptosis signal-regulating kinase 1. Proc Natl Acad Sci U S A. 2017;114(11):E2096–e105. doi:10.1073/pnas.1620813114
83. Du K, Farhood A, Jaeschke H. Mitochondria-targeted antioxidant mito-tempo protects against acetaminophen hepatotoxicity. Arch Toxicol. 2017;91(2):761–773. doi:10.1007/s00204-016-1692-0
84. Yan ZZ, Huang YP, Wang X, et al. Integrated omics reveals tollip as an regulator and therapeutic target for hepatic ischemia-reperfusion injury in mice. Hepatology. 2019;70(5):1750–1769. doi:10.1002/hep.30705
85. Alchera E, Chandrashekar BR, Clemente N, et al. Ischemia/reperfusion injury of fatty liver is protected by A2AR and Exacerbated by A1R stimulation through opposite effects on ASK1 activation. Cells. 2021;10(11). doi:10.3390/cells10113171.
86. Boldorini R, Clemente N, Alchera E, et al. DUSP12 acts as a novel endogenous protective signal against hepatic ischemia-reperfusion damage by inhibiting ASK1 pathway. Clin Sci. 2021;135(1):161–166. doi:10.1042/CS20201091
87. Li TT, Luo YH, Yang H, et al. FBXW5 aggravates hepatic ischemia/reperfusion injury via promoting phosphorylation of ASK1 in a TRAF6-dependent manner. Int Immunopharmacol. 2021;99:107928.
88. Qin JJ, Mao W, Wang X, et al. Caspase recruitment domain 6 protects against hepatic ischemia/reperfusion injury by suppressing ASK1. J Hepatol. 2018;69(5):1110–1122. doi:10.1016/j.jhep.2018.06.014
89. Zhou J, Guo L, Ma T, et al. N-acetylgalactosaminyltransferase-4 protects against hepatic ischemia/reperfusion injury by blocking apoptosis signal-regulating kinase 1 N-terminal dimerization. Hepatology. 2022;75(6):1446–1460. doi:10.1002/hep.32202
90. Ding MJ, Fang HR, Zhang JK, et al. E3 ubiquitin ligase ring finger protein 5 protects against hepatic ischemia reperfusion injury by mediating phosphoglycerate mutase family member 5 ubiquitination. Hepatology. 2022;76(1):94–111. doi:10.1002/hep.32226
91. Xia ZP, Sun L, Chen X, et al. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461(7260):114–119. doi:10.1038/nature08247
92. Lei CQ, Wu X, Zhong X, et al. USP19 Inhibits TNF-α- and IL-1β-Triggered NF-κB activation by deubiquitinating TAK1. J Immunol. 2019;203(1):259–268. doi:10.4049/jimmunol.1900083
93. Zhou J, Qiu T, Wang T, et al. USP4 deficiency exacerbates hepatic ischaemia/reperfusion injury via TAK1 signalling. Clin Sci. 2019;133(2):335–349. doi:10.1042/CS20180959
94. Tao Q, Tianyu W, Jiangqiao Z, et al. Tripartite motif 8 deficiency relieves hepatic ischaemia/reperfusion injury via TAK1-dependent signalling pathways. Int J Biol Sci. 2019;15(8):1618–1629. doi:10.7150/ijbs.33323
95. Wang X, Mao W, Fang C, et al. Dusp14 protects against hepatic ischaemia-reperfusion injury via Tak1 suppression. J Hepatol. 2017;68(1):118–129.
96. Guo WZ, Fang HB, Cao SL, et al. Six-transmembrane epithelial antigen of the prostate 3 deficiency in hepatocytes protects the liver against ischemia-reperfusion injury by suppressing transforming growth factor-β-activated kinase 1. Hepatology. 2020;71(3):1037–1054. doi:10.1002/hep.30882
97. Yang L, Wang W, Wang X, et al. Creg in hepatocytes ameliorates liver ischemia/reperfusion injury in a TAK1-dependent manner in mice. Hepatology. 2019;69(1):294–313. doi:10.1002/hep.30203
98. Liu LM, Liang DY, Ye CG, et al. The UII/UT system mediates upregulation of proinflammatory cytokines through p38 MAPK and NF-κB pathways in LPS-stimulated Kupffer cells. PLoS One. 2015;10(3):e0121383. doi:10.1371/journal.pone.0121383
99. Zhang J, Wieser A, Lin H, et al. Pretreatment with zinc protects Kupffer cells following administration of microbial products. Bio Pharmaco. 2020;127:110208.
100. King J, Brunel SF, Warris A. Aspergillus infections in cystic fibrosis. J Infect. 2016;72:5. doi:10.1016/j.jinf.2016.04.022
101. Sengupta S, Wroblewski K, Aronsohn A, et al. Screening for zinc deficiency in patients with cirrhosis: when should we start? Dig Dis Sci. 2015;60(10):3130–3135. doi:10.1007/s10620-015-3613-0
102. Häcker H, Redecke V, Blagoev B, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439(7073):204–207. doi:10.1038/nature04369
103. Wen J, Bai H, Chen N, et al. USP25 promotes endotoxin tolerance via suppressing K48-linked ubiquitination and degradation of TRAF3 in Kupffer cells. Mol Immunol. 2019;106:53–62.
104. Kandemir FM, Yildirim S, Kucukler S, et al. Protective effects of Morin against acrylamide-induced hepatotoxicity and nephrotoxicity: a multi-biomarker approach. Food Chem Toxicol. 2020;138:111190.
105. Guan MJ, Zhao N, Xie KQ, et al. Hepatoprotective effects of garlic against ethanol-induced liver injury: a mini-review. Food Chem Toxicol. 2018;111:467–473.
106. Nan B, Yang C, Li L, et al. Allicin alleviated acrylamide-induced NLRP3 inflammasome activation via oxidative stress and endoplasmic reticulum stress in Kupffer cells and SD rats liver. Food Chem Toxicol. 2021;148:111937.
107. Crystal GJ, Malik G, Yoon SH, et al. Isoflurane late preconditioning against myocardial stunning is associated with enhanced antioxidant defenses. Acta Anaesthesiol Scand. 2012;56(1):39–47. doi:10.1111/j.1399-6576.2011.02583.x
108. Hofstetter C, Boost KA, Hoegl S, et al. Norepinephrine and vasopressin counteract anti-inflammatory effects of isoflurane in endotoxemic rats. Int J Mol Med. 2007;20(4):597–604.
109. Wang H, Wang L, Li NL, et al. Subanesthetic isoflurane reduces zymosan-induced inflammation in murine Kupffer cells by inhibiting ROS-activated p38 MAPK/NF-κB signaling. Oxid Med Cell Longev. 2014;2014:851692.
110. Su SB, Qin SY, Xian XL, et al. Interleukin-22 regulating Kupffer cell polarization through STAT3/Erk/Akt crosstalk pathways to extenuate liver fibrosis. Life Sci. 2021;264:118677.
111. Peng Y, Sigua CA, Murr MM. Protein kinase C-zeta mediates apoptosis of mouse Kupffer cells via ERK-1/2: a novel mechanism. Surgery. 2011;149(1):135–142. doi:10.1016/j.surg.2010.04.017
112. Yamagoe S, Yamakawa Y, Matsuo Y, et al. Purification and primary amino acid sequence of a novel neutrophil chemotactic factor LECT2. Immunol Lett. 1996;52(1):9–13. doi:10.1016/0165-2478(96)02572-2
113. Lan F, Misu H, Chikamoto K, et al. LECT2 functions as a hepatokine that links obesity to skeletal muscle insulin resistance. Diabetes. 2014;63(5):1649–1664. doi:10.2337/db13-0728
114. Chikamoto K, Misu H, Takayama H, et al. Rapid response of the steatosis-sensing hepatokine LECT2 during diet-induced weight cycling in mice. Biochem Biophys Res Commun. 2016;478(3):1310–1316. doi:10.1016/j.bbrc.2016.08.117
115. Takata N, Ishii KA, Takayama H, et al. LECT2 as a hepatokine links liver steatosis to inflammation via activating tissue macrophages in NASH. Sci Rep. 2021;11(1):555. doi:10.1038/s41598-020-80689-0
116. Pu JL, Huang ZT, Luo YH, et al. Fisetin mitigates hepatic ischemia-reperfusion injury by regulating GSK3β/AMPK/NLRP3 inflammasome pathway. Hepatobiliary Pancreat Dis Int. 2021;20(4):352–360. doi:10.1016/j.hbpd.2021.04.013
117. Huang Z, Mou T, Luo Y, et al. Inhibition of miR-450b-5p ameliorates hepatic ischemia/reperfusion injury via targeting CRYAB. Cell Death Dis. 2020;11(6):455. doi:10.1038/s41419-020-2648-0
118. Shen A, Zheng D, Luo Y, et al. MicroRNA-24-3p alleviates hepatic ischemia and reperfusion injury in mice through the repression of STING signaling. Biochem Biophys Res Commun. 2020;522(1):47–52. doi:10.1016/j.bbrc.2019.10.182
119. Wang X, Maretti-Mira AC, Wang L, et al. Liver-Selective MMP-9 inhibition in the rat eliminates ischemia-reperfusion injury and accelerates liver regeneration. Hepatology. 2019;69(1):314–328. doi:10.1002/hep.30169
120. Huang Z, Zheng D, Pu J, et al. MicroRNA-125b protects liver from ischemia/reperfusion injury via inhibiting TRAF6 and NF-κB pathway. Biosci Biotechnol Biochem. 2019;83(5):829–835.
121. Huang Z, Pu J, Luo Y, et al. FAM49B, restrained by miR-22, relieved hepatic ischemia/reperfusion injury by inhibiting TRAF6/IKK signaling pathway in a Rac1-dependent manner. Mol Immunol. 2022;143:135–146.
122. Li P, Zhang Z, Gong J, et al. S-Adenosylmethionine attenuates lipopolysaccharide-induced liver injury by downregulating the toll-like receptor 4 signal in Kupffer cells. Hepatol Int. 2014;8(2):275–284. doi:10.1007/s12072-014-9528-6
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.