")
Back to Journals » Journal of Blood Medicine » Volume 14
Managing the Cerebrovascular Complications of Sickle Cell Disease: Current Perspectives
Authors Light J, Boucher M, Baskin-Miller J, Winstead M
Received 23 November 2022
Accepted for publication 31 March 2023
Published 14 April 2023 Volume 2023:14 Pages 279—293
DOI https://doi.org/10.2147/JBM.S383472
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Jennifer Light,* Maria Boucher,* Jacquelyn Baskin-Miller,* Mike Winstead*
Pediatric Hematology-Oncology, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
*These authors contributed equally to this work
Correspondence: Mike Winstead, Division of Pediatric Hematology-Oncology, University of North Carolina at Chapel Hill, 101 Manning Drive, Chapel Hill, NC, USA, Tel +1 919-966-1178, Fax +1 919-966-7629, Email [email protected]
Abstract: The importance of protecting brain function for people with sickle cell disease (SCD) cannot be overstated. SCD is associated with multiple cerebrovascular complications that threaten neurocognitive function and life. Without screening and preventive management, 11% of children at 24% of adults with SCD have ischemic or hemorrhagic strokes. Stroke screening in children with SCD is well-established using transcranial Doppler ultrasound (TCD). TCD velocities above 200 cm/s significantly increase the risk of stroke, which can be prevented using chronic red blood cell (RBC) transfusion. RBC transfusion is also the cornerstone of acute stroke management and secondary stroke prevention. Chronic transfusion requires long-term management of complications like iron overload. Hydroxyurea can replace chronic transfusions for primary stroke prevention in a select group of patients or in populations where chronic transfusions are not feasible. Silent cerebral infarction (SCI) is even more common than stroke, affecting 39% of children and more than 50% of adults with SCD; management of SCI is individualized and includes careful neurocognitive evaluation. Hematopoietic stem cell transplant prevents cerebrovascular complications, despite the short- and long-term risks. Newer disease-modifying agents like voxelotor and crizanlizumab, as well as gene therapy, may treat cerebrovascular complications, but these approaches are investigational.
Keywords: stroke, silent cerebral infarction, moyamoya syndrome, transfusion, stem cell transplantation
Plain Language Summary
Sickle cell disease (SCD) is an inherited disease that affects the red blood cells (RBCs). A genetic mutation causes the RBCs to change from a flat, round shape into a long, curved “sickle” shape. Sickled RBCs disrupt blood flow and cause injury to the inner lining of the blood cells. Disrupting blood flow to the brain can lead to a stroke, where areas of the brain stop functioning. Strokes can be fatal or cause permanent difficulty speaking, moving, or learning. SCD can also cause silent infarctions or “silent strokes”, which actually cause symptoms like difficulty learning. Previously, one-quarter of people with SCD had a stroke by age 45, and many of these strokes occurred in children. Today, strokes in children with SCD can be prevented by checking a transcranial Doppler (TCD) ultrasound every year and providing blood transfusions to children with abnormal blood flow on the TCD. If a person with SCD has a stroke, they need an emergency exchange transfusion to remove the “sickled” blood. Preventing stroke requires monthly blood transfusions for life, but there is interest in other treatments. For certain people with SCD, the medication hydroxyurea can prevent stroke. Stem cell or bone marrow transplantation can cure SCD but may not be possible for everyone. Researchers are studying newer medications and gene therapy, but the safety and effectiveness of these treatments are not yet known. Healthcare disparities also make it harder for many people with SCD to get treatment.
Introduction
Cerebrovascular complications of sickle cell disease (SCD) result from widespread vasculopathy. SCD is an autosomal recessive condition caused by a point mutation in the HBB gene, which causes a single amino acid substation in the β-globin chain.1–4 The resulting hemoglobin S (HbS) variant polymerizes in its deoxygenated state, which causes the characteristic “sickle” shape change in erythrocytes when exposed to hypoxia.5 Sickled erythrocytes interfere with the normal rheology of the microvascular circulation,6 leading to vaso-occlusion and reperfusion injury.7 Erythrocytes containing HbS also develop premature senescence8 and have a shorted lifespan in the circulation, resulting in chronic intra- and extravascular hemolysis.9 Other factors like sleep disordered breathing may contribute to intermittent erythrocyte sickling and tissue hypoxia.10,11
Ischemia-reperfusion injury and hemolysis contribute to an inflammatory milieu12,13 and endothelial activation.14 Hypercoagulability results from complex interactions between the coagulation system and the endothelium, including von Willebrand factor release, nitric oxide depletion, upregulation of vascular adhesion molecules like P-selectin,15 interactions with microparticles, and alterations in the activity of intrinsic anticoagulants like protein C and protein S.16–19 Chronic vasculitis and hypercoagulability in the carotid, vertebral, and cerebral arteries lead to acute infarctive and ischemic stroke, as well as chronic complications like carotid stenosis and moyamoya syndrome.20
Stroke and other complications of cerebrovascular disease represent significant sources of morbidity and mortality for people with SCD. In the longitudinal Cooperative Study of SCD, the estimated prevalence of stroke was 11% by age 19 years and 24% by age 45 years, with the highest risk and lowest age at first stroke among people with homozygous hemoglobin S (HbSS) disease and hemoglobin Sβ0 (HbSβ0) thalassemia.21 Stroke risk is lower among people with hemoglobin SC (HbSC) disease21,22 Hemoglobin Sβ+ (HbSβ+) thalassemia and other compound heterozygous forms of SCD have variable stroke risk, which may be related to degree of anemia and hemolysis.23
Strokes in people with SCD frequently involve the distributions of median cerebral arteries, due to chronic narrowing of the internal carotid arteries, but strokes can also involve the vertebrobasilar arteries and other vessels along the Circle of Willis, potentially in association with vascular anomalies like moyamoya syndrome.24–26 Neurocognitive and psychological complications of cerebrovascular disease affect children and adults with SCD,27–30 requiring patients, families, and providers to be aware of screening practices and interventions to prevent life-threatening complications and improve quality of life.
Primary Stroke Prevention
Importance of Transcranial Doppler Screening
Primary stroke prevention identifies patients without a known prior stroke that are at high risk for an event. For people with SCD, the cornerstone of primary stroke prevention is routine screening with transcranial Doppler ultrasound (TCD). A landmark series of 283 TCDs in 190 patients with SCD found that stroke correlated with increasing blood-flow velocity in the median cerebral arteries, a marker of vascular narrowing; strokes occurred in 7 of 23 participants with elevated velocity, compared to none with normal velocity.24 Identifying individuals at high risk allows effective preventive treatment with chronic transfusion therapy. The availability of effective intervention, as well as strong correlation between TCD and more complex studies like cerebral angiography and magnetic resonance imagine (MRI) with magnetic resonance angiography (MRA),31–33 reinforce the utility, reliability, and importance of TCD as a screening technique.
For children with SCD, screening with TCD should be performed annually in patients aged 2 to 16 years with HbSS or HbSβ0 thalassemia.23 TCD is not currently recommended for children with HbSC.23 For children with other forms of heterozygous SCD like HbSβ+ thalassemia, TCD may be indicated if the clinical and laboratory phenotype resembles homozygous SCD.23 TCD measures the time-averaged mean maximum velocity (TAMMV) of blood flow in the bilateral median cerebral arteries; velocities are generally interpreted as normal, conditional, or abnormal. A TAMMV ≥200 cm/s is associated with a 40% increased risk of stroke within 3 years and is the recommended cut-off for abnormal.23,34 Velocities in the conditional range of 170–199 cm/s are at significant risk of rising into the high-risk range35 and should be followed closely. Our usual practice is to repeat TCD within 3 months for velocities of 170–184 cm/s and within 1 month for velocticities of 185–199 cm/s.
These values were established by non-imaging TCD. Imaging TCD uses different technical methods and provides slightly different estimates of velocity.36,37 Current guidelines recommend adjusting the threshold for chronic transfusion based on institutional TCD methods; for imaging TCD, high risk of stroke is indicated by two measurements ≥185 cm/s or any measurement ≥205 cm/s.23
Use of TCD may be limited by difficulty in obtaining adequate sonographic windows, inter-operator variability, and inability to obtain accurate readings in the vertebrobasilar arteries.24,31 For children in whom adequate TCD readings cannot be obtained, despite multiple attempts by experienced operators, our practice is to obtain brain MRI and MRA. In young children these studies usually require sedation, so the treatment team must weigh the risks and benefits collaboratively with the patient’s caregivers; pre-anesthesia transfusion is indicated. Healthcare disparities can also affect the availability of comprehensive care for people with SCD.38 Providers must be aware of societal and racial inequities that affect healthcare for people with SCD.39 At present, most strokes in children with SCD in the United States are associated with inadequate TCD screening or delayed initiation of chronic transfusion, gaps in care influenced by systemic inequity in healthcare resources.25
Even in those with no history of abnormal TCDs, screening MRI may identify silent infarctions. Based on current guidelines, a screening MRI of the brain is appropriate for asymptomatic adults with SCD and for asymptomatic children with SCD who are old enough to undergo MRI without sedation,23 typically around age 10 years in our practice. It is also important to evaluate children for other treatable comorbidities like sleep disordered breathing, which is likely underdiagnosed and may be a risk factor for stroke.10,11
Initiation of Chronic Transfusions
For patients with elevated stroke risk, the standard of care is early initiation of chronic transfusion therapy. The landmark Stroke Prevention Trial in Sickle Cell Anemia (STOP) was a randomized controlled trial that showed the effectiveness of this management strategy. The benefit of chronic transfusion was pronounced, with a 93% reduction in the relative risk of stroke in the transfused group; the trial ended early due to clear evidence of benefit.40,41 Chronic transfusions must be started in collaboration with the patient and family, including a clear discussion of the risks and benefits. Switching from chronic transfusion to hydroxyurea may be feasible for some patients (see “Duration of Transfusion Therapy and the Role of Hydroxyurea” below). For many patients, however, chronic transfusions are a lifelong intervention,42 requiring long-term management of complications like iron overload.
The usual approach to therapy is red blood cell (RBC) transfusion every 3 to 4 weeks, maintaining a hemoglobin concentration of at least 9 g/dl and a fraction of HbS <30% between transfusions.23 This goal is based on the methodologies of STOP and its successor study, STOP2, as well as animal models and blood viscosity studies.40,42–45 It is not necessary to maintain a normal hemoglobin concentration, as people with SCD adapt to chronic anemia and can be vulnerable to increased blood viscosity; our practice is to maintain a post-transfusion hemoglobin concentration of 10 to 11 g/dl.
Patients receiving chronic transfusion therapy may benefit from exchange transfusion, rather than simple transfusion (Table 1).46,47 In exchange transfusion, a predetermined volume of blood is removed while RBCs are being transfused, either by apheresis (automated exchange) or by a medical provider (manual exchange).47 Exchange transfusion provides greater control over the end-transfusion hemoglobin concentration and HbS and can also decrease iron overload, although patients receiving exchange transfusion still require long-term monitoring of iron status. Exchange transfusion is more resource-intensive than simple transfusion. Manual exchange (see “Management of Acute Stroke” below) requires one-to-one care for the duration of the procedure. Automated exchange requires an apheresis device and trained transfusion-medicine staff; priming the apheresis machine requires additional transfusion volume, and the procedure is generally not indicated for small children. Venous access for automated exchange transfusion may require an apheresis-compatible central venous access device; infection risk, MRI compatibility of the access device, and education for patients and caregivers are important in this context.
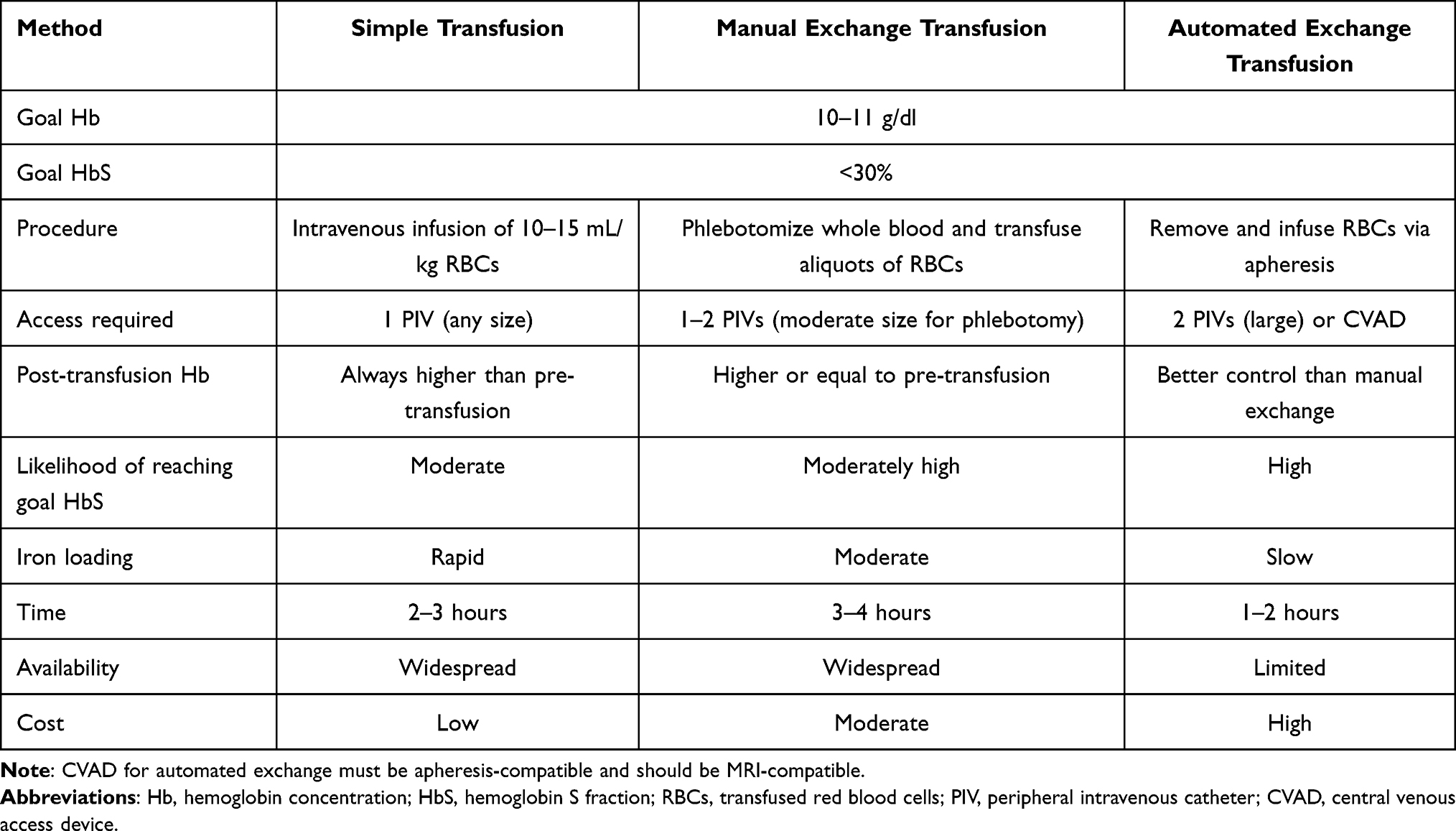 |
Table 1 Approaches to Chronic Transfusion Therapy |
Transfusion Complications and Iron Overload
Chronic transfusion can be life-saving for people with SCD at high risk of stroke, but there are risks, particularly transfusion reactions and iron overload.46 Immune-mediated transfusion reactions include acute and delayed hemolytic reactions, as well as alloimmunization. In people with SCD, severe delayed hemolytic transfusion reactions can involve reticulocytopenia and “bystander” hemolysis, leading to profound anemia and a risk of cerebral ischemia.48,49 Close collaboration between the patient, the hematology team, and the transfusion medicine service is essential for prompt recognition and management of transfusion reactions, as well as prevention.
Individuals with SCD receiving chronic transfusions are at high risk of alloantibody formation, due to repeated exposure to minor antigens on donor RBCs. Alloimmunization can occur to any minor RBC antigen, and it is essential that a person with a history of alloimmunization not be exposed to the mismatched antigen again. The antiglobulin tests of alloimmunized individuals may normalize over time, so it is essential that hematology and transfusion medicine carefully track transfusion reactions.
In addition to standard matching of the ABO antigens and the Rhesus-D antigen, people receiving chronic transfusions require more extensive matching of minor antigens. The most immunogenic antigens for people with SCD are in Rhesus group (C, c, E, e) and the Kell group (K).50 All RBC transfusions for people with SCD should be matched at C, E, and K.46,51 More extended matching of minor RBC blood groups like Duffy (Fya, Fyb), Kidd (Jka, Jkb), and MNSs may further reduce the risk of alloimmunization, but this practice limits the pool of blood donors, and the overall benefit is unclear.44,46,52 For recently-transfused patients, molecular studies of the peripheral blood are not accurate indicators of the patient’s RBC phenotype. In this situation, genetic studies are an alternative. There can be discrepancies between genotype and RBC phenotype.53
Iron overload is a ubiquitous complication of chronic transfusions, including exchange transfusion. Because the body does not have a physiological mechanism for eliminating large amounts of iron, the iron from transfused RBCs accumulates in the endocrine organs, liver, and myocardium, which can lead to life-limiting organ dysfunction.54,55 Serum ferritin levels increase with iron burden55,56 and should be monitored at least quarterly in patients receiving chronic transfusions. Ferritin levels alone, however, do not predict liver iron concentration (LIC) or other sequelae of iron overload.56 Additional monitoring of iron burden should include MRI of the liver and of the heart with T2* sequences (which have generally replaced liver biopsy57) every 1 to 2 years, particularly in those with a ferritin concentration >1000 μg/l.46 Chelation therapy is generally indicated for patients with evidence of cardiac iron loading (T2* <20 ms), LIC of at least 3.5 mg/g, or serum ferritin consistently above 1000 μg/l, although other thresholds may be considered.54,55
Chelation therapy must be individualized and carefully monitored for efficacy and tolerability. Deferoxamine is highly effective but must be given either intravenously or as a daily subcutaneous infusion, which makes it challenging for home use. Deferasirox is an oral chelating agent that has similar efficacy to deferoxamine and is often more feasible for chronic outpatient use.58,59 Personal, social, and economic factors may interfere with a person’s ability to take their chelating agent as prescribed, and careful follow-up is needed to ensure adherence.60 Patients taking deferoxamine or deferasirox should be monitored for ocular, hepatic, or renal toxicity, and there is a small risk of opportunistic infection.61,62 Deferiprone, another effective oral chelating agent, has been studied more recently in people with SCD, although its association with neutropenia may limit its use.63,64
Duration of Transfusion Therapy and the Role of Hydroxyurea
For people at high risk of stroke based on childhood TCD, primary stroke prophylaxis is a lifelong undertaking. In the STOP2 trial, stopping transfusion (without other prophylaxis) significantly increased the risks of rising TCD velocity and overt stroke.42 More recently, the TCD with Transfusions Changing to Hydroxyurea (TWiTCH) trial found that hydroxyurea (hydroxycarbamide) can effectively stabilize TCD velocities in carefully selected patients (Table 2).65 Participants in TWiTCH did not have a history of stroke, TIA, or severe vasculopathy (defined as carotid stenosis on MRA). All participants received chronic transfusion therapy for at least 12 months, and transfusions overlapped with hydroxyurea until reaching the maximum tolerated hydroxyurea dose. Although the TWiTCH trial included participants who had abnormal TCDs while receiving chronic transfusions, our practice is to only consider changing to hydroxyurea in patients whose TCDs have normalized.
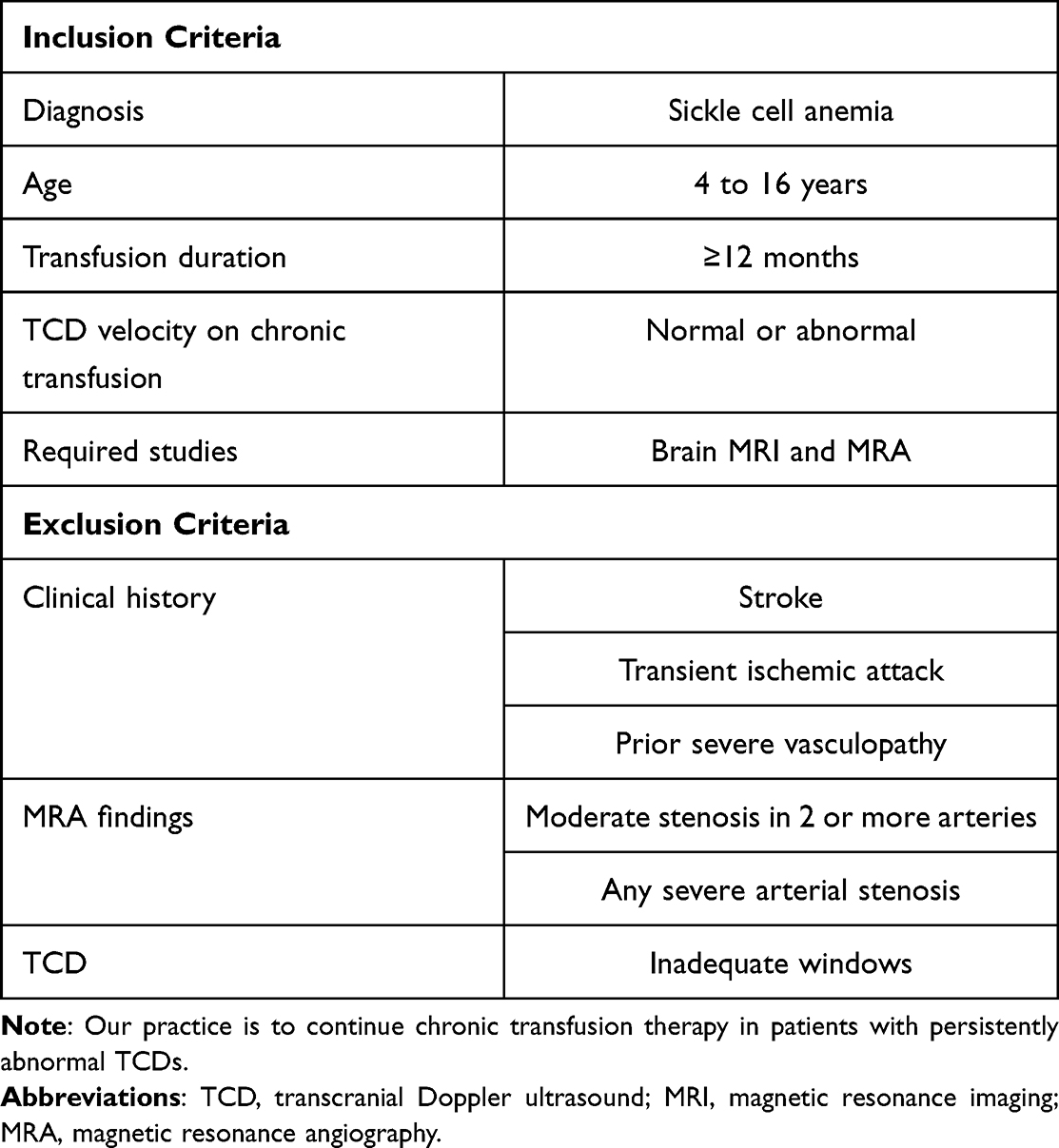 |
Table 2 Inclusions and Exclusions for Changing Primary Stroke Prevention from Chronic Transfusion to hydroxyurea65 |
When considering the transition from chronic transfusions to hydroxyurea, the care team must be cautious and partner with the patient and other caregivers. Like transfusion, primary stroke prevention with hydroxyurea must be continued lifelong. Because hydroxyurea is given at home, it is essential that the patient and family understand the need for strict adherence and also that the care team is aware of social factors that may complicate the patient’s ability to obtain and take their medication.66–68 Continued TCD monitoring is necessary. Hydroxyurea appears to be safe for long-term use, but study of its secondary effects, such as infertility, is ongoing.69–71
In the randomized, controlled Pediatric Hydroxyurea Phase 3 Clinical Trial (BABY HUG) trial, starting hydroxyurea in early childhood was associated with a slower rise in TCD, compared to placebo.72,73 An ongoing clinical trial is evaluating the up-front use of hydroxyurea for primary stroke prophylaxis in regions without access to chronic transfusion.74
Silent Cerebral Infarction
Silent cerebral infarction (SCI) is an abnormality on MRI that does not correlate with a permanent, focal neurological deficit.75–77 Based on observational studies with surveillance MRI, the prevalence of SCI rises with age; around 39% of children and SCD and more than 50% of adults with SCD have silent infarctions.23,76 Despite its name, SCI does manifest clinically as neurocognitive decline, difficulty with school and work performance, and other neuropsychological problems.27,76 SCI is also a significant risk factor for overt stroke.75 Recent reports suggest that SCI in people with HbSC disease may be more common than previously recognized.78,79
Once SCI is identified, it requires ongoing radiographic and clinical evaluation. Management of SCI is an individual process. The Silent Cerebral Infarct Multicenter Clinical Trial (SIT) showed that chronic transfusion therapy in children with SCI reduced the risks of overt stroke and extension of SCI, with 6% of transfused participants and 14% of control participants developing one of these outcomes.77 Given the relatively high number needed to treat and the significant transfusion burden, the decision to initiate chronic transfusions for SCI must be individualized.23 For children with SCI and significant schooling difficulty, for example, the benefits of chronic transfusion may outweigh the risks, although there is no standardized practice. Hydroxyurea may help prevent development and progression of SCI, although evidence is limited.80–82
Acute Stroke
Diagnosis
Because individuals with sickle cell disease (especially HbSS and HbSβ0 thalassemia) are at high risk of stroke, it is important to have a high index of suspicion of stroke in this population. Clinicians should suspect a stroke or transient ischemic attack (TIA) in any patient with an acute neurological deficits, such as hemiparesis, ataxia, or dysphasia; altered consciousness, including new-onset seizures; or acute headache.83–85 While an emergent, non-contrast-enhanced CT scan of the brain is necessary to evaluate for intracranial hemorrhage, the best evaluation of stroke is MRI with MRA, which can delineate the affected areas and also distinguish other conditions that mimic stroke.86 Because strokes can arise from the vertebrobasilar circulation, MRA must include the vertebral arteries. Treatment should be initiated within 2 hours of symptom onset.23 If emergent MRI and MRA are not available, treatment should not be delayed.
Management of Acute Stroke
Due to the pathophysiology of stroke in sickle cell disease, the treatment differs from stroke in other patients. The main goal is to decrease the fraction of HbS to less than 30% using emergent RBC transfusion. Ideally, this is done through an emergency exchange transfusion, which decreases the HbS fraction more efficiently than a simple transfusion and is associated with a five-fold lower risk of secondary stroke.46,87,88 Emergent exchange transfusion may be complicated by the location and resources of the initial treating facility, as well as vascular access and patient acuity. Rather than delaying treatment, it may be necessary to perform an initial simple transfusion using C, E, and Kell-matched blood, targeting a hemoglobin concentration of 10–11 g/dl, followed by exchange transfusion as soon as possible.43,46,86 Other medical therapies for stroke like tissue plasminogen activator are controversial in patients with SCD23 and are not substitutes for transfusion.
Any patient with SCD and an acute stroke should be transferred to an intensive care unit equipped to manage neurological disorders as safely and quickly as possible. Exchange transfusion for acute stroke is best performed by automated apheresis.46 Depending on local resources, however, manual exchange may be more feasible to avoid delays. For a manual exchange, the volumes of blood to remove and transfuse can be calculated using the patient’s body weight, starting hemoglobin/hematocrit, and starting HbS percentage (assume 100% if unavailable).
Our approach to manual exchange (Table 3) is to estimate the patient’s total blood volume (Step 1) and then calculate the desired change in HbS percentage (Step 2) to reach 30% or less. Multiplying these values (Step 3) gives the volume of whole blood to remove. Because transfused RBCs are more concentrated than RBCs in vivo, the erythrocyte volume of the whole blood must be calculated, based on the patient’s hematocrit (Step 4); this erythrocyte volume can then be converted to an equivalent transfusion volume using the hematocrit of transfused RBCs, typically around 60% (Step 5). To raise the final hemoglobin concentration requires adding approximately 5 mL/kg of transfused RBCs for every 1 g/dl change in hemoglobin concentration desired (Steps 6, 7). To prevent fluid shifts and cardiopulmonary decompensation, boluses of crystalloid are given before and after exchange, and blood is removed in aliquots over several hours, alternating with transfusion.
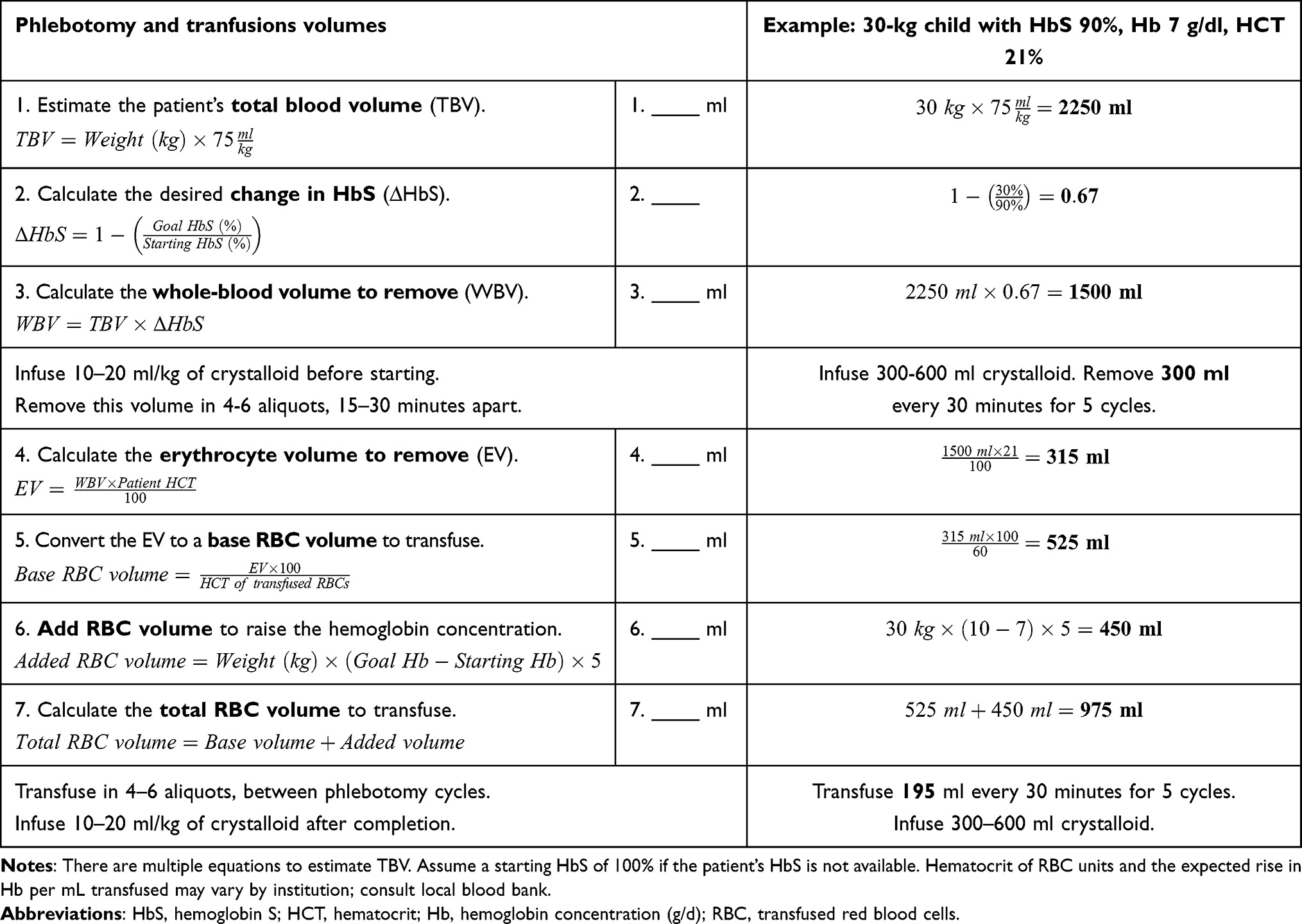 |
Table 3 Sample Calculations for Manual Exchange Transfusion |
For acute ischemic stroke in patients with SCD, there is little role for surgical intervention. Neurosurgical or endovascular intervention may rarely be indicated for intracranial hemorrhage.89 For patients with critical carotid stenosis or moyamoya syndrome, revascularization procedures may be indicated after recovery from the acute stroke (see “Surgical Revascularization” below).
Other Cerebrovascular Complications Presenting Similarly to Stroke
While emergent management of stroke is appropriate for patients with SCD and acute neurological findings, other disorders can present similarly. Recently, it has been reported that people with SCD are at increased risk of posterior reversible encephalopathy syndrome (PRES).90 PRES involves altered mental status, headache, vision changes, and a characteristic finding on MRI of posterior cerebral white matter edema.91 Risk factors for PRES are overrepresented among people with SCD, including hypertension, renal disease, immunosuppressant therapy, and blood transfusion,92,93 Sickle cell disease is a hypercoagulable state,19 with protein C and protein S dysregulation, and individuals with SCD may also be at an increased risk of cerebral venous sinus thrombosis or thrombotic complications of hospitalization.94,95
Stroke Aftercare
Secondary Stroke Prevention
Secondary prophylaxis refers to the prevention of subsequent strokes in patients with a history of stroke. Without preventive treatment after a first stroke, 67% of people with SCD in one series had an additional stroke.83 The earliest reports of chronic transfusion therapy in SCD were for secondary stroke prevention in the 1970s,96 an approach supported by subsequent studies.97,98 Due to the lack of equipoise and the danger of withholding prophylaxis, this practice has not been the subject of controlled trials.99 As with primary prevention, chronic transfusion for secondary stroke prevention is generally performed every 3 to 4 weeks, maintaining a hemoglobin >9 g/dl and an HbS <30% between transfusions.23 Even with chronic transfusions, there is a significant risk of subsequent strokes, as well as other neurocognitive complications.98
In contrast to primary prophylaxis, hydroxyurea alone cannot be used for secondary stroke prevention. The Stroke With Transfusions Changing to Hydroxyurea (SWiTCH) trial, a randomized controlled trial of hydroxyurea versus chronic transfusion for secondary stroke prevention, studied stroke risk and iron overload.100 The trial was closed due to futility, after interim analysis found a 10% prevalence of stroke in the hydroxyurea group, compared to 0% in the transfused group, with no difference in iron burden. Chronic transfusion remains the standard of care for secondary stroke prevention in people with SCD.
Meeting the Needs of Stroke Survivors
Overwhelmingly, the evidence points to a consistent cognitive decline among stroke survivors.30 Routine neurocognitive evaluations are necessary for all patients with a prior stroke,23 and for children it is important to monitor school performance. Children can succeed in school after strokes, but they may require individualized education plans, and their caregivers may require advocacy and support from the medical team. For young adults with SCD, the transition from pediatric to adult care is associated with a significant risk of worsening disease and death.101 While death rates from SCD have consistently declined among children, they have risen among adults, likely reflecting socioeconomic obstacles to obtaining access to care.102
Adult stroke survivors require continuity of high-quality SCD care, including secondary prophylaxis. Cognitive deficits and psychological comorbidities are associated with greater risk during the transition period101,103 Recognizing that the transition to adult care is not a single event but an ongoing process that begins during adolescence and continues through early adulthood,104,105 it is essential that stroke survivors, their caregivers, and their providers actively collaborate to ensure continuity of care, regular neurocognitive assessment, continuation of chronic transfusions, and management of iron overload.
Other Therapies to Modify Stroke Risk
Surgical Revascularization
Neurosurgical consultation is indicated for patients with evidence of a severe vasculopathy, including critical arterial stenosis or moyamoya syndrome on MRA. Moyamoya syndrome is a complication of chronic internal carotid artery occlusion, in which friable collateral vessels develop along the base of the brain; cerebral angiography demonstrates the characteristic “wisp of smoke” enhancement in the collateral vessels.106 Even with chronic transfusion therapy, the rate of stroke and TIA in people with moyamoya was 41% in one series.107
Indirect revascularization procedures like encephaloduroarteriosynangiosis (EDAS) mobilize blood vessels from the scalp into the calvarium to perfuse areas distal to the stenotic vessels; published experience suggests a low risk of serious surgical complications like hemorrhage, perioperative stroke, or functional impairment.23,108,109 As an adjunct to chronic transfusion therapy, surgical intervention may decrease stroke risk, and a recent review of pediatric patients found that EDAS facilitated return to school.110 However, there are no controlled studies of neurosurgical intervention, and the specific revascularization procedure used typically depends on institution and surgeon preference. Any consideration of neurosurgical intervention requires a multidisciplinary collaboration between hematology, neuroradiology, neurosurgery, and the patient.23
Hematopoietic Stem Cell Transplantation
Hematopoietic stem cell transplantation (HSCT) for SCD decreases stroke risk, and a history of stroke is a strong indication to evaluate for HSCT.111,112 HSCT replaces the patient’s hematopoietic progenitor cells and immune system with those of a related or unrelated donor. Cerebrovascular benefits are established by multiple studies, with improved stenosis scores, improved TCDs, and reduction in strokes post-transplant.113,114 Given the availability of effective disease-modifying therapy, however, the risks of HSCT must be carefully considered.115–117
Complications of HSCT include effects of pre-HSCT conditioning therapy, infection, graft rejection, and acute or chronic graft-versus-host disease (GVHD).118 Late effects of conditioning agents like busulfan and cyclophosphamide include infertility and organ dysfunction.111 GVHD, which involves tissue destruction by donor lymphocytes, can be life-threatening or interfere considerably with quality of life, especially after unrelated-donor HSCT.119 Research is ongoing to mitigate these risks, including the use of lower-intensity conditioning regimens and enhanced GVHD prophylaxis with agents like abatacept.120 Interim analysis from an ongoing clinical trial (Clinicaltrials.gov identifier NCT04018937) of HSCT with reduced-intensity conditioning for young children with SCD and matched sibling donors has reported 100% disease-free survival and no severe GVHD,121 emphasizing the viability of early HSCT for SCD.
Risks of HSCT are higher among adolescents and adults with SCD, compared to young children. Data from the Sickle Cell Transplant to Prevent Disease Exacerbation (STRIDE) study, however, found that related- or unrelated-donor HSCT is feasible in adults with SCD and severe comorbidities, with 82% event-free survival in this exceptionally high-risk population.122 The ongoing STRIDE2 trial (Clinicaltrials.gov identifier NCT02766465) will compare the long-term outcomes of HSCT to standard therapy.123 For adults with matched sibling donors, the National Institutes of Health and other centers successfully piloted HSCT with very low-intensity conditioning, which was associated with 87% to 92% disease-free survival.124,125 This approach is currently being studied in children (ClinicalTrials.gov Identifier NCT03587272).126 Regardless of the approach taken, any patient with SCD and a history of stroke or other cerebrovascular disease should be evaluated for HSCT at a center with experience treating SCD.
Investigational Therapies
Voxelotor and Crizanlizumab
Ongoing clinical trials are evaluating voxelotor and crizanlizumab, disease-modifying drugs recently approved in the United States for SCD management, for their effects on cerebrovascular complications (Table 4). Crizanlizumab (SelG1) downregulates endothelial expression of P-selectin, thereby reducing inflammation-mediated cell adhesion.15,127,128 In the Study to Assess Safety and Impact of SelG1 with or Without Hydroxyurea Therapy in Sickle Cell Disease Patients with Pain Crises (SUSTAIN), crizanlizumab decreased vaso-occlusive episodes and acute chest syndrome; there was one stroke in the study group, but cerebrovascular complications were not a primary or secondary endpoint.129
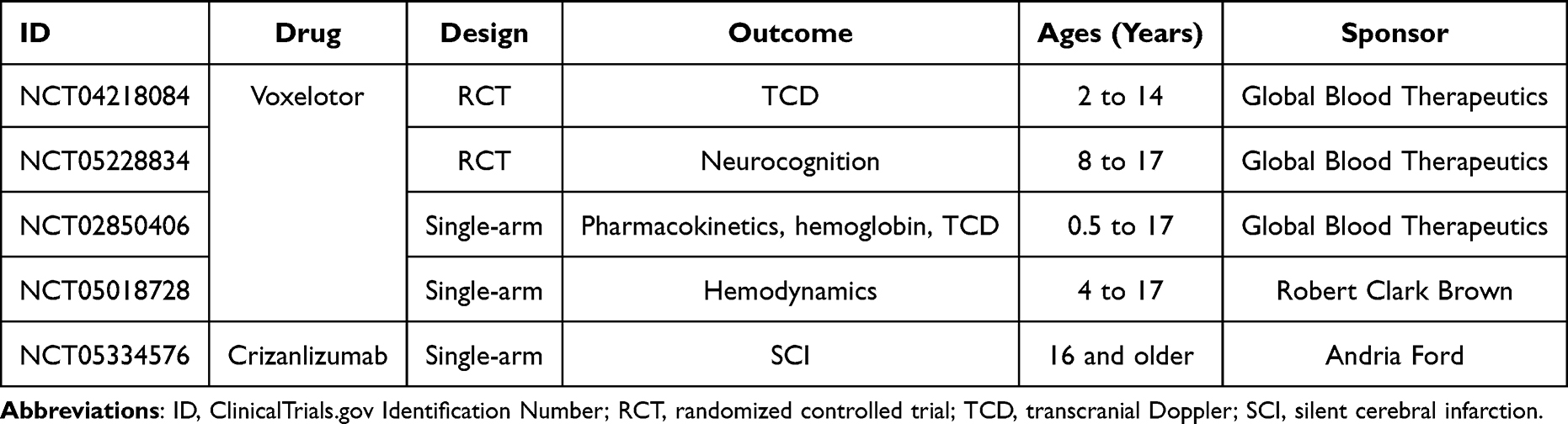 |
Table 4 Active Trials of Disease-Modifying Therapy for Cerebrovascular Disease in the United States |
Voxelotor stabilizes HbS in the oxygenated state, which inhibits polymerization and erythrocyte sickling.127,130 Voxelotor is approved in the United States for patients with SCD at least 12 years of age; the Hemoglobin Oxygen Affinity Modulation to Inhibit HbS Polymerization (HOPE) study did not measure stroke incidence.131 Stroke was a concern during the development of the drug, due to initial concerns that increasing oxygen affinity would reduce oxygen extraction in sensitive tissues like the brain.132 The HOPE trial did not report any strokes in the control or voxelotor groups, nor did interim analysis of HOPE Kids (ClinicalTrials.gov Identifier NCT02850406), an ongoing pediatric voxeloator trial that includes TCD velocity as a primary endpoint.133 TCD velocity is also the primary study question of the HOPE Kids 2 study (ClinicalTrials.gov Identifier NCT04218084).
Gene Therapy
As a monogenic disorder caused by a point mutation, SCD has long been an appealing target for gene therapy.134 Current approaches to gene therapy involve collecting a patient’s hematopoietic progenitor cells via marrow harvest or apheresis; genetically modifying the cells ex vivo by inserting a new transgene or, more recently, direct gene editing; and reinfusing the genetically-modified progenitors after high-dose chemotherapy conditioning.135 In contrast to allogeneic HSCT, there is no need for a donor and no risk of GVHD. Although gene therapy figures prominently in recent media coverage of SCD,136,137 this approach to treatment is investigational, and its risks and benefits are still being established. As of November, 2022 multiple publicly- and privately-funded studies of gene therapy for SCD were actively recruiting in the United States; most of these studies exclude people with cerebrovascular disease (Table 5).
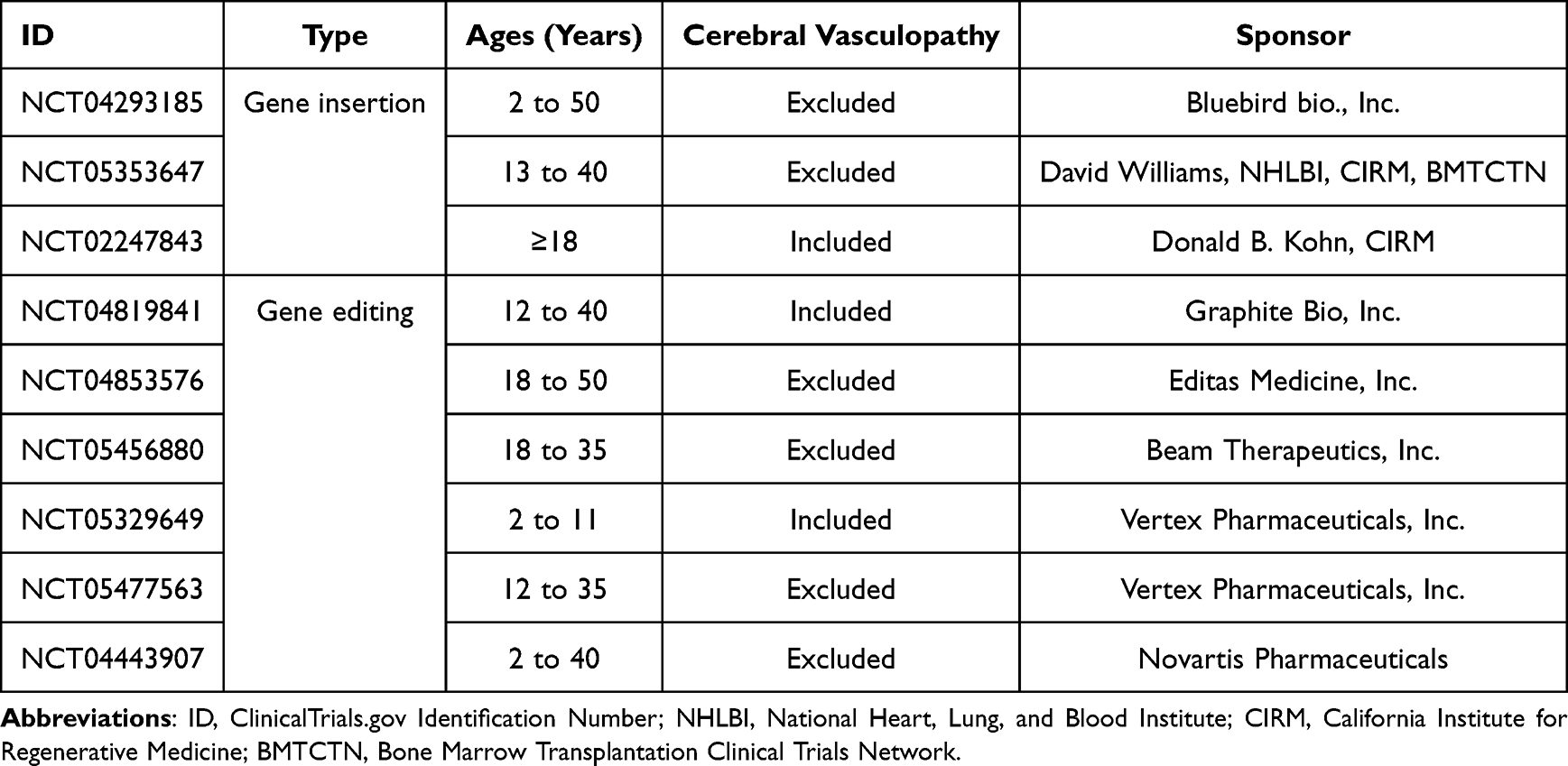 |
Table 5 Active Trials of Gene Therapy for Sickle Cell Disease in the United States |
Ongoing research will be necessary to evaluate the potential risk and benefits of gene therapy for reducing stroke risk. The largest clinical trial of gene therapy for SCD to date was conducted by the biotechnology corporation bluebird bio, using transgene insertion.138 Among 35 participants, there was sustained improvement in hemoglobin concentrations, with decreased vaso-occlusive events and no strokes after treatment, including participants with a prior history of stroke. Two participants subsequently developed worsening anemia and cytogenetic abnormalities, and one participant developed leukemia five years after gene therapy.138,139 In addition to short- and long-term toxicities of conditioning chemotherapy, gene therapy can potentially lead to “genotoxicity”, or malignant transformation of the genetically-modified progenitors,140,141 emphasizing the imperative for long-term follow-up research in this promising field.
Conclusion
Stroke is a devastating contributor to mortality, disability, and loss of neurocognitive function for people with SCD. With access to effective TCD screening and chronic transfusion therapy, which can be substituted for hydroxyurea in selected cases, people with SCD can avoid these complications. Stroke prophylaxis contributes to the overall burden of illness, requiring lifelong management of secondary complications like iron overload, as well as monitoring for neurocognitive complications, vascular malformations, and SCI. People with SCD and cerebrovascular disease can have complex medical and social needs, which require lifelong, comprehensive, patient-centered, multidisciplinary care. Screening and prophylaxis are not available to all people with SCD, due in part to structural disparities in healthcare that must be addressed through policy and advocacy.
There is an ongoing need for research into cerebrovascular complications of SCD, both to address the burdens and limitations of standard treatment and to develop new treatments in a safe but expeditious manner. The past several years have seen a sharp increase in new therapies for SCD, and ongoing clinical trials will evaluate the efficacy of medications like voxelotor and crizanlizumab for stroke prevention. Concurrently, clinical trials in HSCT have the potential to further increase the availability of safe, effective, curative therapy for people with SCD. Gene therapy is a promising area of research and development, which is drawing attention from the media and public; both the long-term benefits of gene therapy for people with cerebrovascular complications of SCD and the long-term risks of genetically modifying marrow progenitors require ongoing study.
As we look toward the future of SCD management, we must not lose sight of the immense suffering caused by cerebrovascular disease for people with SCD and their families; the racial, economic, and geographic barriers to equitable management of these complications; and, above all, the fact that collaboration between policymakers, non-governmental organizations, industry, researchers, medical providers, and people with SCD can overcome this devastating burden of illness.
Disclosure
Dr Maria Boucher reports personal fees from Forma, personal fees from GBT, outside the submitted work. The authors report no conflict of interest in this work.
References
1. Allison AC. Notes on sickle-cell polymorphism. Ann Hum Genet. 1954;19(1):39–51. doi:10.1111/j.1469-1809.1954.tb01261.x
2. Piel FB, Patil AP, Howes RE, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. 2010;1:104. doi:10.1038/ncomms1104
3. Esoh K, Wonkam A. Evolutionary history of sickle-cell mutation: implications for global genetic medicine. Hum Mol Genet. 2021;30(R1):R119–R128. doi:10.1093/hmg/ddab004
4. Wailoo K. Drawing Blood: Technology and Disease Identity in Twentieth-Century America. Johns Hopkins University Press; 1999.
5. Steinberg MH. Pathophysiology of sickle cell disease. Baillieres Clin Haematol. 1998;11(1):163–184. doi:10.1016/s0950-3536(98)80074-7
6. Carden MA, Fasano RM, Meier ER. Not all red cells sickle the same: contributions of the reticulocyte to disease pathology in sickle cell anemia. Blood Rev. 2020;40:100637. doi:10.1016/j.blre.2019.100637
7. Hebbel RP. Ischemia-reperfusion injury in sickle cell anemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain. Hematol Oncol Clin North Am. 2014;28(2):181–198. doi:10.1016/j.hoc.2013.11.005
8. Kuypers FA. Hemoglobin s polymerization and red cell membrane changes. Hematol Oncol Clin North Am. 2014;28(2):155–179. doi:10.1016/j.hoc.2013.12.002
9. Nouraie M, Lee JS, Zhang Y, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica. 2013;98(3):464–472. doi:10.3324/haematol.2012.068965
10. Liguoro I, Arigliani M, Tan HL, Gupta A. The burden of sleep disordered breathing in children with sickle cell disease. Pediatr Pulmonol. 2021;56(12):3607–3633. doi:10.1002/ppul.25632
11. Tantawy A, El-Sherif N, Makkeyah S, et al. Sleep disordered breathing and its relation to stroke and pulmonary hypertension in children with sickle cell disease: a single-center cross-sectional study. Ann Hematol. 2023;102(2):271–281. doi:10.1007/s00277-023-05099-4
12. Vingert B, Tamagne M, Habibi A, et al. Phenotypic differences of CD4(+) T cells in response to red blood cell immunization in transfused sickle cell disease patients. Eur J Immunol. 2015;45(6):1868–1879. doi:10.1002/eji.201445187
13. van Beers EJ, Yang Y, Raghavachari N, et al. Iron, inflammation, and early death in adults with sickle cell disease. Circ Res. 2015;116(2):298–306. doi:10.1161/CIRCRESAHA.116.304577
14. Hoppe CC. Inflammatory mediators of endothelial injury in sickle cell disease. Hematol Oncol Clin North Am. 2014;28(2):265–286. doi:10.1016/j.hoc.2013.11.006
15. Kutlar A, Embury SH. Cellular adhesion and the endothelium: p-selectin. Hematol Oncol Clin North Am. 2014;28(2):323–339. doi:10.1016/j.hoc.2013.11.007
16. Piccin A, Murphy C, Eakins E, et al. Circulating microparticles, protein C, free protein S and endothelial vascular markers in children with sickle cell anaemia. J Extracell Vesicles. 2015;4:28414. doi:10.3402/jev.v4.28414
17. Piccin A, Murphy C, Eakins E, et al. Insight into the complex pathophysiology of sickle cell anaemia and possible treatment. Eur J Haematol. 2019;102(4):319–330. doi:10.1111/ejh.13212
18. Lim MY, Ataga KI, Key NS. Hemostatic abnormalities in sickle cell disease. Curr Opin Hematol. 2013;20(5):472–477. doi:10.1097/MOH.0b013e328363442f
19. Ataga KI, Key NS. Hypercoagulability in sickle cell disease: new approaches to an old problem. Hematology. 2007;2007(1):91–96. doi:10.1182/asheducation-2007.1.91
20. Runge A, Brazel D, Pakbaz Z. Stroke in sickle cell disease and the promise of recent disease modifying agents. J Neurol Sci. 2022;442:120412. doi:10.1016/j.jns.2022.120412
21. Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288–294.
22. Maduakor C, Alakbarzade V, Sammaraiee Y, et al. The epidemiology of neurological complications in adults with sickle cell disease: a retrospective cohort study. Front Neurol. 2021;12:744118. doi:10.3389/fneur.2021.744118
23. DeBaun MR, Jordan LC, King AA, et al. American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv. 2020;4(8):1554–1588. doi:10.1182/bloodadvances.2019001142
24. Adams R, McKie V, Nichols F, et al. The use of transcranial ultrasonography to predict stroke in sickle cell disease. N Engl J Med. 1992;326(9):605–610. doi:10.1056/NEJM199202273260905
25. Kwiatkowski JL, Voeks JH, Kanter J, et al. Ischemic stroke in children and young adults with sickle cell disease in the post-STOP era. Am J Hematol. 2019;94(12):1335–1343. doi:10.1002/ajh.25635
26. Stotesbury H, Kawadler JM, Hales PW, Saunders DE, Clark CA, Kirkham FJ. Vascular instability and neurological morbidity in sickle cell disease: an integrative framework. Front Neurol. 2019;10:871. doi:10.3389/fneur.2019.00871
27. Schatz J, White DA, Moinuddin A, Armstrong M, DeBaun MR. Lesion burden and cognitive morbidity in children with sickle cell disease. J Child Neurol. 2002;17(12):890–894. doi:10.1177/08830738020170122401
28. Vichinsky EP, Neumayr LD, Gold JI, et al. Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA. 2010;303(18):1823–1831. doi:10.1001/jama.2010.562
29. Prussien KV, Jordan LC, DeBaun MR, Compas BE. Cognitive function in sickle cell disease across domains, cerebral infarct status, and the lifespan: a meta-analysis. J Pediatr Psychol. 2019;44(8):948–958. doi:10.1093/jpepsy/jsz031
30. Portela GT, Butters MA, Brooks MM, Candra L, Rosano C, Novelli EM. Comprehensive assessment of cognitive function in adults with moderate and severe sickle cell disease. Am J Hematol. 2022;97(9):E344–E346. doi:10.1002/ajh.26643
31. Adams RJ, Nichols FT, Figueroa R, McKie V, Lott T. Transcranial Doppler correlation with cerebral angiography in sickle cell disease. Stroke. 1992;23(8):1073–1077. doi:10.1161/01.str.23.8.1073
32. Siegel MJ, Luker GD, Glauser TA, DeBaun MR. Cerebral infarction in sickle cell disease: transcranial Doppler US versus neurologic examination. Radiology. 1995;197(1):191–194. doi:10.1148/radiology.197.1.7568822
33. Silva GS, Vicari P, Figueiredo MS, Carrete H, Idagawa MH, Massaro AR. Brain magnetic resonance imaging abnormalities in adult patients with sickle cell disease: correlation with transcranial Doppler findings. Stroke. 2009;40(7):2408–2412. doi:10.1161/STROKEAHA.108.537415
34. Adams RJ, McKie VC, Brambilla D, et al. Stroke prevention trial in sickle cell anemia. Control Clin Trials. 1998;19(1):110–129.
35. Hankins JS, Fortner GL, McCarville MB, et al. The natural history of conditional transcranial Doppler flow velocities in children with sickle cell anaemia. Br J Haematol. 2008;142(1):94–99. doi:10.1111/j.1365-2141.2008.07167.x
36. Jones AM, Seibert JJ, Nichols FT, et al. Comparison of transcranial color Doppler imaging (TCDI) and transcranial Doppler (TCD) in children with sickle-cell anemia. Pediatr Radiol. 2001;31(7):461–469. doi:10.1007/s002470100427
37. McCarville MB, Li C, Xiong X, Wang W. Comparison of transcranial Doppler sonography with and without imaging in the evaluation of children with sickle cell anemia. Am J Roentgenol. 2004;183(4):1117–1122. doi:10.2214/ajr.183.4.1831117
38. Lee L, Smith-Whitley K, Banks S, Puckrein G. Reducing health care disparities in sickle cell disease: a review. Public Health Rep. 2019;134(6):599–607. doi:10.1177/0033354919881438
39. Wailoo K. Dying in the city of the blues: sickle cell anemia and the politics of race and health. Studies in social medicine. University of North Carolina Press; 2001. Available from: http://www.loc.gov/catdir/samples/unc041/00062865.html.
40. Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339(1):5–11. doi:10.1056/NEJM199807023390102
41. Lee MT, Piomelli S, Granger S, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood. 2006;108(3):847–852. doi:10.1182/blood-2005-10-009506
42. Abboud MR, Yim E, Musallam KM, Adams RJ. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood. 2011;118(4):894–898. doi:10.1182/blood-2010-12-326298
43. Wayne AS, Kevy SV, Nathan DG. Transfusion management of sickle cell disease. Blood. 1993;81(5):1109–1123.
44. Wahl S, Quirolo KC. Current issues in blood transfusion for sickle cell disease. Curr Opin Pediatr. 2009;21(1):15–21. doi:10.1097/MOP.0b013e328321882e
45. Marouf R. Blood transfusion in sickle cell disease. Hemoglobin. 2011;35(5–6):495–502. doi:10.3109/03630269.2011.596984
46. Chou ST, Alsawas M, Fasano RM, et al. American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv. 2020;4(2):327–355. doi:10.1182/bloodadvances.2019001143
47. Howard J. Sickle cell disease: when and how to transfuse. Hematology Am Soc Hematol Educ Program. 2016;2016(1):625–631.
48. Talano JAM, Hillery CA, Gottschall JL, Baylerian DM, Scott JP. Delayed hemolytic transfusion reaction/hyperhemolysis syndrome in children with sickle cell disease. Pediatrics. 2003;111(6 Pt 1):e661–665.
49. Win N. Hyperhemolysis syndrome in sickle cell disease. Expert Rev Hematol. 2009;2(2):111–115. doi:10.1586/ehm.09.2
50. Yazdanbakhsh K, Ware RE, Noizat-Pirenne F. Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood. 2012;120(3):528–537. doi:10.1182/blood-2011-11-327361
51. Linder GE, Chou ST. Red cell transfusion and alloimmunization in sickle cell disease. Haematologica. 2021;106(7):1805–1815. doi:10.3324/haematol.2020.270546
52. LaSalle-Williams M, Nuss R, Le T, et al. Extended red blood cell antigen matching for transfusions in sickle cell disease: a review of a 14-year experience from a single center (CME). Transfusion. 2011;51(8):1732–1739. doi:10.1111/j.1537-2995.2010.03045.x
53. Casas J, Friedman DF, Jackson T, Vege S, Westhoff CM, Chou ST. Changing practice: red blood cell typing by molecular methods for patients with sickle cell disease. Transfusion. 2015. doi:10.1111/trf.12987
54. Coates TD, Wood JC. How we manage iron overload in sickle cell patients. Br J Haematol. 2017;177(5):703–716. doi:10.1111/bjh.14575
55. Wilson SR, Sears M, Williams E, et al. Gaps in the diagnosis and management of iron overload in sickle cell disease: a “real-world” report from the GRNDaD registry. Br J Haematol. 2021;195(5):e157–e160. doi:10.1111/bjh.17762
56. Adamkiewicz TV, Abboud MR, Paley C, et al. Serum ferritin level changes in children with sickle cell disease on chronic blood transfusion are nonlinear and are associated with iron load and liver injury. Blood. 2009;114(21):4632–4638. doi:10.1182/blood-2009-02-203323
57. Wood JC. Estimating tissue iron burden: current status and future prospects. Br J Haematol. 2015;170(1):15–28. doi:10.1111/bjh.13374
58. Vichinsky E, Onyekwere O, Porter J, et al. A randomised comparison of deferasirox versus deferoxamine for the treatment of transfusional iron overload in sickle cell disease. Br J Haematol. 2007;136(3):501–508. doi:10.1111/j.1365-2141.2006.06455.x
59. Vichinsky E, Pakbaz Z, Onyekwere O, et al. Patient-reported outcomes of deferasirox (Exjade, ICL670) versus deferoxamine in sickle cell disease patients with transfusional hemosiderosis. Substudy of a randomized open-label Phase II trial. Acta Haematol. 2008;119(3):133–141. doi:10.1159/000125550
60. Treadwell MJ, Law AW, Sung J, et al. Barriers to adherence of deferoxamine usage in sickle cell disease. Pediatr Blood Cancer. 2005;44(5):500–507. doi:10.1002/pbc.20290
61. Novartis. Exjade (Deferasirox). United States Food and Drug Administration; 2013. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/021882s019lbl.pdf.
62. Velasquez J, Wray AA. Deferoxamine. In: StatPearls. StatPearls Publishing; 2022. Available from: http://www.ncbi.nlm.nih.gov/books/NBK557654/.
63. Calvaruso G, Vitrano A, Di Maggio R, et al. Deferiprone versus deferoxamine in sickle cell disease: results from a 5-year long-term Italian multi-center randomized clinical trial. Blood Cells Mol Dis. 2014;53(4):265–271. doi:10.1016/j.bcmd.2014.04.004
64. Elalfy MS, Hamdy M, El Beshlawy A, et al. Deferiprone for transfusional iron overload in sickle cell disease and other anemias: open-label study of up to 3 years. Blood Adv. 2022:
65. Ware RE, Davis BR, Schultz WH, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial Doppler flow velocities in children with sickle cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial. Lancet. 2016;387(10019):661–670. doi:10.1016/S0140-6736(15)01041-7
66. Treadwell MJ, Du L, Bhasin N, et al. Barriers to hydroxyurea use from the perspectives of providers, individuals with sickle cell disease, and families: report from a U.S. regional collaborative. Front Genet. 2022;13:921432. doi:10.3389/fgene.2022.921432
67. Vick L, Potts M, Jaskowiak M, Gibson RW. Hydroxyurea adherence strategies for persons with sickle cell disease: a systematic review. J Health Care Poor Underserved. 2021;32(1):99–118. doi:10.1353/hpu.2021.0011
68. Hodges JR, Phillips SM, Norell S, et al. Intentional and unintentional nonadherence to hydroxyurea among people with sickle cell disease: a qualitative study. Blood Adv. 2020;4(18):4463–4473. doi:10.1182/bloodadvances.2020001701
69. Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol. 2010;85(6):403–408. doi:10.1002/ajh.21699
70. Rodriguez A, Duez P, Dedeken L, Cotton F, Ferster A. Hydroxyurea (hydroxycarbamide) genotoxicity in pediatric patients with sickle cell disease. Pediatr Blood Cancer. 2018;65(7):e27022. doi:10.1002/pbc.27022
71. Rankine-Mullings AE, Nevitt SJ. Hydroxyurea (hydroxycarbamide) for sickle cell disease. Cochrane Database Syst Rev. 2022;(9). doi:10.1002/14651858.CD002202.pub3
72. Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377(9778):1663–1672. doi:10.1016/S0140-6736(11)60355-3
73. Lebensburger JD, Miller ST, Howard TH, et al. Influence of severity of anemia on clinical findings in infants with sickle cell anemia: analyses from the BABY HUG study. Pediatr Blood Cancer. 2012;59(4):675–678. doi:10.1002/pbc.24037
74. Smart LR, Ambrose EE, Balyorugulu G, et al. Stroke Prevention with Hydroxyurea Enabled through Research and Education (SPHERE): a Phase 2 primary stroke prevention trial in Sub-Saharan Africa. AHA. 2022;145(6):66. doi:10.1159/000526322
75. Miller ST, Macklin EA, Pegelow CH, et al. Silent infarction as a risk factor for overt stroke in children with sickle cell anemia: a report from the Cooperative Study of Sickle Cell Disease. The Journal of Pediatrics. 2001;139(3):385–390. doi:10.1067/mpd.2001.117580.
76. DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood. 2012;119(20):4587–4596. doi:10.1182/blood-2011-02-272682
77. DeBaun MR, Gordon M, McKinstry RC, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med. 2014;371(8):699–710. doi:10.1056/NEJMoa1401731
78. Guilliams KP, Fields ME, Hulbert ML. Higher-than-expected prevalence of silent cerebral infarcts in children with hemoglobin SC disease. Blood. 2015;125(2):416–417. doi:10.1182/blood-2014-10-605964
79. Sathi BK, Yoshida Y, Weaver MR, et al. Unusually high prevalence of stroke and cerebral vasculopathy in hemoglobin SC Disease: a Retrospective Single Institution Study. Acta Haematol. 2022;145(2):160–169. doi:10.1159/000519360
80. Hasson C, Veling L, Rico J, Mhaskar R. The role of hydroxyurea to prevent silent stroke in sickle cell disease. Medicine. 2019;98(51):e18225. doi:10.1097/MD.0000000000018225
81. Fields ME, Guilliams KP, Ragan D, et al. Hydroxyurea reduces cerebral metabolic stress in patients with sickle cell anemia. Blood. 2019;133(22):2436–2444. doi:10.1182/blood-2018-09-876318
82. Estcourt LJ, Kimber C, Hopewell S, Trivella M, Doree C, Abboud MR. Interventions for preventing silent cerebral infarcts in people with sickle cell disease. Cochrane Database Syst Rev. 2020;4:CD012389. doi:10.1002/14651858.CD012389.pub3
83. Powars D, Wilson B, Imbus C, Pegelow C, Allen J. The natural history of stroke in sickle cell disease. Am J Med. 1978;65(3):461–471.
84. Webb J, Kwiatkowski JL. Stroke in patients with sickle cell disease. Expert Rev Hematol. 2013;6(3):301–316. doi:10.1586/ehm.13.25
85. Rafay MF, Shapiro KA, Surmava AM, et al. Spectrum of cerebral arteriopathies in children with arterial ischemic stroke. Neurology. 2020;94(23):e2479–e2490. doi:10.1212/WNL.0000000000009557
86. Aa K, Na G. How I treat and manage strokes in sickle cell disease. Blood. 2015;125(22). doi:10.1182/blood-2014-09-551564
87. Hulbert ML, Scothorn DJ, Panepinto JA, et al. Exchange blood transfusion compared with simple transfusion for first overt stroke is associated with a lower risk of subsequent stroke: a retrospective cohort study of 137 children with sickle cell anemia. J Pediatr. 2006;149(5):710–712. doi:10.1016/j.jpeds.2006.06.037
88. Hurlet-Jensen AM, Prohovnik I, Pavlakis SG, Piomelli S. Effects of total hemoglobin and hemoglobin S concentration on cerebral blood flow during transfusion therapy to prevent stroke in sickle cell disease. Stroke. 1994;25(8):1688–1692. doi:10.1161/01.str.25.8.1688
89. Strouse JJ, Lanzkron S, Urrutia V. The epidemiology, evaluation and treatment of stroke in adults with sickle cell disease. Expert Rev Hematol. 2011;4(6):597–606. doi:10.1586/ehm.11.61
90. Frye RE. Reversible posterior leukoencephalopathy syndrome in sickle-cell anemia. Pediatr Neurol. 2009;40(4):298–301. doi:10.1016/j.pediatrneurol.2008.10.024
91. Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334(8):494–500. doi:10.1056/NEJM199602223340803
92. Raj S, Killinger J, Overby P. Blood transfusion in sickle cell disease leading to posterior reversible encephalopathy syndrome (PRES). J Child Neurol. 2013;28(10):1284–1286. doi:10.1177/0883073812453497
93. Vargas A, Testai FD. Posterior reversible encephalopathy syndrome in adult sickle-cell patients: case series and literature review. J Clin Neurosci. 2019;70:249–250. doi:10.1016/j.jocn.2019.08.070
94. Sidani CA, Ballourah W, El Dassouki M, et al. Venous sinus thrombosis leading to stroke in a patient with sickle cell disease on hydroxyurea and high hemoglobin levels: treatment with thrombolysis. Am J Hematol. 2008;83(10):818–820. doi:10.1002/ajh.21261
95. Rahimi Z, Parsian A. Sickle cell disease and venous thromboembolism. Mediterr J Hematol Infect Dis. 2011;3(1):e2011024. doi:10.4084/MJHID.2011.024
96. Lusher JM, Haghighat H, Khalifa AS. A prophylactic transfusion program for children with sickle cell anemia complicated by CNS infarction. Am J Hematol. 1976;1(2):265–273. doi:10.1002/ajh.2830010210
97. Russell M, Goldberg H, Hodson A, et al. Effect of transfusion therapy on arteriographic abnormalities and on recurrence of stroke in sickle cell disease. Blood. 1984;63(1):162–169. doi:10.1182/blood.V63.1.162.162
98. Pegelow CH, Adams RJ, McKie V, et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. J Pediatr. 1995;126(6):896–899. doi:10.1016/S0022-3476(95)70204-0
99. Estcourt LJ, Kohli R, Hopewell S, Trivella M, Wang WC. Blood transfusion for preventing primary and secondary stroke in people with sickle cell disease. Cochrane Database Syst Rev. 2020;7:CD003146. doi:10.1002/14651858.CD003146.pub4
100. Ware RE, Helms RW. Stroke With Transfusions Changing to Hydroxyurea (SWiTCH). Blood. 2012;119(17):3925–3932. doi:10.1182/blood-2011-11-392340
101. Kayle M, Docherty SL, Sloane R, et al. Transition to adult care in sickle cell disease: a longitudinal study of clinical characteristics and disease severity. Pediatr Blood Cancer. 2019;66(1):e27463. doi:10.1002/pbc.27463
102. Lanzkron S, Carroll CP, Haywood C. Mortality rates and age at death from sickle cell disease: u.S., 1979–2005. Public Health Rep. 2013;128(2):110–116. doi:10.1177/003335491312800206
103. Anie KA, Telfair J; Sickle Cell Disease Transition Study Working Group. Multi-site study of transition in adolescents with sickle cell disease in the United Kingdom and the United States. Int J Adolesc Med Health. 2005;17(2):169–178. doi:10.1515/ijamh.2005.17.2.169
104. Treadwell M, Telfair J, Gibson RW, Johnson S, Osunkwo I. Transition from pediatric to adult care in sickle cell disease: establishing evidence-based practice and directions for research. Am J Hematol. 2011;86(1):116–120. doi:10.1002/ajh.21880
105. Treadwell M, Johnson S, Sisler I, et al. Development of a sickle cell disease readiness for transition assessment. Int J Adolesc Med Health. 2016;28(2):193–201. doi:10.1515/ijamh-2015-0010
106. Currie S, Raghavan A, Batty R, Connolly DJA, Griffiths PD. Childhood moyamoya disease and moyamoya syndrome: a pictorial review. Pediatr Neurol. 2011;44(6):401–413. doi:10.1016/j.pediatrneurol.2011.02.007
107. Dobson SR, Holden KR, Nietert PJ, et al. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood. 2002;99(9):3144–3150.
108. Griessenauer CJ, Lebensburger JD, Chua MH, et al. Encephaloduroarteriosynangiosis and encephalomyoarteriosynangiosis for treatment of moyamoya syndrome in pediatric patients with sickle cell disease. J Neurosurg Pediatr. 2015:1–10. doi:10.3171/2014.12.PEDS14522
109. Winstead M, Sun PP, Martin K, et al. Encephaloduroarteriosynangiosis (EDAS) in young patients with cerebrovascular complications of sickle cell disease: single-institution experience. Pediatr Hematol Oncol. 2017;34(2):100–106. doi:10.1080/08880018.2017.1313917
110. Alamri A, Hever P, Cheserem J, Gradil C, Bassi S, Tolias CM. Encephaloduroateriosynangiosis (EDAS) in the management of Moyamoya syndrome in children with sickle cell disease. Br J Neurosurg. 2019;33(2):161–164. doi:10.1080/02688697.2017.1339227
111. Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2016. doi:10.1182/blood-2016-10-745711
112. Khemani K, Katoch D, Krishnamurti L. Curative therapies for sickle cell disease. Ochsner J. 2019;19(2):131–137. doi:10.31486/toj.18.0044
113. Verlhac S, Gabor F, Paillard P, et al. Improved stenosis outcome in stroke-free sickle cell anemia children after transplantation compared to chronic transfusion. Br J Haematol. 2021;193(1). doi:10.1111/bjh.17178
114. Thurn S, Kleinschmidt K, Kovacic I, et al. Transcranial Doppler sonography and the effect of haematopoietic stem cell transplantation in sickle cell disease. Neurol Res Pract. 2022;4(1):12. doi:10.1186/s42466-022-00175-y
115. Wailoo K, Pemberton S. The Troubled Dream of Genetic Medicine: Ethnicity and Innovation in Tay-Sachs, Cystic Fibrosis, and Sickle Cell Disease. Johns Hopkins University Press; 2006. Available from: http://ebookcentral.proquest.com/lib/unc/detail.action?docID=3318266.
116. DeBaun MR, Clayton EW. Primum non nocere: the case against transplant for children with sickle cell anemia without progressive end-organ disease. Blood Adv. 2017;1(26):2568–2571. doi:10.1182/bloodadvances.2017007690
117. Fitzhugh CD, Walters MC. The case for HLA-identical sibling hematopoietic stem cell transplantation in children with symptomatic sickle cell anemia. Blood Adv. 2017;1(26):2563–2567. doi:10.1182/bloodadvances.2017007708
118. Stenger E, Xiang Y, Wetzel M, et al. Long-term organ function after HCT for SCD: a report from the sickle cell transplant advocacy and research alliance. Transplant Cell Ther. 2022:S2666-6367(22)01708–0. doi:10.1016/j.jtct.2022.10.012
119. Shenoy S, Eapen M, Panepinto JA, et al. A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood. 2016;128(21):2561–2567. doi:10.1182/blood-2016-05-715870
120. Ngwube A, Shah N, Godder K, Jacobsohn D, Hulbert ML, Shenoy S. Abatacept is effective as GVHD prophylaxis in unrelated donor stem cell transplantation for children with severe sickle cell disease. Blood Adv. 2020;4(16):3894–3899. doi:10.1182/bloodadvances.2020002236
121. Kasow KA, Krajewski J, Haines H, et al. Curing children with sickle hemoglobinopathies of varying severity early in life with HLA-matched sibling donor (MSD) hematopoietic cell transplantation (HCT) following a reduced intensity conditioning (RIC) regimen: a Sickle Transplant, Advocacy, and Research (STAR) trial. Transplant Cell Ther. 2021;29(2S):S293.
122. Krishnamurti L, Neuberg DS, Sullivan KM, et al. Bone marrow transplantation for adolescents and young adults with sickle cell disease: results of a prospective multicenter pilot study. Am J Hematol. 2019;94(4):446–454. doi:10.1002/ajh.25401
123. Stimpson SJ, DeBaun M. Comparing outcomes for young adults with severe SCD treated with HLA-identical sibling and matched unrelated HCT versus standard supportive care. Hematologist. 2016;13:3. doi:10.1182/hem.V13.3.5469
124. Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA. 2014;312(1):48–56. doi:10.1001/jama.2014.7192
125. Saraf SL, Oh AL, Patel PR, et al. Nonmyeloablative stem cell transplantation with alemtuzumab/low-dose irradiation to cure and improve the quality of life of adults with sickle cell disease. Biol Blood Marrow Transplant. 2016;22(3):441–448. doi:10.1016/j.bbmt.2015.08.036
126. Nickel RS, Chiang K, Hardy SJ, et al. Nonmyeloablative HLA-identical sibling donor transplantation for children and young adults with sickle cell disease: interim results of the SUN multicenter phase II trial. Blood. 2021;138:1799. doi:10.1182/blood-2021-144454
127. Carden MA, Little J. Emerging disease-modifying therapies for sickle cell disease. Haematologica. 2019;104(9):1710–1719. doi:10.3324/haematol.2018.207357
128. Migotsky M, Beestrum M, Badawy SM. Recent advances in sickle-cell disease therapies: a review of voxelotor, crizanlizumab, and L-glutamine. Pharmacy. 2022;10(5):123. doi:10.3390/pharmacy10050123
129. Ataga KI, Kutlar A, Kanter J, et al. SUSTAIN: a multicenter, randomized, placebo-controlled, double-blind, 12-month study to assess safety and efficacy of SelG1 with or without hydroxyurea therapy in sickle cell disease patients with sickle cell-related pain crises. Blood. 2016;128(22):1. doi:10.1182/blood.V128.22.1.1
130. Safo MK, Kato GJ. Therapeutic strategies to alter the oxygen affinity of sickle hemoglobin. Hematol Oncol Clin North Am. 2014;28(2):217–231. doi:10.1016/j.hoc.2013.11.001
131. Ki JH, Rc A. Voxelotor in adolescents and adults with sickle cell disease (HOPE): long-term follow-up results of an international, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Haematol. 2021;8:5. doi:10.1016/S2352-3026(21)00059-4
132. Hebbel RP, Hedlund BE. Sickle hemoglobin oxygen affinity-shifting strategies have unequal cerebrovascular risks. Am J Hematol. 2018;93(3):321–325. doi:10.1002/ajh.24975
133. Estepp JH, Kalpatthi R, Woods G, et al. Safety and efficacy of voxelotor in pediatric patients with sickle cell disease aged 4 to 11 years. Pediatr Blood Cancer. 2022;69(8):e29716. doi:10.1002/pbc.29716
134. Bank A, Markowitz D, Lerner N. Gene transfer. A potential approach to gene therapy for sickle cell disease. Ann N Y Acad Sci. 1989;565:37–43. doi:10.1111/j.1749-6632.1989.tb24147.x
135. Abraham AA, Tisdale JF. Gene therapy for sickle cell disease: moving from the bench to the bedside. Blood. 2021;138(11):932–941. doi:10.1182/blood.2019003776
136. Kolata G. Pioneering gene therapy freed her of sickle cell. Is a cure at hand? The New York Times; 2021. Available from: https://www.nytimes.com/2021/09/14/health/sickle-cell-cure.html.
137. Stein R. First sickle cell patient treated with CRISPR gene-editing still thriving. NPR; 2021. Available from: https://www.npr.org/sections/health-shots/2021/12/31/1067400512/first-sickle-cell-patient-treated-with-crispr-gene-editing-still-thriving.
138. Kanter J, Walters MC, Krishnamurti L, et al. Biologic and clinical efficacy of lentiglobin for sickle cell disease. N Engl J Med. 2022;386(7):617–628. doi:10.1056/NEJMoa2117175
139. Goyal S, Tisdale J, Schmidt M, et al. Acute myeloid leukemia case after gene therapy for sickle cell disease. N Engl J Med. 2022;386(2):138–147. doi:10.1056/NEJMoa2109167
140. Kohn DB, Sadelain M, Glorioso JC. Occurrence of leukaemia following gene therapy of X-linked SCID. Nat Rev Cancer. 2003;3(7):477–488. doi:10.1038/nrc1122
141. Jones RJ, DeBaun MR. Leukemia after gene therapy for sickle cell disease: insertional mutagenesis, busulfan, both, or neither. Blood. 2021;138(11):942–947. doi:10.1182/blood.2021011488
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.