")
Back to Journals » Infection and Drug Resistance » Volume 14
Managing Fungal Infections in Cystic Fibrosis Patients: Challenges in Clinical Practice
Authors Magee LC, Louis M, Khan V, Micalo L, Chaudary N
Received 26 December 2020
Accepted for publication 25 February 2021
Published 22 March 2021 Volume 2021:14 Pages 1141—1153
DOI https://doi.org/10.2147/IDR.S267219
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Lauren C Magee,1 Mariam Louis,2 Vaneeza Khan,3 Lavender Micalo,3 Nauman Chaudary3
1Department of Pharmacy, Virginia Commonwealth University Health System, Richmond, VA, USA; 2Department of Pulmonary, Critical Care and Sleep Medicine, University of Florida, Jacksonville, FL, USA; 3Division of Pulmonary Disease and Critical Care Medicine, Department of Medicine, Virginia Commonwealth University, Richmond, VA, USA
Correspondence: Lauren C Magee; Nauman Chaudary
VCU Adult Cystic Fibrosis Center, Division of Pulmonary Disease and Critical Care Medicine, 1200 East Broad Street, Box 980050, Richmond, VA, 23298, USA
Tel +1 804 628-5046; +1 804 828-1579
Fax +1 804 828-2578
Email [email protected]; [email protected]
Abstract: Cystic Fibrosis (CF) is an autosomal recessive disease characterized by a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Impairment of the CFTR protein in the respiratory tract results in the formation of thick mucus, development of inflammation, destruction of bronchial tissue, and development of bacterial or fungal infections over time. CF patients are commonly colonized and/or infected with fungal organisms, Candida albicans or Aspergillus fumigatus, with prevalence rates ranging from 5% to 78% in the literature. Risk factors for acquiring fungal organisms include older age, coinfection with Pseudomonas aeruginosa, prolonged use of oral and inhaled antibiotics, and lower forced expiratory volume (FEV1). There are limited data available to differentiate between contamination, colonization, and active infection. Furthermore, the pathogenicity of colonization is variable in the literature as some studies report a decline in lung function associated with fungal colonization whereas others showed no difference. Limited data are available for the eradication of fungal colonization and the treatment of active invasive aspergillosis in adult CF patients. In this review article, we discuss the challenges in clinical practice and current literature available for laboratory findings, clinical diagnosis, and treatment options for fungal infections in adult CF patients.
Keywords: cystic fibrosis, fungal infection, colonization, Candida, Aspergillus, ABPA, Aspergillus bronchitis
Introduction
Cystic Fibrosis (CF) is an autosomal recessive disease characterized by a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) protein. The CFTR protein is responsible for balancing salt and water homeostasis throughout the body including the respiratory tract. Impairment in the CFTR protein results in the formation of thick mucus, development of inflammation, destruction of bronchial tissue, and development of bacterial or fungal infections over time.1
CF care involves aggressive monitoring and treatment of infectious exacerbations to prevent a decline in lung function. Only 75% of CF patients return to within 90% of their baseline lung function, as measured by the forced expiratory volume (FEV1), with treatment.2 Clinicians involved in the management of CF patients should complete routine microbiological assessments of expectorated sputum at a minimum annually but preferably on a quarterly basis.3 Routine surveillance for infectious organisms includes bacterial cultures, antibiotic susceptibility testing, and acid fast bacillus (AFB) cultures. However, certain patients may require screening for fungal infections especially those who have lack benefit from routine antibiotics in recovering lung function. Other considerations for fungal infection treatment include abnormal imaging, new fever, change in sputum characteristics, and hemoptysis. As the prevalence of fungal organisms continues to rise, the clinical significance of detecting fungal species in the CF airway remains unclear.4 The fungal species detected, the clinical context in which it is isolated, as well as the duration of fungal species in sputum all play a role in determining colonization versus infection.
Conflicting data exist regarding the consequences of detecting Aspergillus in respiratory samples, with some studies suggesting a worse clinical outcome, and others having found no difference.5–7 However, patients diagnosed with allergic bronchopulmonary aspergillosis (ABPA) should be treated based on their symptoms and classification. In addition, patients in whom invasive fungal infections are suspected should also be treated. Criteria for differentiating invasive fungal infections are not clearly defined. However, several groups have published recommendations for criteria for “highly probable” invasive fungal infections in CF patients (versus colonization) that would require prompt treatment.4,8
In view of the current state of diagnostic challenges for fungal infections in patients with CF, the microbiological, immunological and clinical context of the patient must all be integrated to determine the best treatment pathway for the patient.
Pathology of Fungal Infections in Cystic Fibrosis
CFTR dysfunction has been linked to an increased risk of Aspergillus spp. colonization.9 The dysfunction of this protein may cause a profound Th2 response to inhaled A. fumigatus, which results in hyperinflammatory immune responses leading to the development of ABPA.10 The importance of the CFTR protein in fungal colonization is further supported in a study by Chaudhary et al examing the role of CFTR in the interactions between epithelial cells and A. fumigatus.11 This study showed that when CFTR knockout mice (CFTR-/-) were challenged with inhaled A. fumigatus, the mice developed aberrant pulmonary inflammation and also exhibited poor mucociliary clearance of fungus. Furthermore, Knutsen et al attributed the impaired mucociliary clearance mechanism to the thick and viscous mucus found in CF airways, which in turn created a perfect environment for Aspergillus colonization.12 Thus, the dysfunction of the CFTR protein and resulting impairment in mucociliary clearance predispose CF airways to fungal infections.
The high incidence of bacterial infections in CF patients is also a risk factor for developing fungal infections. The chronic usage of antibiotics to treat bacterial infections has been associated with increased fungal colonization in CF.13 This finding is supported by a prospective evaluation of 56 CF patients showing that chronic use of antibiotics, oral and inhaled steroids predisposed patients to oral colonization of Candida spp.14
The notion that treatment of bacterial infections may lead to enhanced growth of fungal species is an interesting one. This may mean that fungi are not harmful to the host as they are usually accompanied by a rise in FEV1. However, it is not clear whether fungi hasten the return of Pseudomonas aeruginosa over time because of a potential interaction between Candida spp. and P. aeruginosa resulting in biofilm formation.15 It is also thought that several strains of fungi may coexist in the same individual and that variable susceptibilities to these strains exist within the same individual.16 This highlights the complexity of fungal colonies in chronically infected CF patients in addition to the uncertainty of which exogenous and environmental risk factors play a role in the evolution of fungi.
Risk Factors for Fungal Infections
As the understanding of fungal epidemiology in CF airways continues to evolve, various risk factors have been identified for the development of fungal organisms in CF specimens.
A retrospective study investigating the epidemiologic trend of fungal infections in CF patients found that older age was a risk factor for fungal isolation, specifically adults over the age of 18 years old were 51% more likely to grow filamentous fungi.17 Sudfeld et al also found that fungal colonization onset was significantly associated with older age and the use of inhaled antibiotics.
The use of inhaled tobramycin as an intermittent regimen for chronic maintenance therapy in CF patients has been linked to increased isolation of fungal species, such as C. albicans and Aspergillus spp.13 Jubin et al also found a strong association between long-term use of azithromycin and Aspergillus spp. colonization in CF patients.18 This finding is supported by Bargon et al who showed a significant relationship between inhaled and oral antibiotics with Aspergillus colonization.19 They attributed this association to the antagonistic relationship between P. aeruginosa and Aspergillus species. It has been reported that P. aeruginosa species inhibit A. fumigatus filamentation by releasing extracellular molecules that inhibit intracellular communication therefore limiting fungal growth.20
In a retrospective review of 16,095 patients from 2006 to 2012 Cystic Fibrosis Foundation (CFF) registry data, 9.6% of patients (n=1541) persistently grew Aspergillus. Upon further examination, the following risk factors were identified for persistent Aspergillus isolation: chronic inhaled antibiotics (Odds Ratio [OR] 1.33; 95% confidence interval 1.21– 1.46, p<0.001), macrolides (OR 1.23, 95% CI 1.14–1.32, p<0.001), and inhaled corticosteroids (OR 1.14, 95% CI 1.09– 1.20, p=0.001).9
Aspergillus spp. can coexist with many bacteria. Stenotrophomonas maltophilia and Aspergillus spp. coexistence is actually quiet common.7 However, A. fumigatus and P. aeruginosa are the most common pathogens in CF airways and coexistence with these pathogens is associated with poorer FEV1 scores.21 Patients with co-colonization are also associated with increased risk of exacerbations and hospitalizations.7
Prevalence and Pathogenicity of Fungal Organisms
A wide spectrum of fungi have been isolated in patients with CF. The most common fungal organisms detected in CF are Candida albicans, Aspergillus fumigatus, Scedosporium spp. and Exophilia dermatitidis.22,23 [Table 1] Figure 1 shows these different fungal organisms grouped together based on chronicity and pathogenicity.4
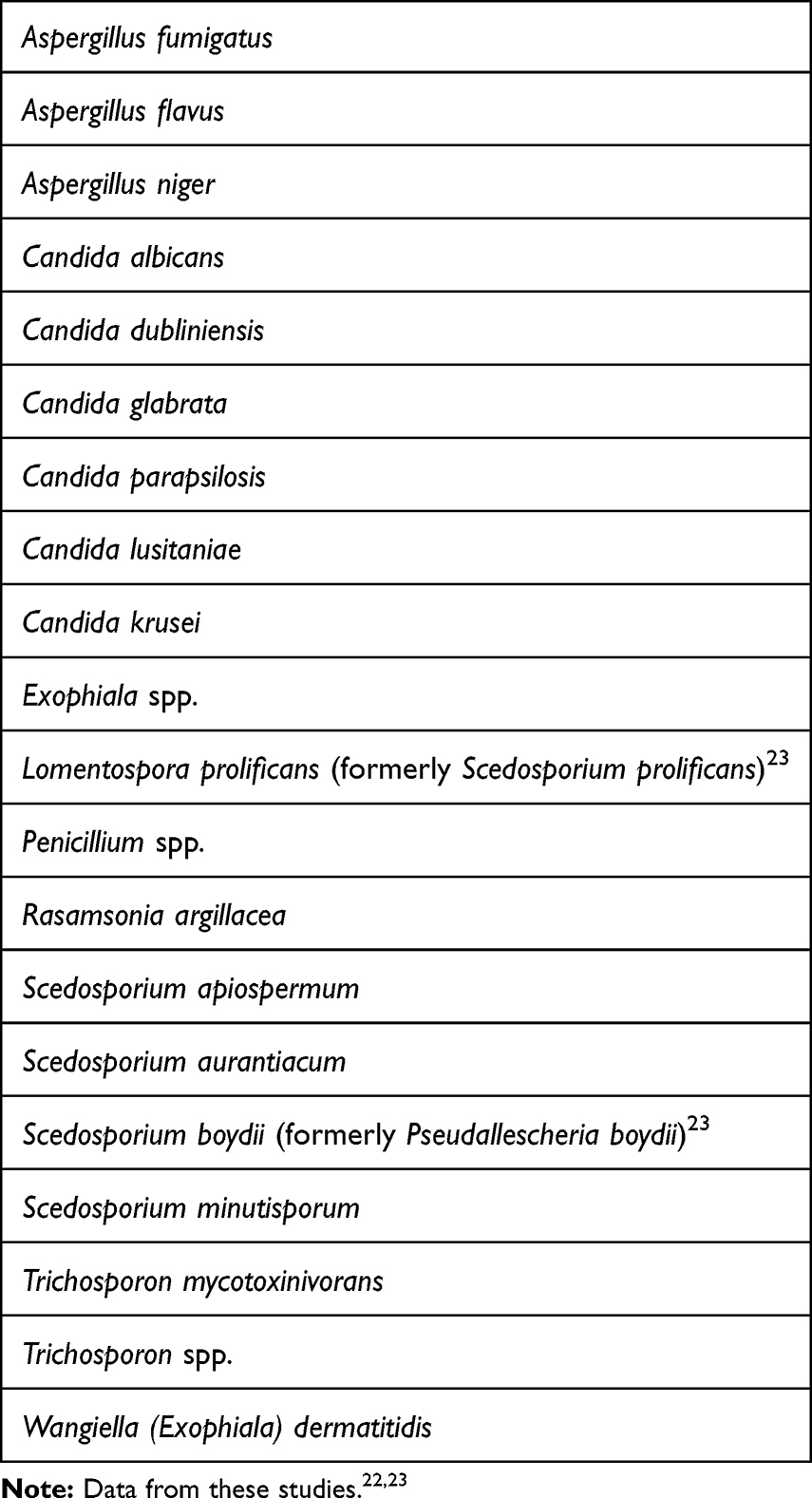 |
Table 1 Common Fungal Organisms in Cystic Fibrosis Patients |
 |
Figure 1 Chronicity and pathogenicity of fungal organisms in cystic fibrosis. Data from Tracy and Moss.4 |
Candida spp
Candida spp. are fungi commonly found in the oral cavity. Candida spp. are identified as dead yeast in sputum cultures following the release of a phenazine toxin.24 Yeast are known to have a high chronicity (ie persist for long durations with difficulty eradicating) due to chronic use of antibiotics and steroids; however, their pathogenicity, or ability to cause active infection, remains a matter of debate.
In a 2016 evaluation of nearly 26,000 sputum samples of CF patients over a 5-year period, Candida spp. was detected in nearly 75% of samples with the most prevalent species being C. albicans (38%).22 A retrospective analysis of 3235 patients from nine European CF centers found geographic variations in the prevalence of Candida spp. C. albicans was reportedly the most prevalent yeast in every center with prevalence rates ranging from 33.8% up to 77.9%. Other Candida species were also detected at lower and more diverse frequencies across each center.25 The effect of Candida on CF disease progression, however, is not well understood.
A prospective observational study in 89 CF patients (3916 sputum samples over 11 years) showed that 49.4% of patient were colonization with C. albicans. Furthermore, colonization with C. albicans was associated with significantly increased rates of hospital-treated pulmonary exacerbations (P=0.004) and FEV1 decline (P<0.001).26
A similar study was conducted in 91 CF patients in Isreal (4244 sputum samples over 7 years). Patients were stratified into three groups: chronic, intermittent or no C. albicans colonization. Patients in the chronic C. albicans colonization group (n=34) had lower FEV1% predicted (74.3 ±23.1% vs. 93.9 ±22.2%) and a higher annual rate of FEV1 decline (−1.9 ±4.2% vs. 0.7 ±4.5%) compared to the non-colonized group (n=27).27
C. dubliniensis has also been associated with a decline in lung function over time.28,29 A 16-year retrospective study on fungal prevalence in Sweden (n=133) showed that CF patients with positive C. dubliniensis cultures for three consecutive years experienced a 7.6% reduction in percent predicted forced expiratory volume in the first second (ppFEV1; p=0.001).28
Microbial colonization and corresponding lung function were characterized for 770 adolescent CF patients in Europe. The results were conflicting as those colonized with C. albicans demonstrated a protective effect on lung function compared to those colonized with C. glabrata who demonstrated an overall decline in FEV1 from baseline.30
Given high prevalence rates and conflicting clinical findings, distinguishing between colonization and pathogenicity, or active infection, can be quite challenging.
Aspergillus spp
Another fungal species that has been isolated in sputum cultures of CF patients is Aspergillus spp. The most common organisms detected are A. fumigatus and A. terreus; these organisms have been identified to be both chronic colonizers and sources of active infection in CF patients. A. flavus, A. niger, and A. nidulans have also been commonly detected in respiratory cultures.31
A. fumigatus spores are 2–4 µm in diameter and inhaled with ease throughout the environment.32 The spores settle on the mucous membranes of the airway and lungs, and as they grow cause the failure of mucociliary clearance. The cells secrete proteolytic enzymes (eg proteases, phosphatases) that inhibit phagocytosis and further facilitate adhesion and colonization within the airways in the presence of melanin and class I hydrophobins.24
Aspergillus spp. have been described in the literature in various classifications: ABPA, colonization, sensitization, bronchitis, and invasive aspergillosis.33 The prevalence of A. fumigatus colonization is widely variable in the literature ranging from 5% to 78%.7, 34–36 In a 2016 evaluation of nearly 26,000 sputum samples of CF patients over a 5-year period, Aspergillus spp. was detected in 35% of samples with the most prevalent species being A. fumigatus (29%).22 Schwartz et al performed a retrospective analysis of 66,616 samples from 3235 CF patients in nine European CF centers. They reported A. fumigatus dominance in most of the centers with prevalence rates ranging from 3.9% up to 42.4%.25
Several studies demonstrate that colonization with A. fumigatus results in declining pulmonary function.7,30,36–38 A case–control study of 20 CF patients chronically colonized with A. fumigatus matched against 60 control patients showed an FEV1 that was 8.66% lower over a 7-year time period compared to those never colonized with A. fumigatus (p=0.020). This study also found that control patients with lower baseline FEV1 values had a higher risk of chronic colonization with A. fumigatus.36
A characterization of microbial organisms in adolescent CF patients showed that patients colonized with A. fumigatus had lower lung function as demonstrated by a decline in FEV1 compared to non-colonized patients (P<0.0001) and an increased risk of colonization with P. aeruginosa.30
In an evaluation of 238 pediatric CF patients, those with persistent A. fumigatus infections had an FEV1 3.61% lower than uninfected patients (P≤0.0001) and an increased risk of pulmonary exacerbations requiring hospitalization (Relative Risk [RR] = 1.94, P=0.0002).7
However, these findings of a decline in pulmonary function are conflicting in the literature.38,39 In an evaluation of 259 CF patients, colonization with A. fumigatus was not associated with lower lung function nor more severe lung function decline over a 5-year period.38 Given these conflicting findings, it is difficult to determine the necessity of eradication therapy for chronic colonization with Aspergillus spp.
Pneumocystis jirovecii
P. jirovecii was detected in a bronchoalveolar lavage (BAL) fluid sample from a non-HIV-infected 15 week-old child diagnosed with CF presenting with pneumonia.40 Following this diagnosis, other countries evaluated the prevalence of Pneumocystis colonization in cystic fibrosis patients. Prevalence rates range from 1.3% to 21.6% throughout Europe.41 The difference in prevalence rate for different countries may be due to the difference in population density or climate factors.41 The seasonal changes in winter months appear to be associated with maximal respiratory infectious diseases; however, the data is conflicting as German researchers found opposing weather patterns with maximal respiratory observed in summer months. Incidental acquisition of P. jirovecii has also been postulated in individuals frequently hospitalized and sharing rooms with patients actively infected with P. jirovecii.41
Exophiala dematitidis
The prevalence of E. dermatitidis varies significantly around the world. A German study reported prevalence rates of 4% in comparison to 17% in Sweden.24 The Swedish study showed that patients presenting with E. dermatitidis were more often colonized with non-tuberculous mycobacteria (NTM) and had lower than predicted FEV1.24 E. dermatitidis infections are more frequently reported in patients with pancreas failure and advanced disease.
Several of the studies described above aimed to address the question of whether colonization carries morbidity, leads to worsening pulmonary function, or in effect, infection. The high prevalence of fungal organisms in the CF population brings to forefront the importance of distinguishing between colonization and active infection.42 Given conflicting results of the overall pathogenicity of fungal infections, it is imperative to rely on diagnostic tools to aid in the diagnosis of infection and ultimately determine if treatment is necessary.
Diagnostic Criteria
Diagnosis of active pulmonary fungal infection is challenging because there are no guidelines nor consensus statements addressing diagnostic criteria, specifically in the CF population.4 Aspergillus spp. have been described in the literature in various states: colonization, ABPA, sensitization, bronchitis, and invasive aspergillosis.
Baxter et al performed extensive laboratory research to develop immunologic classifications of aspergillosis in adult patients with cystic fibrosis34,35 [Table 2].
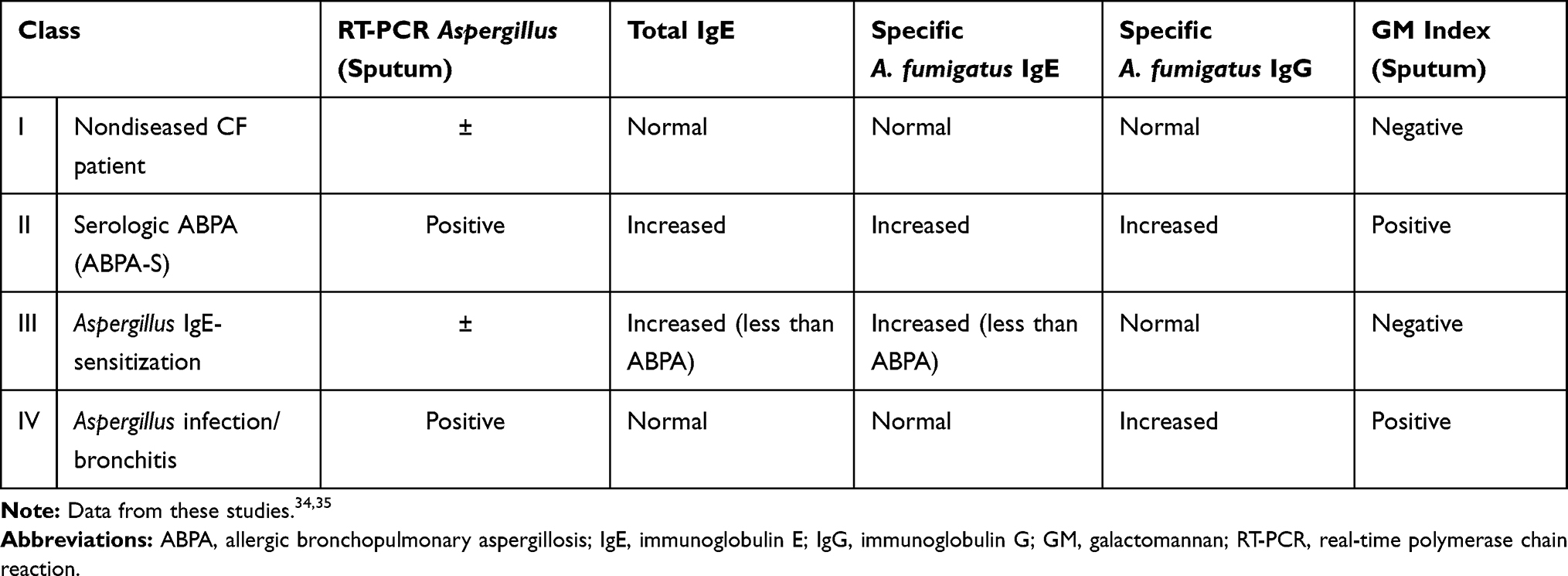 |
Table 2 Immunologic Classification of Aspergillosis in Adult Cystic Fibrosis |
ABPA
ABPA is caused by an allergic response to antigens of Aspergillus species. Pathophysiologically, ABPA is a result of Th2 CD4+ mediated response causing IgG and IgE-specific to Aspergillus spp. This can cause persistent inflammation which leads to bronchiectasis and fibrosis. Clinical features are characterized by wheezing, pulmonary infiltrates on imaging and recurrent exacerbations. The prevalence of ABPA in CF is reported to range from 3% to 25%.34
Both the CFF Consensus Conference and the International Society for Human and Animal Mycology (ISHAM) have published guidelines for the screening and diagnosis of ABPA in CF patients, as there is a higher incidence of ABPA in this patient population.43,44 The CFF Consensus Conference proposed the following diagnostic criteria for ABPA in CF:
- Acute or subacute clinical deterioration not attributable to another etiology.
- Serum total IgE concentration >1000 IU/mL.
- Immediate cutaneous reactivity to Aspergillus or in vitro presence of serum IgE antibody to A. fumigatus.
- Precipitating antibodies to A. fumigatus or serum IgG antibody to A. fumigatus by an in vitro test.
- New or recent abnormalities on chest radiography or chest CT (ie bronchiectasis) that do not resolve with therapy.43
Routine screening for ABPA in CF includes yearly evaluations of serum IgE level in children over the age of 6 years to help determine if further testing and treatment is required.
Active Fungal Infection
Diagnostic criteria have been proposed for “highly probable” invasive pulmonary fungal infection in an effort to distinguish from fungal colonization:
- Increased sputum production.
- Multiple isolation of the same fungal species from sputum or bronchoalveolar lavage (BAL) (at least two culture-positive samples in 6 months).
- Pulmonary infiltrate(s) on chest CT scan or X-ray.
- Treatment failure with antibiotic therapy (two and more antibiotic treatments, duration two or more weeks).
- Unexplained lung function decline (exclusion of new CF-related disease).
- Exclusion of new/other bacteria (eg non-tuberculous mycobacteria or P. aeruginosa).
- Exclusion of ABPA.8
Alternatively, authors from a recently published review article proposed diagnostic criteria for differentiating fungal colonization from active infection in CF lung disease following a thorough review of available literature4 [Table 3].
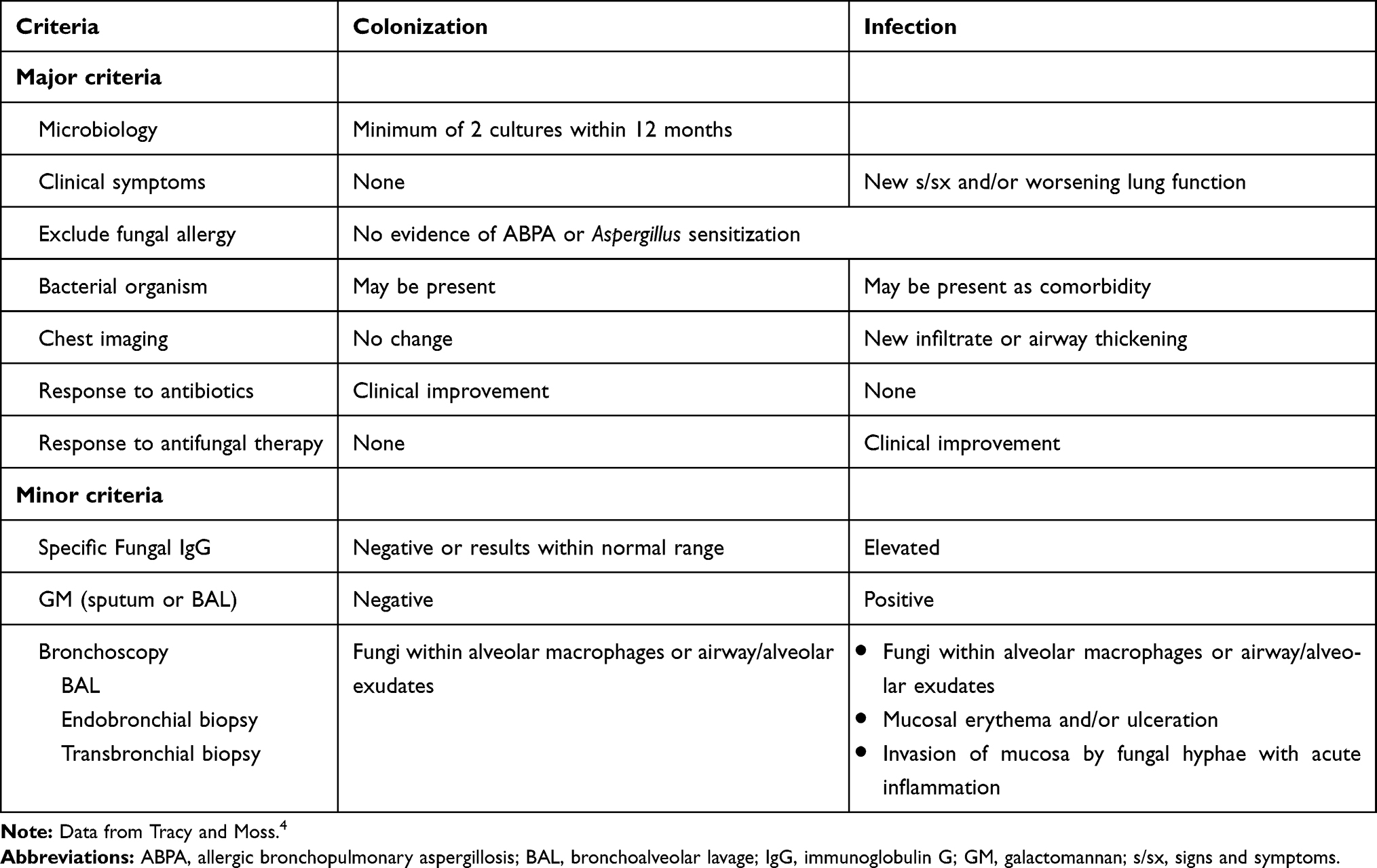 |
Table 3 Proposed Diagnostic Criteria for Fungal Colonization and Active Infection |
Diagnostic Methods
The diagnosis of fungal disease in patients with CF can be challenging. This was demonstrated when patients from the same CF cohort had specimens processed at two centers with different prevalence rates for fungal infections. This leads the authors to conclude that methodological differences in specimen analysis rather than geographic variation are likely the reason for the disparity.45 This was also demonstrated in another study, in which routine processing procedures for isolating filamentous fungi from respiratory sputum samples underestimated fungal prevalence in patients with COPD.46 Culture-based detection methods are the primary approach for isolating fungi. Unfortunately, processing guidelines are not standardized for the several variables that include differences in 1) sample collections, 2) sample processing, 3) and methods of detection.
Sample collection can significantly impact the detection of fungal species. First, the source of the specimen appears to affect yield, with BAL samples detecting more A fumigatus in 29% of specimens, compared to 14% of sputum specimens, and 0.8% of cough swabs in one pediatric study.47 Second, the actual volume of respiratory samples used for culture may be a possible confounder in comparing prevalence rates across centers, with the isolation of Aspergillus being more successful with a higher volume of specimen plated.48 Third, the frequency of sampling may be another confounder. In one study, in which a single sputum specimen collection was compared to a repeated longitudinal design, the recovery of fungal species was more than doubled in the frequent sampling.49
In addition, the methodology used to process the samples may contribute to further disparities in detecting fungal species in patients with CF. For example, due to the viscous quality of the sputum of patients with CF, chemical homogenization of specimens is recommended. This can increase the sensitivity of culture detection from approximately 72% to 96% in one study.50 The addition of sonication to the chemical homogenization increases sensitivity with using PCR testing.51
Finally, the choice of media can also influence the sensitivity of testing for fungal species. In one study, standard culture yielded a positive culture in only 18% of patients in one study, compared to the use of DNA extraction detected fungi in 100% of patients.52 Similar results were reported in a cohort study that compared selective fungal media to traditional bacterial culture for the identification of several fungal species. The authors found that of the 184 samples identified with one or more of these fungal species, bacterial culture detected fungal species in only 26% of these samples, with three other fungal selective media having much higher recovery rates, from 63% to 65.8%.53 In another study, mucolytically treated sputum from 243 patients with CF were plated on 6 different semi-selective media. The authors found that by taking into account individual performances of each medium, they were able to propose a standardized protocol to identify both Aspergillus and non-aspergillus fungal species.54 Another more recent group also compared the performance of different media for fungal isolation in CF patients to accurately assess the respiratory mycobiome in this patient population.55
Given these numerous challenges in correctly identifying fungal species in culture, molecular approaches, especially PCR performed on respiratory samples, are increasingly being applied in conjunction with traditional diagnostic methods. These include panfungal PCR assays, multiplex/pathogen-directed assays, real-time PCR, and probe-based assays. Real-time sputum PCR was shown to have a sensitivity of 74% compared to 46% galactomannan for detection of Aspergillus in patients with CF.35 In addition, molecular approaches may play a critical role in the correct identification of the evolving fungal microbiome of CF patient lungs.16 This is especially important in lung transplant recipients, in which invasive aspergillosis is a feared complication.56 While promising, these newer technologies have limited clinical applications at this time due to variations in assays. An excellent review of these newer modalities is described in the article by Chen et al.57 Additionally, there are emerging data identifying recombinant proteins specific to Scedosporium spp. that would allow for differentiation from Aspergillus spp.; identification and detection of recombinant proteins may lay the groundwork for the development of standardized serological testing.58
Management
Candida spp
As previously described, treatment of Candida spp. in sputum cultures is controversial given a lack of distinction between airway colonization and active infection. The 2016 Infectious Diseases Society of America clinical practice guidelines for the management of candidiasis states that, “growth of Candida from respiratory secretions usually indicates colonization and rarely requires treatment with antifungal therapy”.59 However, given a reported association between Candida colonization and decline in lung function,26 one may consider eradication of colonization in patients with worsening FEV1. Fluconazole is traditionally the drug of choice for C. albicans, C. parapsilosis, C. lusitaniae, and C. dubliniensis. Fluconazole should not be used to treat C. krusei due to intrinsic resistance and caution is advised when treating C. glabrata due to dose-dependent susceptibility.60 The pharmacokinetic profile for fluconazole can be found in Table 4.63,64,76 In the setting of fluconazole resistance, echinocandins (eg caspofungin, anidulafungin, micafungin) can be used to treat Candida spp. Echinocandins are only available in parenteral formulations due to poor gastrointestinal absorption and are generally well tolerated.60
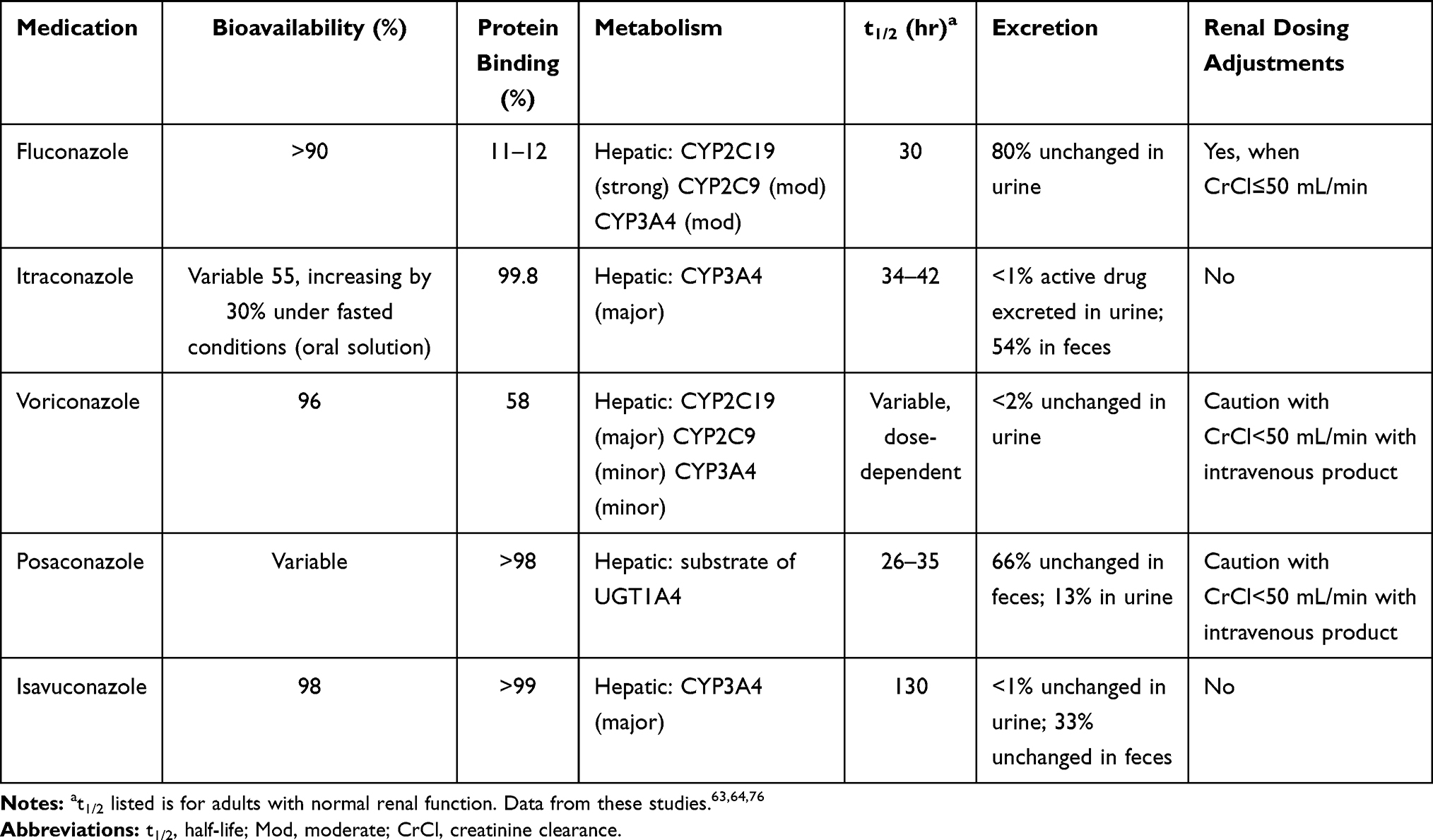 |
Table 4 Pharmacokinetics and Pharmacodynamic Properties of Triazole Antifungals |
ABPA
Because ABPA is caused by an allergic response to antigens of Aspergillus spp, the mainstay of treatment for ABPA focuses on the reduction of inflammation or immunological activity with corticosteroids.33,43 The optimal dosing schedule for oral steroids and duration of treatment for ABPA remains unclear. The most common initial dosing for prednisolone is 0.5 mg/kg/day for 2 weeks.61 Subsequent dosing and steroid taper are dependent upon symptoms, IgE levels, and radiologic fundings. Given the long-term side effects associated with systemic steroid use, dose tapering is recommended based on symptom control with the ultimate goal of corticosteroid discontinuation.
Treatment of ABPA also aims to reduce the antigen burden arising from fungal colonization of the bronchial tree by using antifungal agents. A systematic review of antifungal treatment in ABPA showed that antifungal agents, itraconazole and voriconazole most commonly demonstrated improvement in symptoms, frequency of exacerbations, and lung function. Additionally, antifungals showed a positive impact on biomarkers and radiological pulmonary infiltrates. Given the low quality of evidence and the adverse effects associated with triazoles, the use of antifungal agents remains controversial.62
Aspergillus spp
The mainstay of treatment for Aspergillus spp. is triazoles including voriconazole, posaconazole, and itraconazole63,64,76 [Table 4].
Itraconazole has been used for eradication therapy in a randomized placebo-controlled trial for CF patients chronically colonized with A. fumigatus. Thirty-five patients received oral itraconazole 5 mg/kg/day or placebo for 24 weeks. There were no differences in the rate of respiratory exacerbations requiring intravenous antibiotics nor change in FEV1. However, this study was limited by a small sample size and subtherapeutic itraconazole concentrations.65
Eradication therapy for Aspergillus colonization has become the mainstay of therapy in lung transplant recipients due to an increased risk of morbidity. In a single-center retrospective cohort study of cystic fibrosis-lung transplant recipients, 70% of patients had pretransplant Aspergillus colonization (n=93). Thirty-six patients had positive intraoperative Aspergillus cultures and these patients had a four-fold higher risk of developing invasive aspergillosis post-lung transplant (OR 4.36, 95% CI 1.35–14.05, p=0.01).66
Due to an increased risk of invasive aspergillosis in an immunosuppressed population, voriconazole has been used preemptively in CF lung transplant patients colonized with Aspergillus as well as inhaled amphotericin B.66,67 In an evaluation of 328 lung transplant recipients, preemptive antifungal treatment resulted in a lower rate of invasive pulmonary aspergillosis compared with patients who did not receive 3 months of preemptive antifungal treatment (0% vs 18%, OR=0.8, P=0.003).67
As previously discussed, there are no guidelines for the prevention or treatment of fungal infections in patients with cystic fibrosis. However, the Infectious Diseases Society of America published guidelines for the management of aspergillosis in 2016.68 The treatment recommendations in this guideline are not specific to CF patients but are often used as a reference in the treatment of aspergillosis in CF. The 2016 IDSA guideline supports the use of voriconazole and other triazoles as first-line treatment options. Although not discussed in detail in this publication, triazoles are also used to treat other fungal organisms known to colonize or infect CF patients: Lomentospora prolificans (formerly Scedosporium prolificans), Exophiala dermatitidis, and Trichosporon mycotoxinivorans.22,69
Pharmacology
Triazoles have significant side effect profiles including gastrointestinal upset, skin rash, hepatotoxicity, visual disturbances, and neurological toxicities. Most triazoles cause QTc prolongation; however, isavuconazole can cause QTc shortening.63 Liver function tests and therapeutic drug monitoring are required to monitor and minimize some of the above-mentioned side effects. Additionally, the absorption of the different dosage forms may be impacted by food and/or acid-suppressing medications [Table 5]63,64; thus, therapeutic drug monitoring may be favorable in settings where medication absorption is impacted63,64 [Table 6].
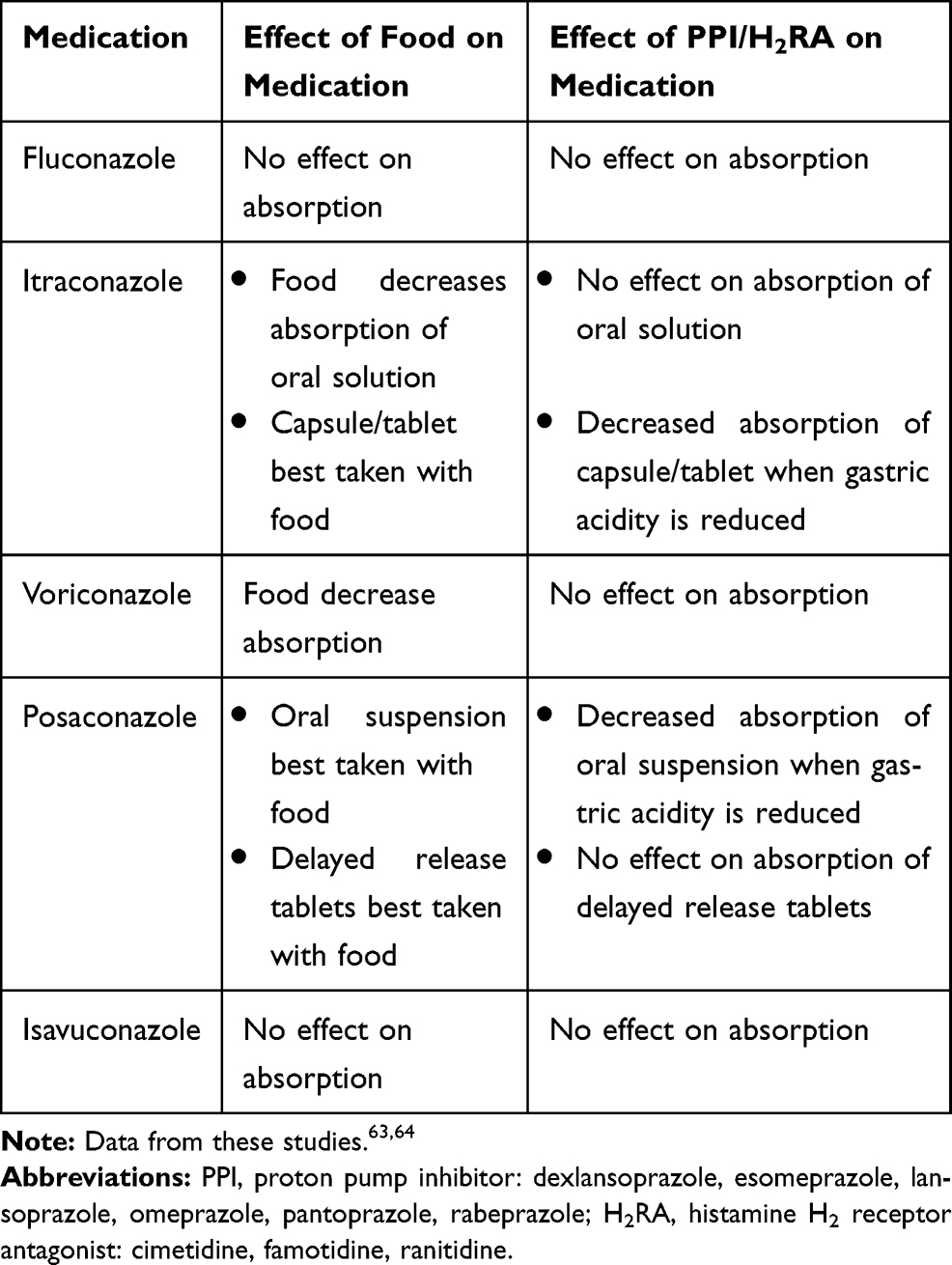 |
Table 5 Food and Acid-Suppressing Medication Interactions with Triazole Antifungals |
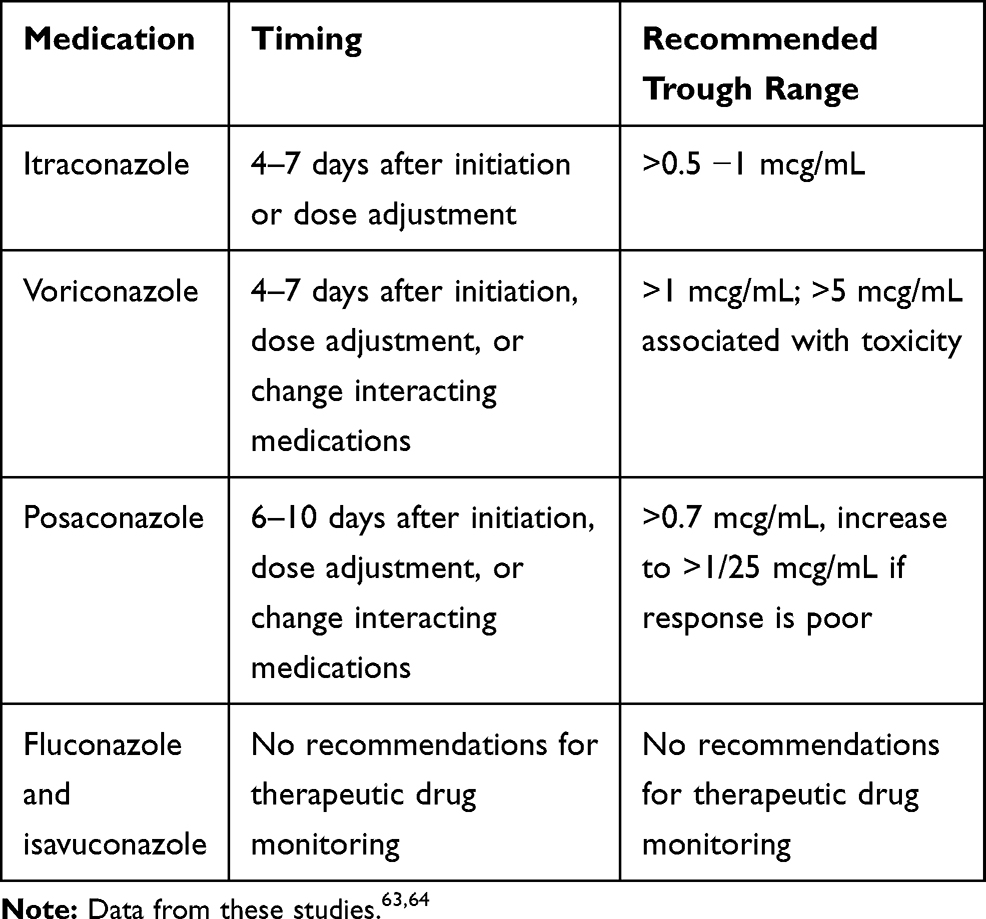 |
Table 6 Therapeutic Drug Monitoring of Triazole Antifungals |
Triazoles have significant drug–drug interactions with other medications via the cytochrome P450 pathway. One of the most notable drug–drug interactions exists between triazoles and Cystic Fibrosis Transmembrane Conductance Receptor (CFTR) modulators. With the introduction of CFTR modulator elexacaftor/tezacaftor/ivacaftor and ivacaftor in October 2019, eligibility for CFTR modulators expanded significantly to nearly 90% of the CF population given that patients need only one copy of the F508del coupled with any other mutation to qualify for treatment.70 Drug–drug interactions between triazoles and elexacaftor/tezacaftor/ivacaftor and ivacaftor require significant dose adjustments to the CFTR modulator63,71–73 [Table 7].
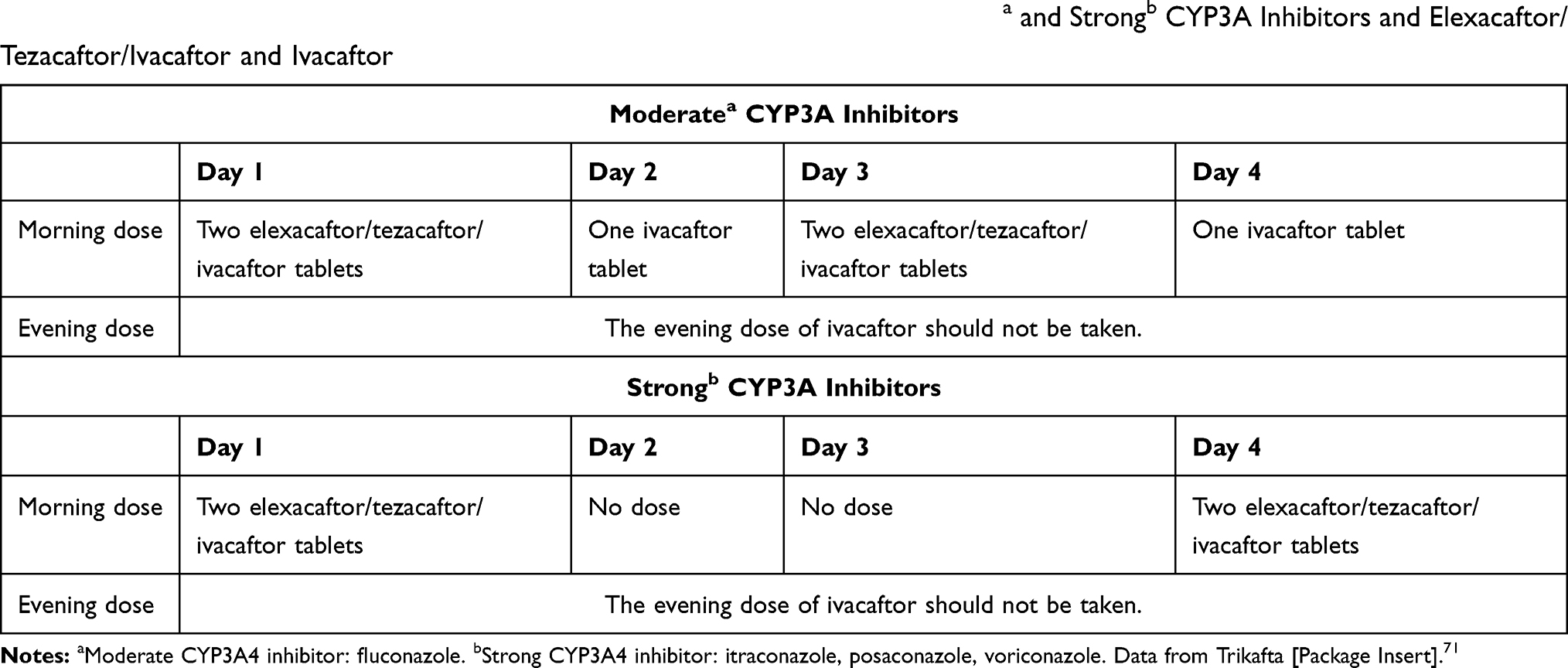 |
Table 7 Dose Adjustments Required for Drug–Drug Interactions Between Moderatea and Strongb CYP3A Inhibitors and Elexacaftor/Tezacaftor/Ivacaftor and Ivacaftor |
Due to growing azole-resistant aspergillosis, amphotericin B and echinocandins can also be considered for the management of aspergillosis.8,74,75 Unfortunately, efficacy of both agents, specifically in the CF population, is restricted to case reports.75 Amphotericin B is available in the United States as three different formulations: conventional, liposomal, and lipid complex. All formulations of amphotericin B have toxicities that limit their use: nephrotic, hepatic, hematologic, infusion-related, and electrolyte abnormalities. However, the lipid-based formulations are associated with significantly less nephrotoxicity.60,76 Inhaled amphotericin B provides targeted administration directly to the lungs thus minimizing systemic exposure and subsequent toxicity. Amphotericin B is not commercially available as an inhaled product. The intravenous solution must be administered via nebulization. Coordination with specialty pharmacies is advisable for medication and supply fulfillment, insurance authorization, and patient education.
Conclusion
CF airways have complex microbial environments predominantly occupied by bacteria. Through newer culture techniques and molecular methods, we now recognize that fungi are increasingly common in CF airways as filamentous organisms and yeast in both adults and children. Whether they play an active role in disease or are microbial bystanders remains unclear. This conundrum has forced clinicians to decide whether to continue with surveillance cultures or initiate antifungal therapy. Although CF management has historically focused on treating the usual suspects like Pseudomonas aeruginosa, Staphylococcus aureus or nontuberculosis mycobacterium, there is emerging evidence that filamentous fungi like Aspergillus and yeast may play an active role in the progression of the disease. Candida species are commensal fungi in the human microbiome as well as considered opportunistic pathogens particularly in the immunocompromized host, so it becomes unclear to clinicians how to interpret the presence of Candida in the sputum: contaminant or active pathogen. Similarly, Aspergillus is very common in soil and air and can become an opportunistic pathogen in the right host.
Future studies are needed to address several key areas such as standardization of diagnostic criteria for fungal organisms, determining whether bacterial and fungal co-infections have additive effects on the lungs, evaluating the immune system’s ability to manage fungal and bacterial coinfection, and determining whether bacteria and fungi alter each other’s pathophysiology.
Thus, it is imperative to establish laboratory and clinical diagnostic criteria to differentiate between benign and active pathogens. This distinction will aid in determining whether treatment of the potential fungal infection is warranted. Treatment with antifungal agents requires a careful understanding of pharmacology, drug–drug interactions, and potential side effect of therapies.
Funding
There is no funding to report.
Disclosure
Vaneeza Khan and Lavender Micalo work as research volunteers with Dr. Chaudary. Dr. Chaudary is a Professor of Medicine and Director of the Adult Cystic Fibrosis Center at Virginia Commonwealth University Medical Center. The authors have received no financial compensation for this review article. Dr. Chaudary was involved in the Continued Medical Education (CME) activity for Michael J. Hennessy (MJH) Events, reports clinical non-profit service grant from CFF, is part of the advisory board for PARI, provided CME talk for Abcomm inc, outside the submitted work. The authors have no other conflicts of interest with any companies or organizations whose products or services may be discussed in this article.
References
1. Davies JC, Alton E, Bush A. Cystic fibrosis. BMJ. 2007;335(7632):1255–1259. doi:10.1136/bmj.39391.713229.AD
2. Sanders DB, Bittner RCL, Rosenfeld M, Hoffman LR, Redding GJ, Goss CH. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am J Respir Crit Care Med. 2010;182(5):627–632. doi:10.1164/rccm.200909-1421OC
3. Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D. Cystic fibrosis adult care: consensus conference report. Chest. 2004;125(1 Suppl):1S–39S.
4. Tracy MC, Moss RB. The myriad challenges of respiratory fungal infection in cystic fibrosis. Pediatr Pulmonol. 2018;53(S3):S75–S85. doi:10.1002/ppul.24126
5. Kraemer R, Delosea N, Ballinari P, Gallati S, Creameri R. Effect of allergic bronchopulmonary aspergillosis on lung function in children with cystic fibrosis. Am J Respir Crit Care Med. 2006;174(11):1211–1220. doi:10.1164/rccm.200603-423OC
6. Harun SN, Wainwright CE, Grimwood K, Hennig S. Aspergillus and progression of lung disease in children with cystic fibrosis. Thorax. 2019;74(2):125–131. doi:10.1136/thoraxjnl-2018-211550
7. Amin R, Dupuis A, Aaron SD, Ratjen F. The effect of chronic infection with Aspergillus fumigatus on lung function and hospitalization in patients with cystic fibrosis. Chest. 2010;137(1):171–176. doi:10.1378/chest.09-1103
8. Schwarz C, Hartl D, Eickmeirir O, et al. Progress in definition, prevention and treatment of fungal infections in cystic fibrosis. Mycopathologia. 2018;183:21–32. doi:10.1007/s11046-017-0182-0
9. Hong G, Psoter KJ, Jennings MT, et al. Risk factors for persistent Aspergillus respiratory isolation in cystic fibrosis. J Cyst Fibros. 2018;17(5):624–630. doi:10.1016/j.jcf.2018.01.008
10. Mueller C, Braag SA, Keeler A, Hodges C, Drumm M, Flotte TR. Lack of cystic fibrosis transmembrane conductance regulator in CD3+ lymphocytes leads to aberrant cytokine secretion and hyperinflammatory adaptive immune responses. Am J Respir Cell Mol Biol. 2011;44(6):922–929. doi:10.1165/rcmb.2010-0224OC
11. Chaudhary N, Datta K, Askin FB, Staab JF, Marr KA. Cystic fibrosis transmembrane conductance regulator regulates epithelial cell response to Aspergillus and resultant pulmonary inflammation. Am J Respir Crit Care Med. 2012;185(3):301–310. doi:10.1164/rccm.201106-1027OC
12. Knutsen AP, Bellone C, Kauffman H. Immunopathogenesis of allergic bronchopulmonary aspergillosis in cystic fibrosis. J Cyst Fibros. 2002;l1(2):76–89. doi:10.1016/S1569-1993(02)00033-4
13. Burns JL, Emerson J, Stapp JR, et al. Microbiology of sputum from patients at cystic fibrosis centers in the United States. Clin Infect Dis. 1998;27(1):158–163. doi:10.1086/514631
14. Muthig M, Hebestreit A, Ziegler U, Seidler M, Müller FC. Persistence of Candida species in the respiratory tract of cystic fibrosis patients. Med Mycol. 2010;48(1):56–63. doi:10.3109/13693780802716532
15. Chen AI, Dolben EF, Okegbe C, et al. Candida albicans ethanol stimulates Pseudomonas aeruginosa WspR-controlled biofilm formation as part of a cyclic relationship involving phenazines. PLoS Pathog. 2014;10(10):e1004480. doi:10.1371/journal.ppat.1004480
16. Kim SH, Clark ST, Surendra A. Global analysis of the fungal microbiome in cystic fibrosis patients reveals loss of function of the transcriptional repressor Nrg1 as a mechanism of pathogen adaptation. PloS Pathog. 2015;11(11):e1005308. doi:10.1371/journal.ppat.1005308
17. Sudfeld CR, Dasenbrook EC, Merz WG, Carroll KC, Boyle MP. Prevalence and risk factors for recovery of filamentous fungi in individuals with cystic fibrosis. J Cyst Fibros. 2010;9(2):110–116. doi:10.1016/j.jcf.2009.11.010
18. Jubin V, Ranque S, Le Bel NS, Sarles J, Dubus J. Risk factors for Aspergillus colonization and allergic bronchopulmonary aspergillosis in children with cystic fibrosis. Pediatr Pulmonol. 2010;45(8):764–771. doi:10.1002/ppul.21240
19. Bargon J, Dauletbaev N, Köhler B, Wolf M, Posselt HG, Wagner TO. Prophylactic antibiotic therapy is associated with an increased prevalence of Aspergillus colonization in adult cystic fibrosis patients. Respir Med. 1999;93(11):835–838. doi:10.1016/S0954-6111(99)90270-6
20. Mowat E, Rajendran R, Williams C, et al. Pseudomonas aeruginosa and their small diffusible extracellular molecules inhibit Aspergillus fumigatus biofilm formation. FEMS Microbiol Lett. 2010;313(2):96–102. doi:10.1111/j.1574-6968.2010.02130.x
21. Lee TWR, Brownlee KG, Conway SP, Denton M, Littlewood JM. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J Cyst Fibros. 2003;2(1):29–34. doi:10.1016/S1569-1993(02)00141-8
22. Ziesing S, Suerbaum S, Sedlacek L. Fungal epidemiology and diversity in cystic fibrosis patients over a 5-year period in a national reference center. Med Mycol. 2016;54:781–786. doi:10.1093/mmy/myw035
23. Lackner M, de Hoog GS, Yang L, et al. Proposed nomenclature for Pseudallescheria, Scedosporium and related genera. Fungal Divers. 2014;67:1–10. doi:10.1007/s13225-014-0295-4
24. Williams C, Ranjendran R, Ramage G. Pathogenesis of fungal infections in cystic fibrosis. Curr Fungal Infect Rep. 2016;10(4):163–169. doi:10.1007/s12281-016-0268-z
25. Schwarz C, Bouchara JP, Buzina W, et al. Organization of patient management and fungal epidemiology in cystic fibrosis. Mycopathologia. 2018;183:7–19. doi:10.1007/s11046-017-0205-x
26. Chotirmall SH, O’Donoghue E, Bennett K, et al. Sputum Candida albicans presages FEV1 decline and hospital-treated exacerbations in cystic fibrosis. Chest. 2010;138:1186–1195. doi:10.1378/chest.09-2996
27. Gileles-Hillel A, Shoseyov D, Polacheck I, Korem M, Kerem E, Cohen-Cymberknoh M. Association of chronic Candida albicans respiratory infection with a more severe lung disease in patients with cystic fibrosis. Pediatr Pulmonol. 2015;50(11):1082–1089. doi:10.1002/ppul.23302
28. Al Shakirchi M, Klingspor L, Bergman P, Hjelte L, de Monestrol I. A 16-year retrospective study on fungal prevalence and diversity in patients with cystic fibrosis: candida dubliniensis was associated with a decline in lung function. Int J Infect Dis. 2020;96:663–670. doi:10.1016/j.ijid.2020.05.063
29. AbdulWahab A, Salah H, Chandra P, Taj‑Aldeen SJ. Persistence of Candida dubliniensis and lung function in patients with cystic fibrosis. BMC Res Notes. 2017;10:326. doi:10.1186/s13104-017-2656-z
30. Hector A, Kirn T, Ralhan A, et al. Microbial colonization and lung function in adolescents with cystic fibrosis. J Cystic Fibros. 2016;15(3):340–349. doi:10.1016/j.jcf.2016.01.004
31. Chotirmall SH, McElvaney NG. Fungi in the cystic fibrosis lung: bystanders or pathogens? Int J Biochem Cell Biol. 2014;52:161–173. doi:10.1016/j.biocel.2014.03.001
32. Latgé JP. Aspergillus fumigatus and aspergillosis. Clin Microbiol Rev. 1999;12(2):310–350.
33. King J, Brunel SF, Warris A. Aspergillus infections in cystic fibrosis. J Infect. 2016;72:S50–S55. doi:10.1016/j.jinf.2016.04.022
34. Delfino E, Del Puente F, Briano F, Sepulcri C, Roberto Giacobbe D. Respiratory fungal diseases in adult patients with cystic fibrosis. Clin Med Insights Circ Respir Pulm Med. 2019;13:1–6.
35. Baxter CG, Dunn G, Jones AM, et al. Novel immunologic classification of aspergillosis in adult cystic fibrosis. J Allergy Clin Immunol. 2013;132:560–566.e10. doi:10.1016/j.jaci.2013.04.007
36. Noni M, Katelari A, Dimopoulos G, Doudounakis S-E, Tzoumaka-Bakoula C, Spoulou V. Aspergillus fumigatus chronic colonization and lung function decline in cystic fibrosis may have a two-way relationship. Eur J Clin Microbiol Infect Dis. 2015;34(11):2235–2241. doi:10.1007/s10096-015-2474-y
37. Fillaux J, Bremont F, Murris M, et al. Assessment of Aspergillus sensitization or persistent carriage as a factor in lung function impairment in cystic fibrosis patients. Scand J Infect Dis. 2012;44(11):842–847. doi:10.3109/00365548.2012.695454
38. de Vrankrijker AM, van der Ent CK, van Berkhout FT, et al. Aspergillus fumigatus colonization in cystic fibrosis: implications for lung function? Clin Microbiol Infect. 2011;17(9):1381–1386. doi:10.1111/j.1469-0691.2010.03429.x
39. McMahon MA, Chotirmall SH, McCullagh B, Branagan P, McElvaney NG, Logan PM. Radiological abnormalities associated with Aspergillus colonization in a cystic fibrosis population. Eur J Radiol. 2012;81(3):e197–e202. doi:10.1016/j.ejrad.2011.01.114
40. Royce FH, Blumberg DA. Pneumocystis carinii isolated from lung lavage fluid in an infant with cystic fibrosis. Pediatr Pulmonol. 2007;335(7632):235–238. doi:10.1002/(SICI)1099-0496(200003)29:3<235::AID-PPUL12>3.0.CO;2-9
41. Calderón EJ, Friaza V, Dapena FJ, de La Horra C. Pneumocystis jirovecii and cystic fibrosis. Med Mycol. 2010;48(Suppl 1):S17–S21. doi:10.3109/13693786.2010.505205
42. Kondori N, Gilljam M, Lindblad A, Jönsson B, Moore ER, Wennerås C. High rate of exophiala dermatitidis recovery in the airways of patients with cystic fibrosis is associated with pancreatic insufficiency. J Clin Microbiol. 2011;49(3):1004–1009. doi:10.1128/JCM.01899-10
43. Stevens DA, Moss RB, Kurup VP, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis–state of the art: cystic Fibrosis foundation consensus conference. Clin Infect Dis. 2003;37(Suppl 3):S225–S264. doi:10.1086/376525
44. Agarwal R, Chakrabarti A, Shah A, et al. Allergic bronchopulmonary aspergillosis: review of literature and proposal of new diagnostic and classification criteria. Clin Exp Allergy. 2013;43(8):850–873.
45. Borman AM, Palmer MD, Delhaes L, et al. Lack of standardization in the procedures for mycological examination of sputum samples from CF patients: a possible cause for variations in the prevalence of filamentous fungi. Med Mycol. 2010;48:S88–S97. doi:10.3109/13693786.2010.511287
46. Pashley CH, Fairs A, Morley JP, et al. Routine processing procedures for isolating filamentous fungi from respiratory sputum samples may underestimate fungal prevalence. Med Mycol. 2012;50:433–438. doi:10.3109/13693786.2011.615762
47. Saunders R, Modha D, Claydon A, Gaillard E. 146 chronic Aspergillus fumigatus colonisation of the cystic fibrosis airway is common and may be associated with a more rapid decline in lung function. J Cyst Fibros. 2011;10:S37. doi:10.1016/S1569-1993(11)60162-8
48. Fraczek MG, Kirwan MB, Moore CB, Morris J, Denning DW, Richardson MD. Volume dependency for culture of fungi from respiratory secretions and increased sensitivity of Aspergillus quantitative PCR. Mycoses. 2014;57(2):69–78. doi:10.1111/myc.12103
49. Pihet M, Carrere J, Cimon B, et al. Occurrence and relevance of filamentous fungi in respiratory secretions of patients with cystic fibrosis – a review. Med Mycol. 2009;47(4):387–397. doi:10.1080/13693780802609604
50. Masoud-Landgraf L, Badura A, Eber E, Feierl G, Marth E, Buzina W. Modified culture method detects a high diversity of fungal species in cystic fibrosis patients. Med Mycol. 2014;52:179–186. doi:10.3109/13693786.2013.792438
51. Baxter CG, Jones AM, Webb K, Denning DW. Homogenisation of cystic fibrosis sputum by sonication-an essential step for Aspergillus PCR. J Microbiol Methods. 2011;85:75–81. doi:10.1016/j.mimet.2011.01.024
52. Nagano Y, Elborn JS, Millar BC, et al. Comparison of techniques to examine the diversity of fungi in adult patients with cystic fibrosis. Med Mycol. 2010;48(1):
53. Hong G, Miller HB, Allgood S, Lee R, Lechtzin N, Zhang S. Use of selective fungal culture media increases rates of detection of fungi in the respiratory tract of cystic fibrosis patients. J Clin Microbiol. 2017;55(4):1122–1130. doi:10.1128/JCM.02182-16
54. Coron N, Pihet M, Fréalle E, et al. Toward the standardization of mycological examination of sputum samples in cystic fibrosis: results from a French multicenter prospective study. Mycopathologia. 2018;183(1):101–117. doi:10.1007/s11046-017-0173-1
55. Delhaes L, Touati K, Faure-Cognet O, et al. Prevalence, geographic risk factor, and development of a standardized protocol for fungal isolation in cystic fibrosis: results from the international prospective study “MFIP”. J Cyst Fibros. 2019;18(2):212–220. doi:10.1016/j.jcf.2018.10.001
56. Nosotti M, Tarsia P, Corinna Morlacchi L. Infections after lung transplantation. J Thorac Dis. 2018;10(6):3849–3868. doi:10.21037/jtd.2018.05.204
57. Chen SC-A, Meyer W, Pashley CH. Challenges in laboratory detection of fungal pathogens in the airways of cystic fibrosis patients. Mycopathologia. 2018;183(1):89–100. doi:10.1007/s11046-017-0150-8
58. Mina S, Staerck C, Marot A, et al. Scedosporium boydii CatA1 and SODC recombinant proteins, new tools for serodiagnosis of Scedosporium infection of patients with cystic fibrosis. Diagn Microbiol Infect Dis. 2017;89:282–287. doi:10.1016/j.diagmicrobio.2017.08.013
59. Pappas PG, Kauffman CA, Andes DR. Clinical practice guideline for the management of candidiasis: 2016 update by the infectious diseases society of America. Clin Infect Dis. 2016;62(4):e1–e50.
60. Nett JE, Andes DR. Antifungal agents: spectrum of activity, pharmacology, and clinical indications. Infect Dis Clin North Am. 2016;30(1):51–83.
61. Shah A, Panjabi C. Allergic bronchopulmonary aspergillosis: a perplexing clinical entity. Allergy Asthma Immunol Res. 2016;8(4):282–297. doi:10.4168/aair.2016.8.4.282
62. Moreira AS, Silva D, Ferreira AR, Delgado L. Antifungal treatment in allergic bronchopulmonary aspergillosis with and without cystic fibrosis: a systematic review. Clin Exp Allergy. 2014;44(10):1210–1227. doi:10.1111/cea.12333
63. Rivosecchi RM, Samanta P, Demehin M, et al. Pharmacokinetics of azole antifungals in cystic fibrosis. Mycopathologia. 2018;183(1):139–150. doi:10.1007/s11046-017-0189-6
64. Fluconazole, Itraconazole, Voriconazole, Posaconazole, and Isavuconazole. Lexi-drugs. Lexicomp. Wolters Kluwer Health, Inc. Riverwoods, IL. Available from: http://online.lexi.com.
65. Aaron SD, Vandemheen KL, Freitag A, et al. Treatment of Aspergillus fumigatus in patients with cystic fibrosis: a randomized, placebo-controlled pilot study. PLoS One. 2012;7(4):e36077. doi:10.1371/journal.pone.0036077
66. Luong ML, Chaparro C, Stephenson A, et al. Pretransplant Aspergillus colonization of cystic fibrosis patients and the incidence of post-lung transplant invasive aspergillosis. Transplantation. 2014;97:351–357. doi:10.1097/01.TP.0000437434.42851.d4
67. Hosseini-Moghaddam SM, Chaparro C, Luong ML, et al. The effectiveness of culture-directed preemptive anti-Aspergillus treatment in lung transplant recipients at one year after transplant. Transplantation. 2015;99:2387–2393. doi:10.1097/TP.0000000000000743
68. Patterson TF, Thompson GR, Denning DW, et al. Practice guidelines for the diagnosis and management of Aspergillosis: 2016 update by the infectious diseases society of America. Clin Infect Dis. 2016;63(4):e1–e60.
69. Schwarz C, Vandeputte P, Rougeron A, et al. Developing collaborative works for faster progress on fungal respiratory infections in cystic fibrosis. Med Mycol. 2009;47(4):42–59. doi:10.1093/mmy/myx106
70. Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, Phase 3 trial. Lancet. 2019;394:1940–1948.
71. Trikafta [Package Insert]. Boston, MA: Vertex Pharmaceutical Inc.; 2019.
72. Symdeko [Package Insert]. Boston, MA: Vertex Pharmaceutical Inc.; 2018.
73. Orkambi [Package Insert]. Boston, MA: Vertex Pharmaceutical Inc.; 2015.
74. Hamprecht A, Morio F, Bader O, Le Pape P, Steinmann J, Dannaoui E. Azole resistance in Aspergillus fumigatus in patients with cystic fibrosis: a matter of concern? Mycopathologia. 2018;183(1):151–160. doi:10.1007/s11046-017-0162-4
75. Schwarz C, Brandt C, Whitaker P, et al. Invasive pulmonary fungal infections in cystic fibrosis. Mycopathologia. 2018;183(1):33–43. doi:10.1007/s11046-017-0199-4
76. Dodds Ashley ES, Lewis R, Lewis JS, Martin C, Andes D. Pharmacology of systemic antifungal agents. Clin Infect Dis. 2006;43:S28–S39. doi:10.1086/504492
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.