")
Back to Journals » OncoTargets and Therapy » Volume 13
Management of Ewing Sarcoma Family of Tumors: A Short Description of a Rare Primitive Uterine pPNET and Literature Review
Authors Pisconti S, Della Vittoria Scarpati G, Buonerba C , Messinese S, Carella R, Di Marzo M, Di Lorenzo G, Lazzari G , Perri F , Solla R
Received 23 April 2019
Accepted for publication 12 December 2019
Published 14 February 2020 Volume 2020:13 Pages 1179—1184
DOI https://doi.org/10.2147/OTT.S213233
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Takuya Aoki
Salvatore Pisconti,1 Giuseppina Della Vittoria Scarpati,2 Carlo Buonerba,3 Simona Messinese,1 Roberta Carella,1 Massimiliano Di Marzo,4 Giuseppe Di Lorenzo,3 Grazia Lazzari,5 Francesco Perri,6 Raffaele Solla7,8
1Medical Oncology Unit, POC, SS Annunziata, Taranto, Italy; 2Medical Oncology Unit, Pollena Trocchia Hospital, ASL NA3sud, Naples, Italy; 3Department of Clinical Medicine and Surgery, University Federico II of Naples, Naples, Italy; 4Department of Abdominal Surgery, National Tumor Institute of Naples IRCCS G. Pascale, Naples, Italy; 5Department of Radiation Oncology, POC, SS Annunziata, Taranto, Italy; 6Head and Neck Medical Oncology Unit, National Tumor Institute of Naples IRCCS G. Pascale, Naples, Italy; 7Department of Radiation Oncology, University of Naples Federico II, Naples, Italy; 8Italian National Research Council, Institute of Biostructure & Bioimaging, Naples, Italy
Correspondence: Francesco Perri
Head and Neck Medical Oncology Unit, National Tumor Institute of Naples IRCCS G. Pascale, Via Mariano Semmola, Naples 80131, Italy
Tel +390815903362
Email [email protected]
Purpose: To describe the outcome of a patient with a rare primitive uterine pPNET and to perform a review of the available data in literature, leading the clinicians to better face this rare disease.
Methods: We have rescued data regarding the multidisciplinary treatment of pPNET from the PUBMED database, highlighting also issues regarding the pathogenesis and the genetic landscape of the ESFTs (Ewing Sarcoma Family of Tumors).
Results: Ewing sarcoma and primitive neuroectodermal tumors (PNETs) are small round cell tumors presenting with different degrees of neuroectodermal differentiation. PNETs are further divided into central PNET and peripheral PNET (pPNET). Since pPNETs share the same genetic background of Ewing Sarcomas, they are considered to belong to the Ewing Sarcoma Family of Tumors (ESFTs). Multimodality treatment currently represents the best choice to offer to the affected patients.
Conclusion: Although pPNETs are generally diagnosed in children and young adults, an elderly woman aged 85 years came to our attention after a diagnosis of uterine pPNET. Her medical history is presented here, along with a literature review of the subject, highlighting the main biological, pathological and clinical features, with a hypothesis about the possible future therapeutic approaches for these rare malignancies.
Keywords: PNET, pPNET, Ewing Sarcoma, genetics
Background
Ewing sarcoma and primitive neuroectodermal tumors (PNETs) are small round cell tumors originating from fetal neuroectodermal cells and showing various degrees of neuroectodermal differentiation. They are commonly diagnosed during childhood and originate in the bone, the nervous system and in soft tissues.1,2
From a pathological and genetic perspective, PNETs appear closely related to Ewing sarcoma (ES), and for a long time, it was believed that they originated from mesenchymal stem cells. Lately, recent studies on electron microscopy and immunohistochemical techniques have instead found a neuroepithelial origin of Ewing Sarcoma cells so that it is currently considered as a member of the PNETs.3 PNETs show improved neural differentiation if compared to Ewing Sarcoma,3 and include central and peripheral tumors. Peripheral PNETs (pPNETs) occur mostly in the soft tissue of the thoracopulmonary region, of the pelvis, and of lower extremities. Both ES and pPNET show the same chromosomal translocations such as t(11;22)(q24;q12) and were included in the same category of ES family of tumors (ESFTs) by the World Health Organization Classification in 2002. ESFTs include Askin’s tumor also (Table 1), but do not include central PNETs, which have a different genetic background.6 While ES is mostly diagnosed in the bone, pPNETs are commonly reported in soft tissues.4,5
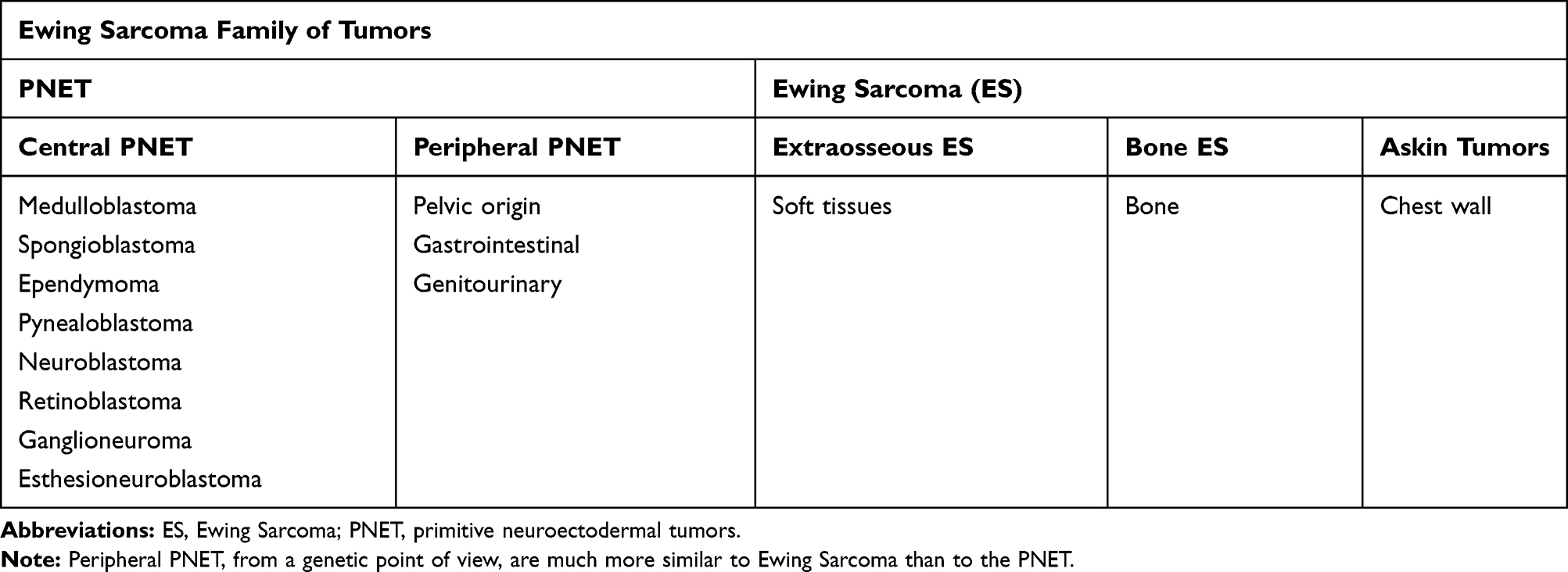 |
Table 1 Classification of Ewing Sarcoma Family of Tumors |
Case Report
An 84-year-old woman came to our attention on August 1, 2017 due to vaginal bleeding.
A large, partially necrotic lesion measuring about 10 cm was reported in the uterine fundus on the whole-body CT (computed tomography) scan with and without contrast, along with multiple enlarged pelvic lymph-nodes (largest short axis, 2 cm). No distant metastases in lung, liver or bone were reported.
On August 22, 2017, the patient underwent total hysterectomy with bilateral salpingo-oophorectomy, pelvic lymphadenectomy and omentectomy.
Histological analysis showed a peripheral primitive neuroectodermal tumor (pPNET) arising from the fundus of uterus and infiltrating the perimetrium and parametrium through the uterine wall (Figure 1). Peritoneal carcinomatosis, but not lymph node metastasis, was also reported.
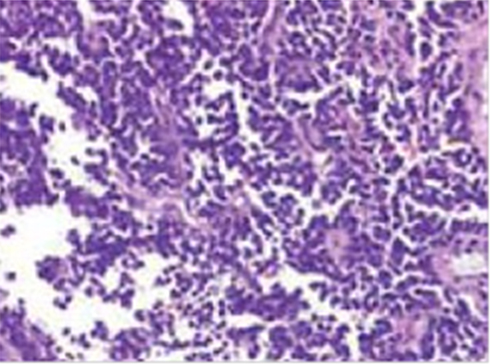 |
Figure 1 Primitive uterine PNET. |
Immunohistochemical analysis showed a strong positivity for CD99, vimentin, CD56, Synaptophysin, WT-1.
Biomolecular analyses revealed the pathognomonic translocation of PNET (11;22) (q24;q12) consisting of the fusion gene of the EWS and FLI1 genes (Figure 2).
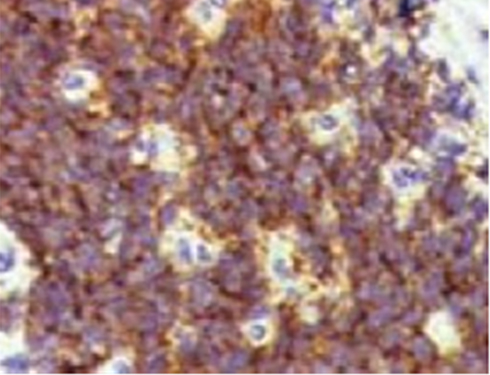 |
Figure 2 IHC for CD99, strongly positive. |
Due to the advanced stage of the disease, ECOG (eastern cooperative oncology group) PS (performance status) of 2 and the advanced age, we decided to administer weekly paclitaxel at the dose of 85 mg/m2 for 3 weeks, followed by a stop of 1 week.
After the first cycle was finished, with no noteworthy toxicity, the patient refused to continue chemotherapy and received best supportive care only. She died on November 12, 2017, 4 months after the first diagnosis, as a result of peritoneal carcinomatosis.
Discussion
Ewing Sarcoma and PNETs are similar from an histological, genetic and a clinical point of view, since they are characterized by small, round blue cells, and importantly by a bad clinical behavior and a poor life expectancy, due to their metastatic potential. Since central PNETs are characterized by different genetic abnormalities, only Ewing sarcoma and pPNET are included in the same family of tumors, named ESFTs.7,8,9
ESFTs are composed of small cells with a high nuclear to cytoplasmic ratio. ESFTs cells show expression of CD99 or MIC2 on immunohistochemistry. Antibody against FLI1 has been shown to be specific for ESFTs. Depending on the degree of neuroectodermal differentiation, they may also express neuron-specific enolase (NSE), synaptophysin, and S-100 protein.10,11
While central PNETs are characterized by mutations of RASSF1A, NOTCH1 and, especially in neuroblastoma, c-MYC amplifications, ESFTs display different genetic changes, which render these lasts histologically and clinically very different from the firsts.12
Ewing sarcoma/PNETs demonstrate, in fact, the characteristic translocation (11;22) (q24;q12) in more than 90% of the cases. Most commonly, this involves a rearrangement of the EWS and FLI1 genes (85%) or EWS and ERG genes (10%). The presence of a (11;22) (q24;q12) translocation is therefore the strongest diagnostic feature able to identify Ewing sarcoma/PNET.
The fusion of EWS gene on 22q12 with the FLI1 gene on 11q24 results in a chimeric fusion transcript EWS-FLI1.12,13
The EWS gene is a part of TET family of genes, which encodes for proteins able to bind RNA and to participate in transcription and RNA processing.
FLI1 gene is expressed in hematopoietic, endothelial cells and also in the mesenchymal cells of neural crest origin during embryonal development; it is well acknowledged to regulate the hematopoiesis and vasculogenesis.14
Moreover, overexpression of FLI1 is observed to promote self-renewal, repress Rb (retinoblastoma) protein, and induce BCL2 expression in erythroid cells with a corresponding enhancement of cell survival.15,16
The expression of EWS-FLI1 in murine NIH-3T3 cells may result in anchorage-independent growth and accelerated tumorigenesis with a tumor phenotype reminiscent of those of the human Ewing sarcoma.17 Based on these characteristics, EWS-FLI1 is able to stimulate oncogenesis and it is responsible for the histological characteristics associated with ESFsT.18,19
Due to the low incidence of ESFTs, epidemiology data are scarce. Surgery represents the mainstay of treatment of ESFTs and it should be carried out whenever possible. Adjuvant chemo and radiotherapy are commonly employed in the adjuvant setting.1
The treatment approach of ESFTs includes the initial cytoreduction, obtained with the neoadjuvant chemotherapy, with the purpose to eradicate micrometastatic disease and to facilitate the effective local control, increasing the probability to obtain pathologic negative margins after the following surgery; definitive radiation or surgical therapy to eradicate all visible disease; and adjuvant treatment based on chemotherapy or radiotherapy, if not previously given. Importantly, neoadjuvant chemotherapy not only may improve the outcome after surgery but it also provides information about the disease chemo-sensitivity.20
During the past 30 years, chemotherapy has increased survival from less than 5% to 65–70% in localized tumors and to 25–30% in primary metastatic tumors, although there is still a lack of consensus on the optimal chemotherapy timing and the regimen to be used, due to the rarity of the disease.21
The most employed chemotherapy regimen lasts about 6–9 months and consists of alternating courses of two chemotherapeutic regimens: (1) vincristine, doxorubicin, and cyclophosphamide and (2) ifosfamide and etoposide. In the presence of disease progression, there are no standardized second-line treatment plans. Considerations need to be made depending on sites of disease recurrence and prior therapy. Chemotherapy combinations such as vincristine/irinotecan/temozolomide seem to be valid options.22,23
ESFT sarcomas have a potential for hematogenous metastasis and the most common sites of metastases include lungs, bones and bone marrow. The chemotherapy regimen and initial treatment for patients with metastatic disease are the same as those for localized disease.
In conclusion, in the case reported here of an elderly female patient with advanced pPNET the main peculiarities lie in the advanced age of presentation and in the anatomic site of origin.
Future Perspectives
As ESFT is a poorly differentiated tumor with both mesenchymal and neuroectodermal histological features, it is unclear if this tumor belongs to mesenchymal or neuroectodermal malignancies, but the latest data seem to confirm its neuroectodermal origin.
With current multimodal treatment options, including surgery, chemotherapy and radiotherapy, the 5-year survival for localized disease is 60%. Nevertheless patients presenting with metastatic disease, have a 5-year survival of 20%.2 The outcome for patients with relapsed or refractory is much more poor, reaching in the best case studies the 10%. Therefore, new treatment strategies are strongly needed.
A pathognomonic genetic feature of ES is a chromosomal translocation generating a fusion protein (WS-FLI1) able to promote cell cycle, angiogenesis and metastasization. The EWS-FLI1 fusion is present only in ESFT cells and does not exist in any normal cell of the body. Thus, ESFTs contain a unique protein generated by tumor-specific translocation with a potential for molecular target.
Unfortunately, no drugs able to target the above-mentioned molecule are currently available in clinical experimentations, due to the poor solubility of the EWS-FLI1 protein, and the consequent difficulty to directly analyze it.24
Currently, the insulin-like growth factor 1 receptor (IGF-1R) pathway and the poly-ADP-ribose-polymerase (PARP) pathway are being investigated for potential targeted therapies.
IGF-1R is a member of the tyrosine kinase receptors linked to an intracellular pathway able to promote cell survival and growth. One of the main intracellular pathways stimulated by IFG-1R dimerization is the MAPK (mitogen-activated kinase) pathway. Phosphorylation of IGFR subunits, after their dimerization, induces, through the downstream signaling proteins IR substrate (IRS), the activation of the phosphoinositide 3-kinase (PI3K) and MAPK pathway, thus resulting in the stimulation of cellular proliferation, cell motility and inhibition of apoptosis.25
Interestingly, the peak incidence of primary bone ES correlates with the increased levels of the IGF ligands in puberty. Starting from 1990, different preclinical trials demonstrated that IGF1 is expressed in ES carrying a t(11;22) translocation and that blocking the IGF-1 loop inhibits cell growth.26,27 Scotlandi et al, using “in vitro” cell models, demonstrated that wild-type cells of ES showing the fusion protein (EWS-FLI1) had a greater degree of ligand-stimulated IRS phosphorylation, thus giving evidence that altered IGF-1R signaling by expression of the EWSR1-FLI1 fusion protein is required to transform the cell lines.28
These data are in favor of a “cross talk” between the two pathways, namely those IGF-1R and those EWS-FLI1 stimulated.
The conclusion is that IGF-1R induction could arrest the EWS-FLI1-induced cell growth.
As IGF-1 is associated with ESFT growth, monoclonal antibodies against this potential target have been evaluated in six different Phase II trials conducted upon a total of 291 patients affected by ES. As a result, in these trials, two (1%) complete responses (CR), 32 (11%) partial responses (PR) of which some durable, and 61 (21%) stable diseases (SD) were observed.29–31
Recently, Annemiek et al tried to interpret the above-mentioned data and concluded that the IGF-1R pathway is an interesting target for ES and should be further explored.32
Conclusions
ES family of tumors (ESFTs) are malignancies that show variable degrees of neural differentiation but also common cytogenetic and molecular features. They comprise ES and pPNET and generally they are diagnosed in the second decade of life, while 20–30% are in the first decade. Occurrences are rare in individuals over 30 years.
Common therapeutic strategies include surgery, chemo and radiotherapy and, generally, a multidisciplinary approach is strongly needed.
Given some unique genetic features, such as the chromosomal translocation (11;22) which leads to the aberrant production of a chimeric protein (EWS-FLI1), lately, some translational study aimed to perform a well-shaped therapy on the basis of the genetics of disease has been carried out.
IGF-1R block may be an interesting future option for patients with tumors bearing chromosomal translocation (11;22). Nevertheless, while there was great enthusiasm for this class of therapy several years ago, different cooperative group trials have not found consistent benefits or any predictive biomarkers that can help with effective use of this agent in larger groups of patients.33
Being disease interesting the young adult and the pediatric age, only few data regarding therapy in the elderly are available, and presently,34 the chemotherapy adapted to the age and ECOG PS of the patients represents an important option. This has been the only reason for which the authors have decided to choose a so little toxic and so manageable therapy in the patient under examination.
Compliance with Ethical Standards
Ethical approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Ethical approval: This article does not contain any studies with animals performed by any of the authors.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Acknowledgments
We would like to thank Dr Monica Pontone and Dr Marialuisa Marciano for helping us in the realization of this work.
Disclosure
All authors declared no conflicts of interest in this work.
References
1. Ewing Sarcoma Treatment (PDQ®): Patient Version. PDQ Pediatric Treatment Editorial Board.Source PDQ Cancer Information Summaries. Bethesda (MD): National Cancer Institute(US); 2002–2017 October 26.
2. Nicholas G, Verma S, Asmis T. Systemic therapy outcomes in adult patients with Ewing sarcoma family of tumors. Case Rep Oncol. 2017;10(2):462–472. doi:10.1159/000475806
3. Biswas B, Rastogi S, Khan SA, et al. Outcomes and prognostic factors for Ewing-family tumors of the extremities. J Bone Joint Surg Am. 2014;96(10):841–849. doi:10.2106/JBJS.M.00411
4. Peng RJ, Sun XF, Xiang XJ, et al. Efficacy and survival of 92 cases of Ewing’s sarcoma family of tumor initially treated with multidisciplinary therapy. Ai Zheng. 2009;28(12):1304–1309. doi:10.5732/cjc.008.10609
5. Lahl M, Fisher VL, Laschinger K. Ewing’s sarcoma family of tumors: an overview from diagnosis to survivorship. Clin J Oncol Nurs. 2008;12(1):89–97. doi:10.1188/08.CJON.89-97
6. Maheshwari AV, Cheng EY. Ewing sarcoma family of tumors. J Am Acad Orthop Surg. 2010;18(2):94–107. doi:10.5435/00124635-201002000-00004
7. Kim SK, Park YK. Ewing sarcoma: a chronicle of molecular pathogenesis. Hum Pathol. 2016;55:91–100. doi:10.1016/j.humpath.2016.05.008
8. Renard C, Ranchère-Vince D. Ewing/PNET sarcoma family of tumors: towards a new paradigm? Ann Pathol. 2015;35(1):86–97. doi:10.1016/j.annpat.2014.11.001
9. Machado I, Navarro L, Pellin A, et al. Defining Ewing and Ewing-like small round cell tumors (SRCT): the need for molecular techniques in their categorization and differential diagnosis. A study of 200 cases. Ann Diagn Pathol. 2016;22:25–32. doi:10.1016/j.anndiagpath.2016.03.002
10. Antonescu C. Round cell sarcomas beyond Ewing: emerging entities. Histopathology. 2014;64(1):26–37. doi:10.1111/his.12281
11. Mariño-Enríquez A, Fletcher CD. Round cell sarcomas - biologically important refinements in subclassification. Int J Biochem Cell Biol. 2014;53:493–504. doi:10.1016/j.biocel.2014.04.022
12. Dagher R, Pham TA, Sorbara L, et al. Molecular confirmation of Ewing sarcoma. J Pediatr Hematol Oncol. 2001;23(4):221–224. doi:10.1097/00043426-200105000-00009
13. Meier VS, Kühne T, Jundt G, Gudat F. Molecular diagnosis of Ewing tumors: improved detection of EWS-FLI-1 and EWS-ERG chimeric transcripts and rapid determination of exon combinations. Diagn Mol Pathol. 1998;7(1):29–35. doi:10.1097/00019606-199802000-00006
14. May WA, Denny CT. Biology of EWS/FLI and related fusion genes in Ewing’s sarcoma and primitive neuroectodermal tumor. Curr Top Microbiol Immunol. 1997;220:143–150.
15. Desmaze C, Brizard F, Turc-Carel C, et al. Multiple chromosomal mechanisms generate an EWS/FLI1 or an EWS/ERG fusion gene in Ewing tumors. Cancer Genet Cytogenet. 1997;97(1):12–19. doi:10.1016/S0165-4608(96)00326-3
16. Kim J, Pelletier J. Molecular genetics of chromosome translocations involving EWS and related family members. Physiol Genomics. 1999;1(3):127–138. doi:10.1152/physiolgenomics.1999.1.3.127
17. May WA, Lessnick SL, Braun BS, et al. The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol. 1993;13(12):7393–7398. doi:10.1128/MCB.13.12.7393
18. Arvand A, Denny CT. Biology of EWS/ETS fusions in Ewing’s family tumors. Oncogene. 2001;20(40):5747–5754. doi:10.1038/sj.onc.1204598
19. Ordóñez JL, Osuna D, Herrero D, de Alava E, Madoz-Gúrpide J. Advances in Ewing’s sarcoma research: where are we now and what lies ahead? Cancer Res. 2009;69(18):7140–7150. doi:10.1158/0008-5472.CAN-08-4041
20. Owens C, Abbott LS, Gupta AA. Optimal management of Ewing sarcoma family of tumors: recent developments in systemic therapy. Paediatr Drugs. 2013;15(6):473–492. doi:10.1007/s40272-013-0037-1
21. Seddon BM, Whelan JS. Emerging chemotherapeutic strategies and the role of treatment stratification in Ewing sarcoma. Paediatr Drugs. 2008;10(2):93–105. doi:10.2165/00148581-200810020-00004
22. Goto T, Kosaku H, Kobayashi H, Hozumi T, Kondo T. Soft tissue sarcoma: postoperative chemotherapy. Gan to Kagaku Ryoho. 2004;31(9):1324–1330.
23. Krikelis D, Judson I. Role of chemotherapy in the management of soft tissue sarcomas. Expert Rev Anticancer Ther. 2010;10(2):249–260. doi:10.1586/era.09.176
24. Subbiah V, Anderson P, Lazar AJ, Burdett E, Raymond K, Ludwig JA. Ewing’s sarcoma: standard and experimental treatment options. Curr Treat Options Oncol. 2009;10(1–2):126–140. doi:10.1007/s11864-009-0104-6
25. LeRoith D, Roberts CT. The insulin-like growth factor system and cancer. Cancer Lett. 2003;195(2):127e37. doi:10.1016/S0304-3835(03)00159-9
26. Yu H, Jin F, Shu XO, et al. Insulin-like growth factors and breast cancer risk in Chinese women. Cancer Epidemiol Biomarkers Prev. 2002;11(8):705e12.
27. Yee D, Favoni RE, Lebovic GS, et al. Insulin-like growth factor I expression by tumors of neuroectodermal origin with the t(11;22) chromosomal translocation. A potential autocrine growth factor. J Clin Invest. 1990;86(6):1806e14. doi:10.1172/JCI114910
28. Scotlandi K, Benini S, Sarti M, et al. Insulin-like growth factor I receptor-mediated circuit in Ewing’s sarcoma/peripheral neuroectodermal tumor: a possible therapeutic target. Cancer Res. 1996;56(20):4570e4.
29. Theisen ER, Pishas KI, Saund RS, Lessnick SL. Therapeutic opportunities in Ewing sarcoma: EWS-FLI inhibition via LSD1 targeting. Oncotarget. 2016;7(14):17616–17630. doi:10.18632/oncotarget.7124
30. van Maldegem AM, Bovée JV, Peterse EF, Hogendoorn PC, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171–180. doi:10.1016/j.ejca.2015.09.009
31. Pappo AS, Vassal G, Crowley JJ, et al. A phase 2trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer. 2014;120(16):2448e56.
32. Annemiek M, van Maldegem A, Judith VMG, et al. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171e180.
33. Bailey K, Cost C, Davis I, et al. Emerging novel agents for patients with advanced Ewing sarcoma: a report from the Children’s Oncology Group (COG) new agents for Ewing Sarcoma task force. F1000Res. 2019;8:F1000 Faculty Rev–493. doi:10.12688/f1000research
34. Kayhan B, Ozer D, Ozaslan E, Ademoğlu E, Akgül A. Ewing sarcoma in a geriatric patient. Aging Clin Exp Res. 2005;17(4):347–349. doi:10.1007/BF03324621
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.