")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Management of epilepsy in patients with Rett syndrome: perspectives and considerations
Authors Krajnc N
Received 19 January 2015
Accepted for publication 21 March 2015
Published 10 June 2015 Volume 2015:11 Pages 925—932
DOI https://doi.org/10.2147/TCRM.S55896
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Natalija Krajnc
Department of Child, Adolescent and Developmental Neurology, University Children’s Hospital, Ljubljana, Slovenia
Abstract: Rett syndrome (RTT) is a common neurodevelopmental disorder that appears in infancy with regression of acquired motor skills, loss of purposeful activity, hand stereotypies, loss of acquired spoken language, and seizures. Epilepsy affects the majority of patients in a specific clinical stage of the disease and is drug resistant in approximately one-third of cases. The association of epilepsy and even drug-resistant epilepsy has been reported in certain genotypes of the methyl-CpG-binding protein 2 mutation, which is present in a majority of patients with classical RTT. The evolution of electroencephalographic abnormalities accompanying the clinical development of the syndrome is well described, but much less is known about the seizure semiology and the effectiveness of specific antiepileptic drugs. The aim of this review is to present the clinical and electrophysiological aspects of epilepsy in RTT and the current treatment approach.
Keywords: Rett syndrome, epilepsy, treatment
Introduction
Rett syndrome (RTT) is a neurodevelopmental disorder first described in the medical literature more than 50 years ago by Andreas Rett. The prevalence is approximately 1:10,000 girls, making it the second major cause of mental retardation in females after Down syndrome.1,2 It has also been documented in normal female carriers, in females with variant forms, and in some males with Klinefelter syndrome, fatal encephalopathy, familial X-linked mental retardation, and males with features of RTT.3,4
The methyl-CpG-binding protein 2 gene (MECP2) mutation, discovered in 1999, is associated with up to 95% of classical RTT patients, but only with 50%–70% of atypical cases of RTT.
Variant forms of RTT are: preserved-speech (Zappella) variant, early-seizure (Hanefeld) variant, and congenital (Rolando) variant. Mutation in MECP2 is found in the majority of cases of the preserved-speech variant, but is less frequently found in the other two variants. The early-seizure variant is frequently associated with mutations in CDKL5 and the congenital variant with mutations in FOXG1.3
Mutations in MECP2 are not synonymous with RTT, since the presence of the mutation alone is not sufficient to make the diagnosis of RTT, nor does its absence exclude it. Therefore, the diagnosis of RTT still remains clinical.
Clinical evolution
The evolution of symptoms usually starts in infancy with regression of acquired motor skills, loss of purposeful activity and acquired spoken language, hand stereotypies, and seizures. Secondary microcrania due to deceleration in the rate of head growth is obvious in the vast majority of patients, but may not be seen in up to 15% of the cases.
The clinical diagnosis of typical RTT is defined by four main criteria: a regression of purposeful hand use and spoken language, the development of gait abnormalities, and hand stereotypies. A period of regression is the necessary criteria, followed by stabilization and some recovery.
Supportive criteria are helpful to establish the diagnosis in atypical or variant RTT but are not necessary to fulfill the diagnosis of classical RTT. Two of the four main criteria and five of the eleven supportive criteria are necessary to establish the diagnosis of atypical RTT but the period of regression is still an obligatory prerequisite. It is necessary to exclude other disorders that may mimic RTT3 (Table 1).
 | Table 1 Diagnostic criteria for Rett syndrome |
The course of the disease, in its classical form, is characterized by four stages:5
Stage I: apparently normal early psychomotor development, but abnormal spontaneous movements may be observed during prolonged video analysis.6
Stage II: begins between the ages of 1 year and 3 years with rapid loss of acquired language and social interaction, loss of purposeful hand skills, appearance of hand stereotypies, hypotonia, gait dyspraxia, deceleration of head growth, respiratory abnormalities, and seizures.
Stage III: begins between the ages of 2 years and 10 years as a pseudo-stationary phase, with some improvement in communication skills but slow motor deterioration, rigidity, and seizures.
Stage IV: further motor deterioration with loss of the ability to walk (IVa), or never ambulant (IVb). (Clinical vignette).
Electroencephalographic abnormality and epilepsy
The electroencephalogram (EEG) is usually normal in the early stage, but slowing of the background activity and epileptiform abnormalities occur later.
The EEG is typically normal or shows some slowing of occipital background activity in the awake state in stage I. Loss of non-REM sleep characteristics and focal spikes or sharp waves in sleep and then in the awake state are observed in stage II. Additional slowing of background activity, absence of non-REM sleep characteristics, multifocal epileptiform discharges, generalized slow spike-wave, and rhythmic delta over the central regions during sleep and in the awake state are present in stage III. Marked slowing of the background activity with delta rhythms, multifocal epileptiform activity in the awake state, and generalized slow spike-wave activity in sleep are frequent in stage IV.
However, the EEG patterns are neither diagnostic nor pathognomonic of RTT, and correlation between clinical and EEG staging is not a constant finding, although frequently observed.1,7 EEG abnormalities also develop in RTT patients who do not have seizures.7
Some features of RTT, including hand stereotypies, episodic hyperventilation and apneas, laughing and screaming spells, staring gaze and gait dyspraxia, may be misinterpreted as seizures if not carefully evaluated by video-EEG recordings. On the other hand, some seizure events may be easily overlooked. In a group of 82 patients, clinical events identified by parents as their child’s typical seizures during EEG monitoring were not associated with EEG seizure discharges in more than 40% of cases, while 15% of patients had EEG seizure discharges, which were not recognized by parents as seizures.8,9
Regarding epilepsy management, it is necessary to critically review the events described as seizures. Home video recordings may help to differentiate paroxysmal events, but in difficult-to-manage cases, video-EEG recording is mandatory. Taken together with the clinical evidence, video-EEG is very helpful in differential diagnosis.
Epilepsy has been reported in 60%–80% of patients with RTT. Seizures usually appear in clinical stage II or III, around 4 years of age, with the maximal seizure frequency occurring between 7 years and 12 years. The incidence seems to be higher in those with early-onset RTT and more severe developmental disabilities. The severity of the epilepsy is considered to be an important contributor to the clinical severity of the RTT phenotype. Seizures have been reported to occur earlier in those patients who do not have MECP2 mutations.10
More than 300 different mutations in MECP2 have been described, 60% of these being in eight hotspot regions. Severe mutations have been found in the earlier or more severe forms of epilepsy. A tendency toward a higher prevalence of epilepsy in specific mutation types has been described, and a higher prevalence of specific mutations in drug-resistant patients has been reported in some studies.11–13 Other studies found no correlation between genotype and phenotype.14–17
The discrepancies between different cohorts may be explained by differences in sample size and different X-inactivation in patients with similar mutations.
Neuropathological and neuroradiological studies revealed reduced Rett brain volume in prefrontal, posterior frontal, anterior temporal regions, and caudate nucleus.18 The size of individual neurons is decreased and the packing density is increased in these regions. Golgi studies exhibited selectively reduced apical and basal dendritic arborizations in the pyramidal neurons of layer III and V in the frontal, motor, and inferior temporal regions in Rett brains.19 There are no specific neuroradiological findings described in RTT to be directly related to seizures as in some other symptomatic epilepsies amenable to surgical treatment.
Seizure semiology
There is no seizure type specific for RTT; all seizure types except for typical absences and clonic seizures were reported. Seizures were most commonly reported as partial complex, followed by tonic-clonic, tonic, and myoclonic in a retrospective analysis of a large Italian cohort.14
Complex partial and generalized tonic-clonic seizures (GTCC) were considered by far the most frequent seizure types, both at the onset and during follow-up by video-EEG recordings of Pintaudi.11
Myoclonic and GTCC, which could be mistaken for breathing dysfunction or hand stereotypies, tremor, and dystonia, were described in clinical video-EEG analysis in three of eight patients. In the same study, ictal onset was determined to be regional (right fronto-centro-temporal) in a patient with GTCC and myoclonic seizures, and non-lateralized (symmetric, bisynchronous) in two patients with myoclonic seizures. Further differentiation between seizures and episodes of motor activity and breathing abnormalities was possible in another four patients in the same group.20 There are no specific triggering factors reported to contribute to seizures in RTT.
Long-term video-EEG monitoring is a safe diagnostic tool that provides a high diagnostic yield. It is essential for characterizing paroxysmal events, differentiating breathing and autonomic dysfunction and motor phenomena, and for evaluating the treatment response to antiepileptic drugs (AED).
Epilepsy treatment
There are many AED currently available and the choice of the most appropriate treatment according to seizure type and a patient’s individual needs can be difficult. There are problems in evaluating the treatment efficiency of AED due to the general lack of comparative studies and the number of different AED used in monotherapy or combination treatment in RTT. As a result, the data, which are mostly provided by retrospective analysis, may be conflicting.
Due to limited experience of epilepsy treatment in RTT, no definite recommendation regarding anticonvulsants is currently available.
Large international Rett databases provide statistically powerful information on the characteristics of epilepsy and the relationships between epilepsy and genotype, but clinically relevant data on the effectiveness of specific medication, seizure type, ictal semiology, side effects, and relapse rates are lacking in large cohorts.
Carbamazepine was reported to be the most effective in the study of Huppke, followed by sulthiame, while valproate was significantly less effective in this cohort.21 Jian reported on the response to valproate and carbamazepine monotherapy, while responses to valproate, lamotrigine, and carbamazepine monotherapy were stated in Krajnc’s series.12,22 Carbamazepine was the most commonly used in Buoni’s series, followed by valproate.14 The five most commonly used drugs were valproate, carbamazepine, lamotrigine, levetiracetam, and topiramate in Bao’s Australian cohort.23 On the contrary, the US study of Glaze reported carbamazepine, lamotrigine, and levetiracetam as the most frequently used drugs, with valproate being only rarely used.13
A number (9–14) of different AED were used in monotherapy or in different treatment combinations in these studies.12,21–23
Only small numbers of patients have been included in reports addressing the effectiveness of newer AED such as lamotrigine, levetiracetam, and topiramate.
There are five reports on therapy with lamotrigine in RTT, in two as monotherapy, with seizure freedom or more than a 50% reduction in seizure frequency in 2 of 4, 2 of 4, and 2 of 2 subjects, respectively.21,22,24 Three other reports found lamotrigine add-on therapy to be successful in 2 of 3, 2 of 4, and 4 of 7 subjects.21,25,26
Levetiracetam was reported to be effective as add-on therapy in 3 of 3 and 8 of 8, but was not effective in 7 of 7 subjects.21,22,27 There is a case report of myoclonic status in RTT successfully treated with levetiracetam.9
Topiramate was mostly effective as an add-on treatment (only three cases as monotherapy) in 2 of 3, 4 of 7, and 7 of 8 subjects, respectively.21,22,28
A beneficial treatment effect was reported in two patients successfully treated with topiramate for severe respiratory dysrhythmia associated with prolonged apneas and convulsions, which were documented by long-term video-EEG recording.29
The treatment of subjects having only EEG abnormalities without clearly documented seizures does not seem to be justified.21,22
Side effects of AED
Reports on adverse events suggest the spectrum and rate of side effects are comparable to that seen in other patients with epilepsy. Huppke reported a 40% frequency of side effects and the need for discontinuation in only a few of his 61 patients.21 Krajnc reported the need for discontinuation due to rash and weight loss in 3 of 16 patients receiving carbamazepine, lamotrigine, and topiramate.22 The five medications with the most frequent side effects reported in 46% of 339 patients in Bao cohort were also the five most frequently used drugs, valproate, carbamazepine, lamotrigine, topiramate, and levetiracetam, with a specific profile of side effects reported for each of the drugs.23
Aggravation of myoclonic seizures by inappropriate treatment with carbamazepine was reported in four patients by D’Orsi.9 (Table 2).
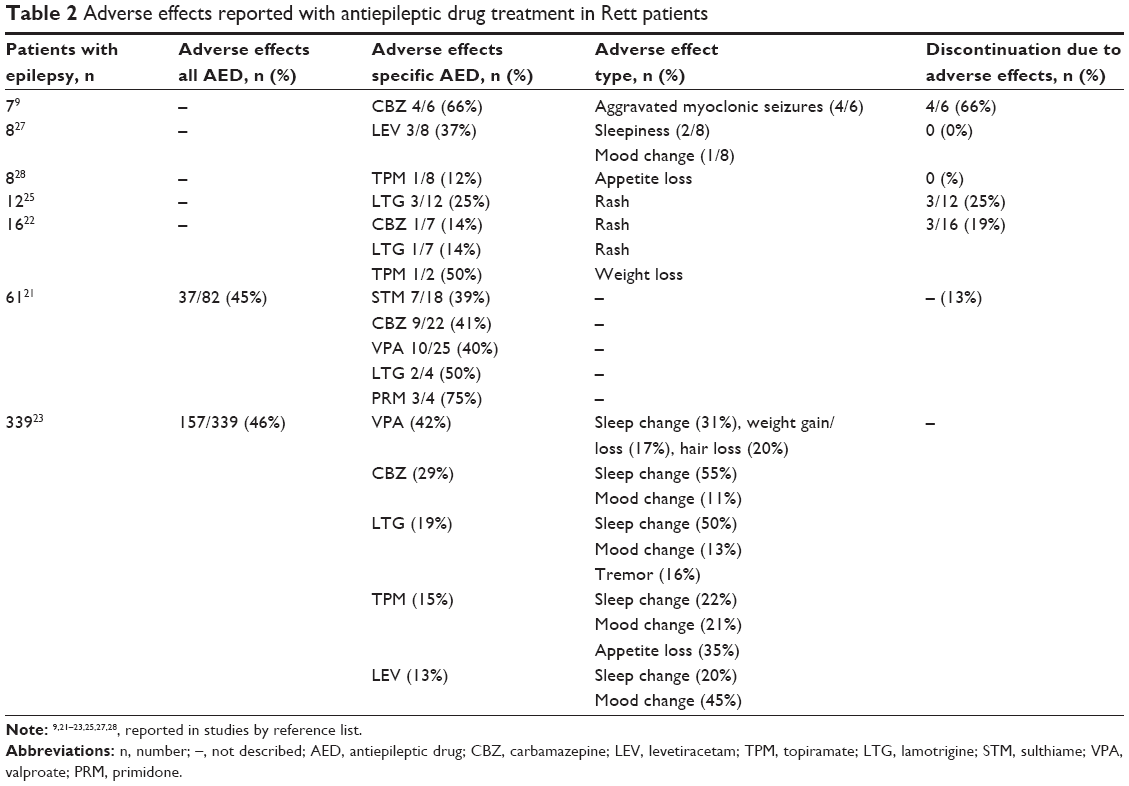 | Table 2 Adverse effects reported with antiepileptic drug treatment in Rett patients |
Leonard reported a threefold increased risk of fractures in RTT when valproate was used for more than a year, supporting the need for regular monitoring of bone density and appropriate nutritional support.30
Drug resistance
Between 60% and 75% of treated RTT patients achieve seizure remission after the first and second monotherapy and even in a group receiving polytherapy (one-fifth of 64 and one-third of 16 patients with active epilepsy in Huppke and Krajnc’s series respectively), seizure remission was achieved in 40% and 100%, respectively.21,22
Jian reported the need for three or more drugs in 19%, Krajnc in 18.5%, and Bao in 28% of RTT.12,22,23
Moser reported 3 of 8 RTT patients as having drug-resistant epilepsy (DRE).1 In Buoni’s cohort of 154 patients, DRE was observed in only 16%, with no significant relationship being found between the clinical severity of DRE and the quantitative or qualitative EEG abnormalities. In addition, no significant relationship was found between DRE and the RTT genotype.14 Pintaudi reported 30% of DRE among the 79% of those patients having epilepsy in a group of 165 RTT patients, while Bao reported 30% of DRE in 339 patients.11,23
These findings suggest that the prevalence of DRE in patients with RTT is about equal to patients with other newly-diagnosed epilepsies, where 20%–40% become refractory to treatment.31
The category of DRE may include patients with nonepileptic paroxysmal events, which are frequent and difficult to differentiate in RTT without long-term video-EEG recording, and these events may be a reason for polytherapy and pseudo-drug resistance.
Other treatment options
There is a single case report of a child with RTT and hypogammaglobulinemia successfully treated with intravenous gamma globulin due to intractable seizures.32
The ketogenic diet and vagal nerve stimulation have been reported to be effective treatment options in some refractory RTT cases.33–37
There are no reports on any definitive or palliative epilepsy surgery in this syndrome.
Treatment discontinuation
Common guidelines for withdrawal of AED should also be implemented in RTT. A minimum period of 2 years of seizure freedom is generally recommended before drug withdrawal, but this can be considerably shorter in children, depending on the epilepsy syndrome.38
Lower epilepsy activity in the late clinical course of RTT is generally reported, with a tendency toward seizure remission in late adolescence, even in those who previously required combination treatment. Therefore, the decision to lower and discontinue AED treatment should be taken when lasting seizure remission is achieved.
Data on discontinuation of AED in RTT are available only for one small series, where 3 of 16 treated patients stopped medication after 1–7 years of treatment without seizure relapse (follow-up 2–4 years). Behavioral deterioration was reported by the same group in one adult patient with RTT after valproate discontinuation following prolonged seizure remission, attributed to the mood-stabilizing effect of this drug.22
There is no data in the literature on the estimated relapse risk of epilepsy in RTT following discontinuation of AED.
Conclusion
The goal of epilepsy treatment is freedom from seizures with as few treatment adverse effects as possible. If seizure control is not achieved, alternative monotherapy should be instituted. If the first drug has some degree of efficacy, adding a second drug to the first one would be a rational approach.39 If success is not achieved with the second drug, the patient should be referred to a tertiary epilepsy center to document seizures and to evaluate further treatment options with AED or other treatment alternatives.
Long-term video-EEG recording is frequently required to differentiate between paroxysmal events in RTT and to provide useful information on treatment effectiveness. It is the standard procedure to further classify seizure semiology and seizure onset, which influence the choice of the most appropriate drug treatment.
Carbamazepine, valproate, and lamotrigine are the most frequently used drugs, but some regional (continental) differences in prescribing are obvious, possibly reflecting concerns about side effects, pricing by manufacturers, marketing, published evidence of effectiveness versus toxicity, the nature of health care systems, and the role of local experts.40
Other treatment alternatives are rarely reported in this syndrome but could be effective in drug-resistant cases.
Some genotypes are reported to be more frequently associated with epilepsy and even with a drug-resistant course, but the information on genotype is generally not used for predicting the clinical course in a given patient.
DRE in RTT is no more frequent than in patients reported with other newly-diagnosed epilepsies and the decision to lower and discontinue AED treatment should be taken when lasting seizure remission is achieved, which is usually expected in late adolescence. Evidence on the relapse rate in RTT is lacking, but the burden of epilepsy is reported to be lower with increasing age.
Despite better understanding of the relationship between the genotype and phenotype of epilepsy in RTT provided by large cohorts, there is a need for comparative studies to provide more data on the effectiveness of AED treatment in this syndrome and to recommend the approach to drug treatment.
Disclosure
The author reports no conflicts of interest in this work.
References
Jordan Moser S, Weber P, Lütschg J. Rett syndrome: clinical and electrophysiologic aspects. Pediatr Neurol. 2007;36:95–100. | ||
Laurvick CL, de Klerk N, Bower C, et al. Rett syndrome in Australia: a review of the epidemiology. J Pediatr. 2006;148:347–352. | ||
Neul JL, Kaufmann WE, Glaze DG, et al; RettSearch Consortium. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68:944–950. | ||
Amir RE, Van der Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. | ||
Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:61–65. | ||
Einspieler C, Kerr AM, Prechtl HF. Is the early development of girls with Rett disorder really normal? Pediatr Res. 2005;57:696–700. | ||
Glaze DG. Neurophysiology of Rett syndrome. J Child Neurol. 2005;20:740–746. | ||
Glaze DG, Schultz RJ, Frost JD. Rett syndrome: characterization of seizures versus non-seizures. Electroencephalogr Clin Neurophysiol. 1998;106:79–83. | ||
D’Orsi G, Demaio V, Minervini MG. Myoclonic status missdiagnosed as movement disorder in Rett syndrome: a video polygraphic study. Epilepsy Behav. 2009;15(2):260–262. | ||
Jian L, Nagarajan L, de Klerk N, et al. Predictors of seizure onset in Rett syndrome. J Pediatr. 2006;149:542–547. | ||
Pintaudi M, Calevo MG, Vignoli A, et al. Epilepsy in Rett syndrome: clinical and genetic features. Epilepsy Behav. 2010;19:296–300. | ||
Jian L, Nagarajan L, de Klerk N, Ravine D, Christodoulou J, Leonard H. Seizures in Rett syndrome: an overview from a one-year calendar study. Eur J Ped Neurol. 2007;11:310–317. | ||
Glaze DG, Percy AK, Skinner S, et al. Epilepsy and the natural history of Rett syndrome. Neurology. 2010;74(11):909–912. | ||
Buoni S, Zannolli R, De Felice C, et al. Drug-resistant epilepsy and epileptic phenotype-EEG association in MECP2 mutated Rett syndrome. Clin Neurophysiol. 2008;119:2455–2458. | ||
Nissenkorn A, Gak E, Vecsler M, Reznik H, Manascu S, Ben Zeev B. Epilepsy in Rett syndrome-the experience of a National Rett Center. Epilepsia. 2010;51:1252–1258. | ||
Cardoza B, Clarke A, Wilcox J, et al. Epilepsy in Rett syndrome: association between phenotype and genotype, and implications for practice. Seizure. 2011;20:646–649. | ||
Bebbington A, Anderson A, Ravine D, et al. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology. 2008;70:868–875. | ||
Naidu S, Kaufmann WE, Abrams MT, et al. Neuroimaging studies in Rett syndrome. Brain Dev. 2001;23:S62–S71. | ||
Duncan Armstrong D. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:47–53. | ||
d’Orsi G, Trivisano M, Luisi C, et al. Epileptic seizures, movement disorders and breathing disturbances in Rett syndrome: diagnostic relevance of video-polygraphy. Epilepsy Behav. 2012;25(3):401–407. | ||
Huppke P, Köhler K, Brockmann K, Stettner GM, Gärtner J. Treatment of epilepsy in Rett syndrome. Eur J Pediatr Neurol. 2007;11: 10–16. | ||
Krajnc N, Župančič N, Oražem J. Epilepsy treatment in Rett syndrome. J Child Neurol. 2011;26(11):1429–1433. | ||
Bao X, Downs J, Kingsley W, Williams S, Leonard H. Using a large international sample to investigate epilepsy in Rett syndrome. Dev Med Child Neurol. 2013;55:553–558. | ||
Kumandas S, Caksen H, Ciftçi A, Oztürk M, Per H. Lamotrigine in two cases of Rett syndrome. Brain Dev. 2001;23:240–242. | ||
Stenbom Y, Tonnby B, Hagberg B. Lamotrigine in Rett syndrome: treatment experience from a pilot study. Eur Child Adolesc Psychiatry. 1998;7:49–52. | ||
Uldall P, Hansen FJ, Tonnby B. Lamotrigine in Rett syndrome. Neuropediatrics. 1993;24:339–340. | ||
Specchio N, Balestri M, Striano P, et al. Efficacy of levetiracetam in the treatment of drug resistant Rett syndrome. Epilepsy Res. 2010;88(2–3):112–117. | ||
Goyal M, O’Riordan A, Wiznitzer M. Effect of topiramate on seizure and respiratory dysrhythmia in Rett syndrome. J Child Neurol. 2004;19: 588–591. | ||
Krajnc N. Severe respiratory dysrhythmia in Rett syndrome treated with topiramate. J Child Neurol. 2014;29:N118. http://jcn.sagepub.com/content/29/10/NP118 | ||
Leonard H, Downs J, Jian L, et al. Valproate and risk of fracture in Rett syndrome. Arch Dis Child. 2010;95:444–448. | ||
French JA. Refractory epilepsy: clinical overview. Epilepsia. 2007;48(suppl 1):3–7. | ||
Sindher SB, Eisenfeld M, Madhok N, Djukic A, Shiozberg J. Effect of gammaglobulin treatment on a child with Rett syndrome, intractable seizures and hypogammaglobulinemia. Poster presented at: Annual Meeting of American College of Allergy, Asthma and Immunology; November 11, 2010; Phoenix, AR. [Annals of Allergy, Asthma and Immunology. 105(5):(pp A 95)]. | ||
Anghelescu C, Butoianu N, Tarta-Arsene O, et al. Could vagus nerve stimulation serve as first line of treatment for epilepsy in Rett syndrome. Poster presented at: 11th European Congress on Epileptology; June 29, 2014; Stockholm. [Epilepsia. 55:(pp 145)]. | ||
Wilfong AA, Schultz RJ. Vagus nerve stimulation for treatment of epilepsy in Rett syndrome. Dev Med Child Neurol. 2006;48:683–686. | ||
Topcu M, Yalnizoglu D, Turanil G, Bilginer B, Akalan N. Vagus nerve stimulation in children with Rett syndrome. 62 American Epilepsy Society Annual Meeting; December 5–9, 2008; Seattle, Washington. Abstract No 2298. | ||
Haas BRH, Rice MA, Trauner DA, Merritt TA. Therapeutic effect of a ketogenic diet in Rett syndrome. Am J Med Genet Suppl. 1986;1:225–246. | ||
Liebhaber GM, Riemann E, Baumeister FA. Ketogenic diet in Rett syndrome. J Child Neurol. 2003;18:74–75. | ||
Beghi E, Giussani G, Grosso S, et al. Withdrawal of antiepileptic drugs: guidelines of the Italian league against epilepsy. Epilepsia. 2013;54(suppl 7):2–12. | ||
Ben-Menachem E. Medical management of refractory epilepsy-practical treatment with novel antiepileptic drugs. Epilepsia. 2014;55(suppl 1):3–8. | ||
Clarke A. How much further can large international databases take Rett syndrome research? Dev Med Child Neurol. 2013;55:490–496. |
Supplementary material
This 32-year-old patient presented at the age of 3.5 years due to a 1-year history of screaming spells occurring 2–3 times daily.
The family history was normal; she had a healthy older brother. She was born after a normal pregnancy with a normal labor and postnatal course. Birth measurements were normal and proportional, including the occipito-frontal circumference. She acquired independent walking at the age of 13 months and used 5–10 meaningful words at the age of 2 years. She did not acquire a pincer-grasp but was able to use a spoon for feeding.
At 2.5 years, her parents noticed a quite sudden behavioral change; she became irritable, with screaming spells lasting up to 1 hour. This was followed by regression of verbal communication, hand stereotypies (clapping and washing movements, circular hand-to-mouth movements), and loss of interest in play and social interaction.
When she presented 1 year later, her neurologic examination revealed a broad-based dyspraxic gait, loss of verbal and nonverbal communication, loss of eye contact, and hand stereotypies. Muscle tone was generally low, and proprioceptive reflexes were normal, with a flexor plantar response. There was no deceleration of head growth.
EEG disclosed irregular background activity in the awake state and preserved sleep structure, but bursts of bilateral spike-wave complexes, more pronounced over the right hemisphere, were recorded.
Six months later she experienced her first epileptic seizure during sleep with eye and mouth-angle deviation to the left side, followed by generalized tonic-clonic convulsions, lasting for 5 minutes. The seizures were repeated over the next few months, so antiepileptic treatment with valproate was started. She has remained seizure-free since the age of 11 years. Total discontinuation of treatment was not possible due to behavioral deterioration on two successive attempts, with her autoaggressive behavior attributed to the mood-stabilizing effect of valproate.
Brain MRI was normal at the age of 7 years. The clinically established diagnosis of RTT was confirmed years later by MECP2 mutation analysis at the age of 20 years, when genetic testing became available.
She did not lose the ability to walk independently in the following years, but her gait was broad-based and dyspraxic. Currently, she has no functional hand use and hand stereotypies are still very prominent. She is not able to communicate verbally but her eye contact is very intense. Episodes of hyperventilation are obvious in the awake state. She has small, cold feet, slight scoliosis, and generally higher muscle tone with brisk reflexes and a flexor plantar response.
She is still on low-dose valproate treatment and oral calcium supplementation due to osteopenia.
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.