")
Back to Journals » OncoTargets and Therapy » Volume 7
Management of cytopenias in patients with myelofibrosis treated with ruxolitinib and effect of dose modifications on efficacy outcomes
Authors Verstovsek S, Gotlib J, Gupta V, Atallah E, Mascarenhas J, Quintas-Cardama A, Sun W, Sarlis N, Sandor V, Levy R, Kantarjian H, Mesa R
Received 21 August 2013
Accepted for publication 20 September 2013
Published 17 December 2013 Volume 2014:7 Pages 13—21
DOI https://doi.org/10.2147/OTT.S53348
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Srdan Verstovsek,1 Jason Gotlib,2 Vikas Gupta,3 Ehab Atallah,4 John Mascarenhas,5 Alfonso Quintas-Cardama,1 William Sun,6 Nicholas J Sarlis,6 Victor Sandor,6 Richard S Levy,6 Hagop M Kantarjian,1 Ruben A Mesa7
1University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Stanford Cancer Institute, Stanford, CA, USA; 3Princess Margaret Cancer Centre, University of Toronto, Toronto, ON, Canada; 4Medical College of Wisconsin, Milwaukee, WI, USA; 5Mount Sinai School of Medicine, New York, NY, USA; 6Incyte Corporation, Wilmington, DE, USA; 7Mayo Clinic, Scottsdale, AZ, USA
Purpose: Ruxolitinib is an oral Janus kinase (JAK) 1/JAK2 inhibitor approved in the US for the treatment of intermediate- or high-risk myelofibrosis (MF). Because thrombopoietin and erythropoietin signal through JAK2, dose-dependent cytopenias are expected with treatment. In the COMFORT-I (COntrolled Myelofibrosis study with Oral JAK inhibitor Treatment I) trial, these cytopenias were effectively managed with dose adjustments. These analyses were conducted to evaluate the relationship between ruxolitinib titrated doses and changes in platelet count and hemoglobin level as well as efficacy measures.
Patients and methods: COMFORT-I was a randomized, placebo-controlled trial in 309 patients with intermediate-2 or high-risk MF and a platelet count ≥100 × 109/L. Ruxolitinib starting doses were 15 and 20 mg twice daily (bis in die [BID]) for patients with baseline platelet counts of 100–200 × 109/L and >200 × 109/L, respectively. Percentage changes from baseline to week 24 in spleen volume and MF-related symptoms were assessed in subgroups defined by final titrated dose (average daily dose during weeks 21 to 24).
Results: The median final titrated doses for patients starting at doses of 15 and 20 mg BID were 10 and 20 mg BID, respectively, at week 24. Most dose reductions occurred in the first 8–12 weeks of treatment and coincided with decreases in platelet count and hemoglobin level. Subsequently, platelet counts stabilized and hemoglobin levels gradually returned to near baseline levels (red blood cell transfusion rates followed a similar trend). Final titrated doses of ≥10 mg BID were associated with clinically meaningful improvements in MF-related symptoms that were comparable across doses, while marginally greater reductions in spleen volume were observed at higher doses.
Conclusion: This COMFORT-I analysis shows that dose-dependent cytopenias were effectively managed with ruxolitinib dose adjustments, and titrated doses of ≥10 mg BID were associated with clinically meaningful reductions in spleen volume and symptom improvement at week 24.
Keywords: COMFORT-I, dose titration, JAK2 inhibitor, myelofibrosis, ruxolitinib, treatment-related cytopenias
Introduction
Myelofibrosis (MF) is a life-threatening chronic myeloproliferative neoplasm (MPN).1 The median age at diagnosis is around 65 years,2,3 and the estimated survival time for patients with advanced MF is 5 years or less, depending on the number of risk factors.4 Although the natural history of MF is highly variable, typical manifestations are splenomegaly, which may be massive; cytopenias, particularly anemia and thrombocytopenia; and debilitating symptoms, which commonly include fatigue, fever, night sweats, pruritus, weight loss, early satiety, and abdominal pain.5,6 MF may develop de novo (primary MF) or evolve from two other MPNs: essential thrombocythemia and polycythemia vera.7 Irrespective of the etiology of MF, the pathobiology of the disease is primarily characterized by dysregulation of the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling pathway.8
The oral JAK1/JAK2 inhibitor ruxolitinib (Jakafi®; Incyte Corporation, Wilmington, DE, USA) is currently the only therapy approved in the US,9 Canada,10 and Europe11 for the treatment of patients with MF. In two randomized Phase III studies, COMFORT (COntrolled Myelofibrosis study with ORal JAK inhibitor Treatment)-I12 and COMFORT-II,13 ruxolitinib demonstrated efficacy in reducing splenomegaly, alleviating MF-related symptoms, and improving quality-of-life measures compared with placebo (COMFORT-I) or best available therapy (COMFORT-II). In addition, a survival advantage in patients randomized to ruxolitinib was observed in both trials.14,15 The most common adverse events were anemia and thrombocytopenia.12,13 Given the essential role of JAK2 in erythropoiesis and thrombopoiesis,16 these dose-dependent cytopenias were expected on-target effects of ruxolitinib. To minimize treatment-related cytopenias, COMFORT-I and COMFORT-II study designs incorporated a patient-management strategy that included the use of different starting doses based on platelet count at baseline, mandatory dose reductions and treatment interruptions based on predefined changes in platelet count and absolute neutrophil count (ANC), and the use of red blood cell (RBC) transfusions for anemic patients at the discretion of the treating investigator. Consequently, discontinuations for anemia or thrombocytopenia were rare.12,13
The present COMFORT-I data analysis was performed to assess the relationship between final titrated doses of ruxolitinib (defined as the average daily dose from week 21 to week 24) and reductions in spleen volume and symptom scores from baseline to week 24, and to describe the time course of platelet counts, hemoglobin levels, and white blood cell (WBC) counts in patients who participated in the COMFORT-I trial.
Patients and methods
Study design and patients
The study design and patient characteristics of the COMFORT-I trial have been described.12 In brief, COMFORT-I was a randomized, placebo-controlled, Phase III trial of ruxolitinib that enrolled 309 patients with intermediate-2 or high-risk MF and a platelet count ≥100 × 109/L. The primary end point was the proportion of patients with a 35% or greater reduction in spleen volume from baseline to week 24. A secondary end point was the proportion of patients who achieved a 50% or greater reduction in total symptom score (TSS) from baseline to week 24.12
Assessments
Spleen volume was measured by magnetic resonance imaging or computed tomography. MF-related symptoms were assessed using the modified Myelofibrosis Symptom Assessment Form (MFSAF) version 2.0 daily diary.12,17 The electronic diary contains seven questions, which pertain to three abdominal symptoms (abdominal discomfort, pain under the ribs on the left side, early satiety), three nonabdominal symptoms (night sweats, pruritus, muscle/bone pain), and inactivity. Symptoms were rated from 0 (absent) to 10 (worst imaginable). TSS is defined as the sum of the scores for all individual symptoms except inactivity. Abdominal TSS and cytokine TSS are defined as total score for all abdominal symptoms and nonabdominal (ie, cytokine-related) symptoms, respectively. Fatigue was assessed using the self-administered Patient-Reported Outcomes Measurement System (PROMIS) fatigue score.17
Dose adjustments and monitoring of blood cell counts
Ruxolitinib starting doses in COMFORT-I were 15 mg twice daily (bis in die [BID]) for patients with a baseline platelet count of 100–200 × 109/L and 20 mg BID for those with a baseline platelet count greater than 200 × 109/L.12 Doses could be increased if patients did not achieve protocol-defined spleen responses and had an adequate platelet count and ANC. Dose reductions were required for protocol-defined decreases in the platelet count. Administration of ruxolitinib or placebo was interrupted if the platelet count decreased to less than 50 × 109/L or ANC was less than 0.5 × 109/L. Treatment could be resumed and doses could be increased after platelet count and/or ANC had recovered to acceptable predefined levels, as described previously.12 Assessment of blood cell counts was recommended at least once a week if the platelet count was lower than 100 × 109/L or ANC was lower than 1 × 109/L, and at least twice a week if the platelet count was lower than 50 × 109/L or the ANC was lower than 0.5 × 109/L. The use of growth factors, such as granulocyte colony-stimulating factor and erythropoietin-stimulating agents, was not permitted during the study.
Analyses
To determine the relationship between ruxolitinib titrated dose and treatment efficacy at week 24, patients randomized to treatment with ruxolitinib were stratified in five subgroups based on the average final titrated dose from week 21 to week 24: 5 mg or less BID (total daily dose ≤10 mg), 10 mg BID (total daily dose >10–20 mg), 15 mg BID (total daily dose >20–30 mg), 20 mg BID (total daily dose >30–40 mg), and 25 mg BID (total daily dose >40 mg). Percentage changes from baseline to week 24 in efficacy parameters, including spleen volume, TSS, abdominal TSS, cytokine TSS, individual symptom scores, and PROMIS fatigue score, were evaluated in these subgroups as well as in the placebo group.
Mean percentage changes in platelet counts, WBC count, ANC, and hemoglobin levels from baseline over time were evaluated in the ruxolitinib group (by starting dose) and in the placebo group. Percentage change in hemoglobin level from baseline over time was also evaluated in patients who did not receive RBC transfusions or dose adjustments during the first 24 weeks of the study.
Results
In COMFORT-I, 155 patients were randomized to ruxolitinib, and 154 were randomized to placebo.12 A total of 136 patients in the ruxolitinib group completed 24 weeks of treatment. Of those patients, 43 had a baseline platelet count of 100–200 × 109/L and started with a dose of 15 mg BID, whereas 93 had a baseline platelet count greater than 200 × 109/L and started with a dose of 20 mg BID.
Ruxolitinib dose adjustments
At week 24, 77% of patients with a baseline platelet count of 100–200 × 109/L and 39% of those with a baseline platelet count greater than 200 × 109/L had a reduced ruxolitinib dose relative to their starting dose. The starting and final titrated doses differed for approximately 70% of all ruxolitinib-treated patients (Figure 1), and dose adjustments typically occurred within the first 8–12 weeks of the study. The median final titrated dose at week 24 was 10 mg BID for those starting at 15 mg BID and 20 mg BID for those starting at 20 mg BID.
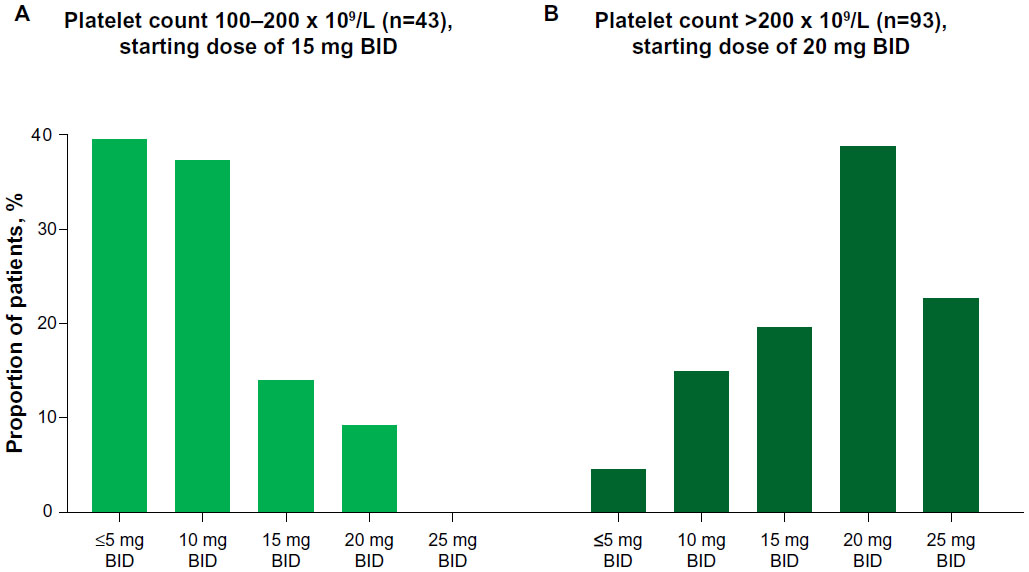 | Figure 1 Distribution of patients per final titrated dose for patients starting ruxolitinib at 15 mg twice daily (bis in die [BID]) (A) versus 20 mg BID (B). |
Relationship between final titrated dose and efficacy of ruxolitinib
Because ruxolitinib dose adjustments occurred mainly during the first 8–12 weeks after treatment initiation, the efficacy of ruxolitinib at week 24 was in large part a reflection of the titrated dose and not the starting dose. Therefore, changes in spleen volume and symptoms were evaluated by the average daily dose received during the 4 weeks prior to the 24-week assessment. This analysis showed that final titrated doses of ruxolitinib of 10 mg BID or higher were associated with median reductions in spleen volume from baseline to week 24 of 31%–41%, with slightly greater efficacy observed at higher doses (Table 1). Patients with final titrated doses of 5 mg BID or less achieved a more modest improvement in spleen volume, with a median reduction from baseline to week 24 of 10.4%. In contrast, patients in the placebo group had a 9.2% median increase in spleen volume (Table 1).
 | Table 1 Median percentage change from baseline to week 24 in spleen volume and total symptom scores by final titrated ruxolitinib dose in COMFORT-I |
Median reductions in TSS were similar at titrated doses of 10 mg BID and higher (60%–71% improvement) (Table 1). Similar results were obtained for abdominal and cytokine TSS (Table 1). Overall, symptom improvements associated with ruxolitinib doses of 5 mg BID or less were more modest than those achieved with doses of 10 mg BID or higher, and patients in the placebo group experienced symptom worsening (Table 1). An analysis of individual MFSAF symptom scores and the PROMIS fatigue score confirmed that a final titrated dose of 10 mg BID was associated with maximal or near-maximal reductions in symptom scores compared with higher doses, with the exception of pruritus, which showed greater improvements at higher doses (Table 2).
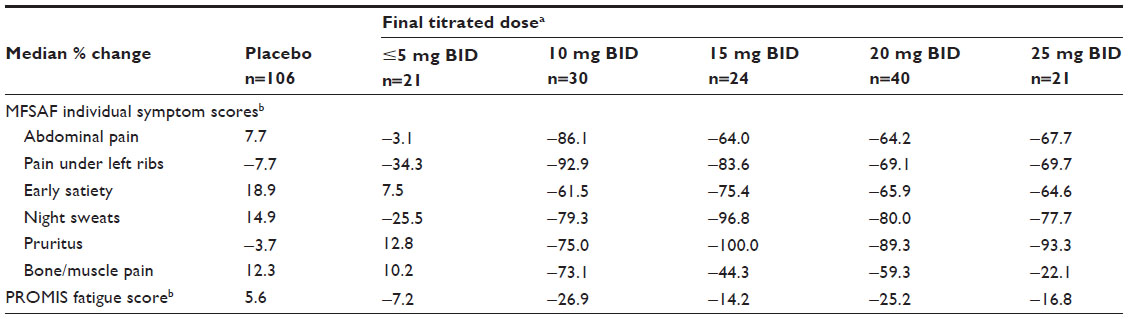 | Table 2 Median percentage change from baseline to week 24 in symptom scores of the MFSAF TSS and PROMIS fatigue scale by final titrated ruxolitinib dose |
Changes in platelet count over time
Individual dose adjustments were implemented in response to changes in platelet count, and as expected, based on the findings reported previously,12 the greatest decrease in platelet count was observed within the first 8 weeks. By week 8, patients with a baseline platelet count of 100–200 × 109/L had a 25% mean decrease in platelet count (mean ± standard deviation, 145±29 × 109/L at baseline to 109±46 × 109/L at week 8), whereas those with a baseline platelet count greater than 200 × 109/L had a 43% mean decrease in platelet count (416±191 × 109/L to 234±146 × 109/L). However, after these initial decreases, platelet counts generally stabilized (Figure 2). Furthermore, with careful patient monitoring (ie, at least weekly hematology parameters for platelet count <100 × 109/L) and dose adjustments, only one patient in the ruxolitinib group discontinued for thrombocytopenia.
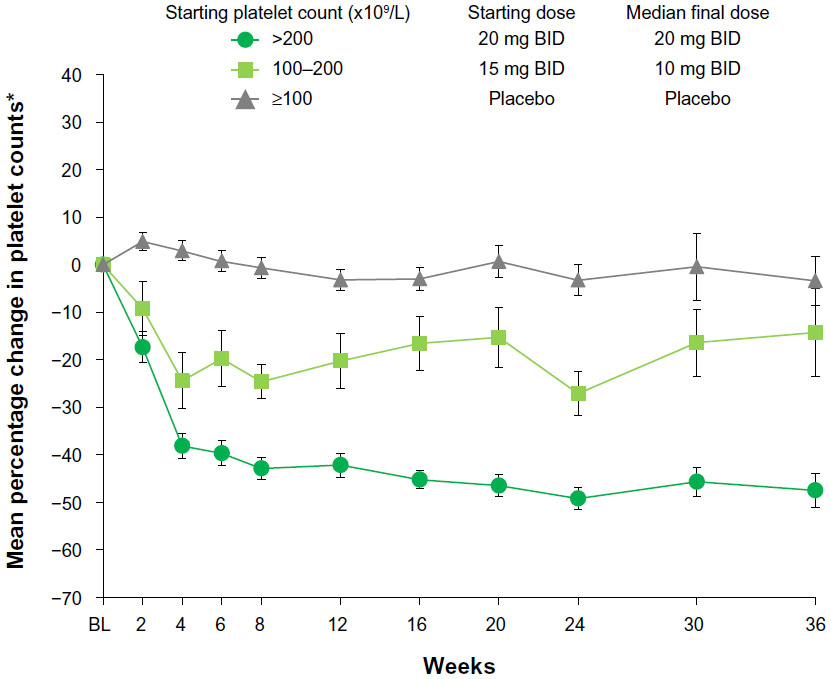 | Figure 2 Mean percentage change from baseline (BL) in platelet counts over time. |
Changes in hemoglobin level over time
Mean changes in hemoglobin level in patients randomized to ruxolitinib were characterized by a temporary decrement with a nadir around weeks 8–12 (Figure 3A). At week 12, hemoglobin levels (mean ± standard deviation) in patients with a baseline platelet count of 100–200 × 109/L and greater than 200 × 109/L decreased from baseline by approximately 10% and 12%, respectively, to 9.4±1.4 g/dL and 9.6±1.7 g/dL. After week 12, hemoglobin values in the two subgroups gradually recovered, reaching new steady-state levels approximately 5% below baseline by weeks 24–36 (Figure 3A). In COMFORT-I, RBC transfusions were optional in patients who developed anemia, and as previously reported, the proportion of patients receiving RBC transfusions increased after treatment initiation and then declined to levels observed in the placebo group.12 To assess the effects of RBC transfusions as well as study discontinuations on hemoglobin levels over time, a separate analysis was conducted that included only patients who received no postbaseline RBC transfusions and completed the first 24 weeks of the study. This analysis included 17 patients with baseline platelet counts of 100–200 × 109/L and 46 patients with baseline platelet counts greater than 200 × 109/L. Mean percentage changes in hemoglobin levels over time in this patient group (Figure 3B) were similar to those seen in the analysis of all patients, regardless of transfusion status or time of discontinuation (Figure 3A). During this time frame of the primary analysis, only one patient discontinued for anemia in the ruxolitinib group.
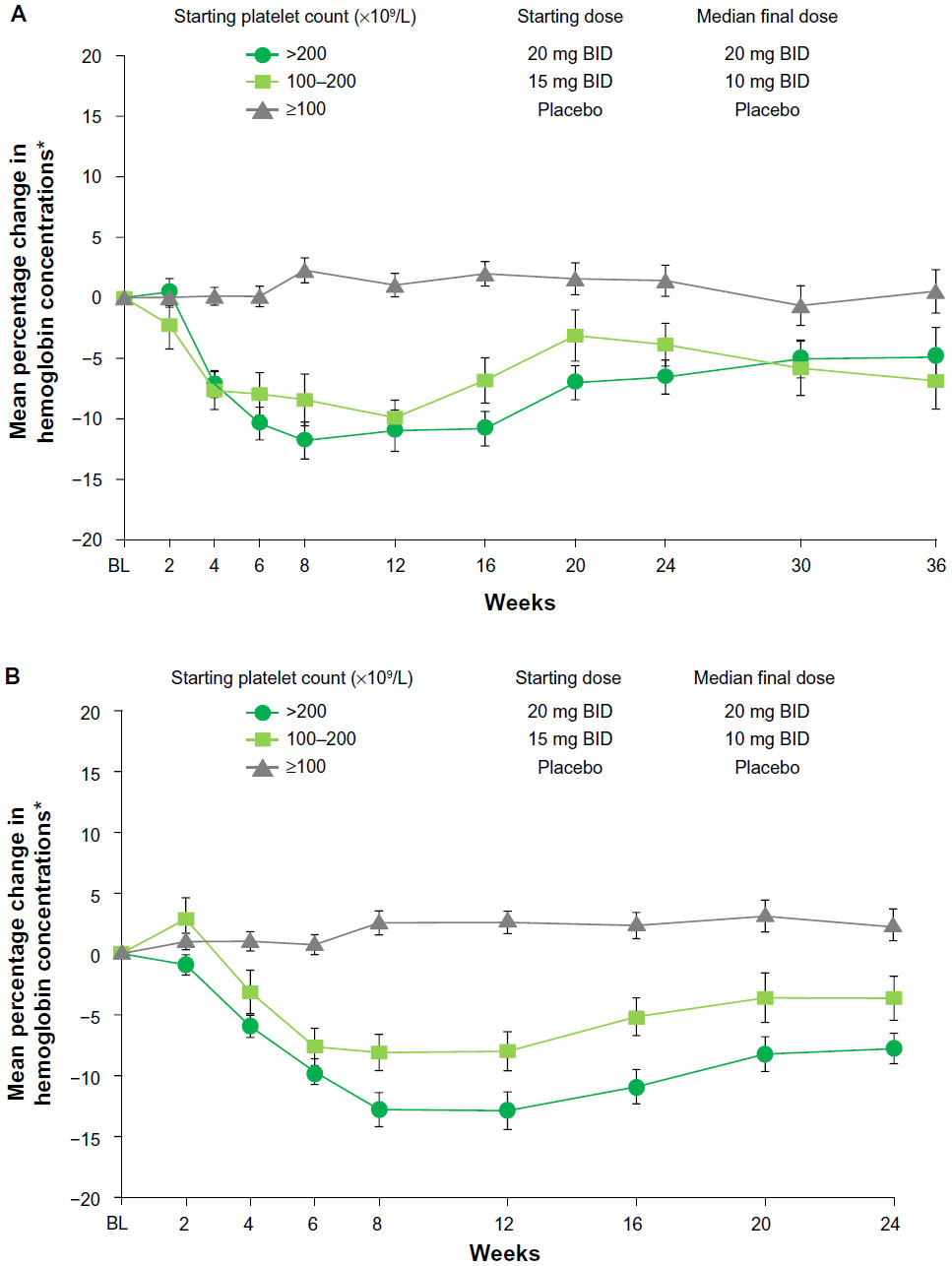 | Figure 3 Mean percentage change from baseline (BL) in hemoglobin levels over time in all patients (A) and in patients who received no red blood cell transfusions and completed the first 24 weeks of the study (B). |
Changes in WBC count and ANC over time
Overall, patients tended to have elevated WBC counts and ANCs at baseline (Figure 4A and B). Few patients had low counts (>90% of patients had grade 0 leukopenia or neutropenia at baseline). In patients with starting platelet counts of 100–200 × 109/L, the mean WBC count (± standard deviation) decreased during the first 4 weeks of ruxolitinib therapy by 23%, from 21.3±22.1 × 109/L at baseline to 16.3±18.1 × 109/L at week 4. In patients with starting platelet counts greater than 200 × 109/L, the mean WBC count (± standard deviation) decreased by 42%, from 22.1±17.8 × 109/L at baseline to 12.8±9.2 × 109/L at week 4. Counts remained stable thereafter (Figure 4A). Changes in ANC followed a similar trend (Figure 4B). Mean WBC count and ANC slightly increased in the placebo group (Figure 4A and B). Because mean WBC count and ANC were elevated at baseline, decreases in counts with ruxolitinib generally resulted in a trend toward normalization. At the time of the primary analysis, 5.8% of patients randomized to ruxolitinib and 2.0% randomized to placebo experienced grade 3 or 4 leukopenia; 7.1% randomized to ruxolitinib and 2.0% randomized to placebo experienced grade 3 or 4 neutropenia.12
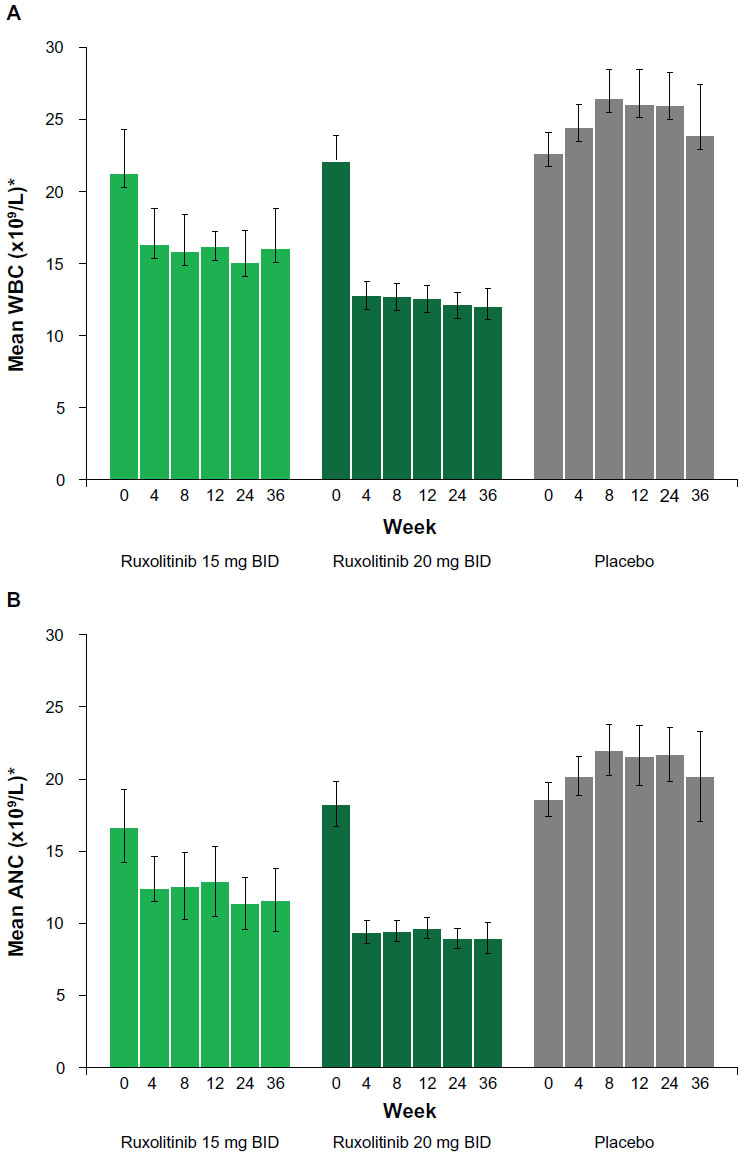 | Figure 4 Mean white blood cell (WBC) count (A) and mean absolute neutrophil count (ANC) (B) over time. |
Discussion and conclusion
In COMFORT-I, dose reductions were part of a strategy to manage treatment-related cytopenias. Although dose adjustments were common early in the course of the study, the vast majority of patients eventually attained stable doses associated with stabilization of platelet counts and recovery of hemoglobin levels to near baseline values. As previously reported, monthly rates of grade 3 or 4 thrombocytopenia and anemia followed a similar trend, increasing during the first 8 weeks and subsequently returning to rates similar to placebo.12 In addition, although RBC transfusion rates increased with ruxolitinib during the first 8 weeks compared with placebo, they subsequently declined and reached rates similar to those seen with placebo by week 36.14
The majority of patients treated with ruxolitinib in COMFORT-I achieved a final titrated dose of 10 mg or higher, including a majority of patients with baseline platelet counts of 100–200 × 109/L. Our results indicate that final titrated doses (average daily dose during weeks 21–24) of 10 mg BID or higher were associated with clinically meaningful reductions in spleen size and improvements in MF-related symptoms, including fatigue, night sweats, pruritus, bone/muscle pain, and abdominal symptoms. In addition, maximal improvement in symptoms (with the exception of pruritus) was seen at final titrated doses of 10 mg BID, whereas further reduction in spleen volume was seen with higher doses. Our findings are supported by preliminary results from an ongoing study of patients with MF and baseline platelet counts of 50–100 × 109/L. After starting with a dose of 5 mg BID, patients could receive higher doses, provided that they maintained adequate platelet counts.18 The majority of patients (with available data) were receiving doses of 10 mg BID or higher by week 24. At this time, the proportions of patients who achieved at least a 35% reduction in spleen volume and at least a 50% reduction in TSS (33% and 36%, respectively) approached the corresponding numbers for the ruxolitinib group in the COMFORT-I study population (42% and 46%, respectively).18
The management of cytopenias in COMFORT-I involved careful monitoring of blood counts, and therefore dose adjustments could be implemented in a way that minimized large declines in hematology parameters that would necessitate dose holds or the receipt of less optimal doses. The utility of this strategy was most evident in the group of patients starting with a lower baseline platelet count. This group experienced the greatest percentage of dose reductions, yet many were able to continue treatment at a stable and efficacious dose. Although initial dose increases were optional in COMFORT-I (for inadequate efficacy), these analyses suggest that aggressive dose increases early in the course of therapy should be reserved for those patients able to maintain adequate platelet counts.
In conclusion, our findings suggest that careful patient monitoring and appropriate ruxolitinib dose titration as needed is an effective approach to optimizing treatment benefit for patients with MF. Because inhibition of JAK2 is expected to result in dose-dependent cytopenias, it is important to consider that new-onset or worsening anemia and thrombocytopenia during ruxolitinib therapy may not be a sign of worsening of the underlying disease. These treatment-related cytopenias tend to occur early in the course of treatment, and appropriate management may contribute to better patient outcomes. Additional follow-up from COMFORT-I will further our understanding of these outcomes with longer-term therapy.
Acknowledgments
The COMFORT-I clinical trial was sponsored by Incyte Corporation. Medical writing support was provided by Roland Tacke, PhD, of Evidence Scientific Solutions, and funded by Incyte Corporation. These data were previously presented at the Eighth Annual Hematologic Malignancies Conference, Houston, TX, October 10–14, 2012.
Disclosure
SV received grant support through his institution from Incyte Corporation, Exelixis, Celgene, NS Pharma, Infinity Pharmaceuticals, SBIO, Lilly Oncology, AstraZeneca, Geron, Bristol-Myers Squibb, YM BioSciences, Gilead, and Roche; JG received consultancy fees, honoraria, and support for travel to meetings for the study from Incyte Corporation; VG received grant support through his institution from Incyte Corporation and Novartis, consulting fees from Incyte Corporation and Novartis, and lecture fees from Novartis; EA received grant or research support and consultancy fees from Incyte; JM received grant or research support from Incyte and Novartis; AQC received consultancy fees from Incyte and Sanofi; WS, NJS, VS, and RSL are employees of Incyte and own stock in Incyte Corporation; HMK received grant support through his institution from Incyte Corporation; and RAM received research funding from Incyte Corporation, Eli Lilly, Sanofi, NS Pharma, and YM Bioscience.
References
Mesa RA, Green A, Barosi G, Verstovsek S, Vardiman J, Gale RP. MPN-associated myelofibrosis (MPN-MF). Leuk Res. 2011;35(1):12–13. | |
Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. | |
Tefferi A, Lasho TL, Jimma T, et al. One thousand patients with primary myelofibrosis: the Mayo Clinic experience. Mayo Clin Proc. 2012; 87(1):25–33. | |
Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–397. | |
Gregory SA, Mesa RA, Hoffman R, Shammo JM. Clinical and laboratory features of myelofibrosis and limitations of current therapies. Clin Adv Hematol Oncol. 2011;9(9 Suppl 22):1–16. | |
Abdel-Wahab OI, Levine RL. Primary myelofibrosis: update on definition, pathogenesis, and treatment. Annu Rev Med. 2009;60:233–245. | |
Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): consensus on terminology by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT). Leuk Res. 2007;31(6):737–740. | |
Mascarenhas J, Mughal TI, Verstovsek S. Biology and clinical management of myeloproliferative neoplasms and development of the JAK inhibitor ruxolitinib. Curr Med Chem. 2012;19(26):4399–4413. | |
Incyte Corporation. Jakafi® [prescribing information]. 2013. Available from: http://www.incyte.com/sites/default/files/Jakafi_PI.pdf. Accessed September 24, 2013. | |
Canadian Newswire. Jakavi the first medication to receive Health Canada approval to treat patients with myelofibrosis [press release]. Dorval, QC: CNW. July 5, 2012. Available from: http://www.newswire.ca/en/story/1003655/-pr-jakavi-the-first-medication-to-receive-health-canada-approval-to-treat-patients-with-myelofibrosis. Accessed July 9, 2013. | |
Novartis. Novartis drug Jakavi first medication to receive European Commission approval to treat patients with myelofibrosis [press release]. Basel, Switzerland: Novartis; August 28, 2012. Available from: http://www.novartis.com/newsroom/media-releases/en/2012/1636508.shtml. Accessed July 9, 2013. | |
Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012; 366(9):799–807. | |
Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. | |
Verstovsek S, Mesa RA, Gotlib J, et al. Efficacy, safety and survival with ruxolitinib treatment in patients with myelofibrosis: results of a median 2-year follow-up of COMFORT-I. Haematologica. Epub Sept 13, 2013. | |
Cervantes F, Kiladjian JJ, Niederwieser D, et al. Long-term safety, efficacy, and survival findings from Comfort-II, a phase 3 study comparing ruxolitinib with best available therapy (BAT) for the treatment of myelofibrosis (MF) [abstract]. Blood. 2012;120(21):801. | |
Ward AC, Touw I, Yoshimura A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood. 2000;95(1):19–29. | |
Mesa RA, Gotlib J, Gupta V, et al. Effect of ruxolitinib therapy on myelofibrosis-related symptoms and other patient-reported outcomes in COMFORT-I: a randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2013;31(10):1285–1292. | |
Talpaz M, Paquette R, Afrin L, et al. Efficacy, hematologic effects, and dose of ruxolitinib in myelofibrosis patients with low starting platelet counts (50–100 × 109/L): A comparison to patients with normal or high starting platelet count [abstract]. Blood. 2012;120(21):176. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.