")
Back to Journals » International Journal of General Medicine » Volume 7
Management of cancer pain: 1. Wider implications of orthodox analgesics
Authors Lee S, Dawson J, Lee J, Osman G, Levitin M, Guzel R, Djamgoz M
Received 30 December 2012
Accepted for publication 21 May 2013
Published 7 January 2014 Volume 2014:7 Pages 49—58
DOI https://doi.org/10.2147/IJGM.S42187
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Susannah K Lee,1 Jill Dawson,2 Jack A Lee,3 Gizem Osman,4 Maria O Levitin,5 Refika Mine Guzel,5 Mustafa BA Djamgoz5,6
1Pomona College, Claremont, CA, USA; 2Healthcare Communications Consultancy, Danville, CA, USA; 3College of Arts and Sciences, Vanderbilt University, Nashville, TN, USA; 4Department of Chemical Engineering, Loughborough University, Loughborough, UK; 5Division of Cell and Molecular Biology, Neuroscience Solutions to Cancer Research Group, South Kensington Campus, Imperial College London, London, UK; 6Cyprus International University, Biotechnology Research Centre, Haspolat, North Cyprus, Mersin, Turkey
Abstract: In this review, the first of two parts, we first provide an overview of the orthodox analgesics used commonly against cancer pain. Then, we examine in more detail the emerging evidence for the potential impact of analgesic use on cancer risk and disease progression. Increasing findings suggest that long-term use of nonsteroidal anti-inflammatory drugs, particularly aspirin, may reduce cancer occurrence. However, acetaminophen may raise the risk of some hematological malignancies. Drugs acting upon receptors of gamma-aminobutyric acid (GABA) and GABA “mimetics” (eg, gabapentin) appear generally safe for cancer patients, but there is some evidence of potential carcinogenicity. Some barbiturates appear to slightly raise cancer risks and can affect cancer cell behavior in vitro. For cannabis, studies suggest an increased risk of squamous cell carcinoma of the tongue, larynx, and possibly lung. Morphine may stimulate human microvascular endothelial cell proliferation and angiogenesis; it is not clear whether this might cause harm or produce benefit. The opioid, fentanyl, may promote growth in some tumor cell lines. Opium itself is an emerging risk factor for gastric adenocarcinoma and possibly cancers of the esophagus, bladder, larynx, and lung. It is concluded that analgesics currently prescribed for cancer pain can significantly affect the cancer process itself. More futuristically, several ion channels are being targeted with novel analgesics, but many of these are also involved in primary and/or secondary tumorigenesis. Further studies are needed to elucidate possible cellular and molecular effects of orthodox analgesics and their possible long-term impact, both positive and negative, and thus enable the best possible clinical gain for cancer patients.
Keywords: NSAIDs, cannabinoids, opioids, GABA-ergic drugs, GABA mimetics, ion channels
Introduction
Cancer incidence worldwide is rising, with a recent estimate predicting an increase in all cancers from 12.7 million new cases in 2008 to 22.2 million by 2030.1 The five most frequently occurring cancers are those of lung, breast, colorectum, stomach, and prostate.2 Cancer represents a broad group of pathophysiologies, typically starting with uncontrolled multiplication of cells, giving rise to a primary tumor. Secondary tumorigenesis (metastasis) may then follow, although this can be independent of the initial proliferative activity. The overall process is strongly epigenetic, in which external factors (chemical, physical, and biological) play significant roles. The chance of developing cancer increases with age, likely due to an overall accumulation of risk factors coupled with a tendency for cellular repair mechanisms to become less effective. The most life-threatening aspect of cancer is metastasis in which tumor cells break away from the primary lesion and spread around the body via the bloodstream or lymphatic system. The cancer cells that survive, ultimately reaching distant sites, either remain dormant as micro-metastases or re-proliferate to form secondary tumors, in organs such as lungs, liver, brain, and bones (Figure 1).3,4 Importantly, metastasis depends upon two-way interactions between the cancer cells themselves and the surrounding stroma. Thus, the biochemical makeup of the tumor micro-environment plays a crucial role in cancer progression.
Pain follows as an integral part of the disruptive nature of cancer growth (primary or secondary). This can seriously diminish patient quality of life and be a major cause of fear.5 Cancer-related pain is estimated to affect some 9 million people worldwide each year either as the direct result of tumor development (75%–80% of patients) or the indirect side-effect of treatment (15%–20%).6 The prevalence of pain in cancer is estimated at 25% (newly diagnosed), 33% (undergoing treatment), more than 75% (advanced disease), and 33% (post-treatment).7 As many as 90% of patients with advanced cancer suffer from debilitating chronic pain, which can be hard to treat, leading to increased morbidity, mental health problems, such as depression, and significantly reduced quality of life.8
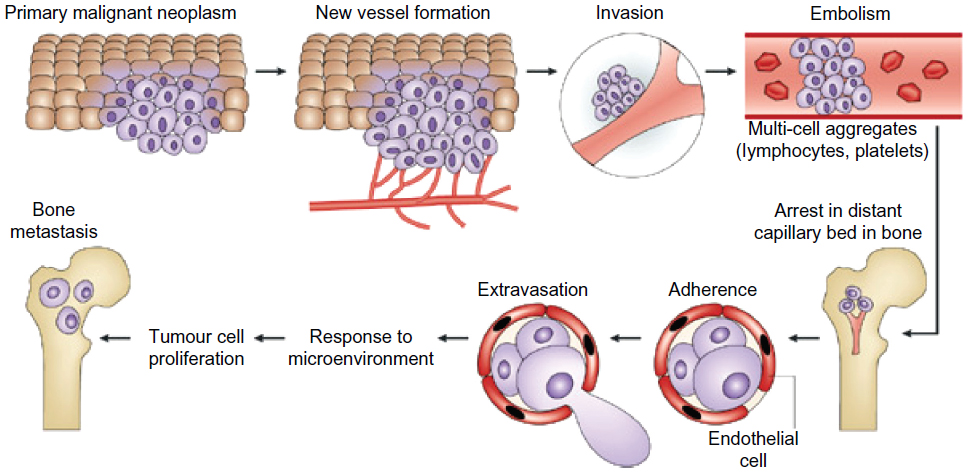 | Figure 1 The steps involved in tumor-cell metastasis from a primary site to the skeleton. Each of these steps represents a potential therapeutic target to reverse or prevent metastatic bone disease. The primary malignant neoplasm promotes new blood-vessel formation, and these blood vessels carry the cancer cells to capillary beds in bone. Aggregates of tumor cells and other blood cells eventually form embolisms that arrest in distant capillaries in bone. These cancer cells can then adhere to the vascular endothelial cells to escape the blood vessels. As they enter the bone, they are exposed to factors of the microenvironment that support growth of metastases. |
The pathophysiology of cancer pain is complex, involving inflammatory, neuropathic, ischemic and compression mechanisms that can occur at multiple sites. Cancer pain can result from the growing primary tumor putting pressure on nerves and bones and/or as nearby tissues are destroyed. Secondary tumors may also cause pain, as in the case of bone metastases, which occur in 90% of patients with advanced cancers of breast, prostate, or lung.9 In particular, cytokines produced by the tumor cells and/or the cells in the bone micro-environment activate osteoclasts and thus contribute to the signaling of cancer-associated pain in bones.10 Compression of the spinal cord occurs when a tumor grows on the spine. Visceral and neuropathic pain are also hallmarks of cancer; visceral pain can result when tumors obstruct the bowel or enlarge the liver capsule. Neuropathic pain arises following injury to peripheral or central neurons. Additional pain may result from diagnostic tests (biopsies, etc) and therapies, including cytotoxic chemotherapy and radiotherapy treatments that often produce peripheral neuropathies and other types of pain.
It is becoming increasingly apparent that there are individual and gender differences in the way pain is perceived, and that its severity does not always correlate with the severity of tissue damage.11 For example, around 20% of patients do not respond to morphine, or experience excessive side effects, or both.12 Such variability highlights the importance of developing more personalized treatments.
The widespread incidence of pain among cancer patients and survivors necessitates its management in as many different ways as possible. This is a two-part review dealing with this topic. In Part 1, we first give an overview of the orthodox pharmaceutical therapies used to treat cancer pain. Then we review in more detail the more recent evidence suggesting that some common analgesics given to cancer patients may also have an impact (negative or positive) upon the cancer process itself. As far as we are aware, this issue is being reviewed broadly for the first time here. In Part 2, we discuss the range of non-pharmaceutical treatments that may be integrated with conventional therapies for control of cancer pain.
Orthodox analgesics given to cancer patients: an overview
A range of pharmacological therapies is available for control of cancer pain. Non-opioid analgesics – acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs), such as aspirin and ibuprofen – are well established and used commonly as single agents to control mild to moderate cancer pain.13 When used in combination with opioids, these products may provide additive pain relief, allowing the opioid dose to be reduced, and resulting in fewer opioid-related side effects.
Gamma-aminobutyric acid (GABA) is a major inhibitory neurotransmitter synthesized and released by neurons mainly in the central nervous system. Benzodiazepines, such as valium, which impinge upon GABAA receptors and enhance GABAergic signaling, are widely used as analgesics.14 Gabapentin, a GABA “mimetic” (ie, it shares some structural homology with GABA), is an effective analgesic commonly prescribed to cancer patients to reduce neuropathic and postoperative pain. Gabapentin is not toxic at high clinical doses, is thought to have few side effects and does not interact adversely with other drugs.15 Although a GABA analog, gabapentin acts mainly on the α2δ subunit of voltage-gated calcium channel (VGCC).16 Much work has been conducted on the effectiveness of gabapentin in reducing neuropathic pain in both animal models and humans. Pregabalin, which has a similar mode of action to gabapentin, is also prescribed extensively to control neuropathic and postoperative pain, and multiple trials have shown that it is safe and effective.17
Cannabinoids, effective against chronic neuropathic pain, can be included among adjuvant medications for palliative care of cancer patients. For example, a novel cannabinoid formulation, nabiximols, may be a useful add-on analgesic for patients with opioid-refractory cancer pain.18 A randomized, double-blind, placebo-controlled, graded-dose study on 360 patients with advanced cancer showed that nabiximols is efficient and safe at low to medium doses. A recent, small-scale study of 12 healthy male volunteers suggests that the psychoactive ingredient of cannabis can make the experience of pain more tolerable rather than reducing the intensity of the pain.19 The authors used functional magnetic resonance imaging to examine the effects of the naturally occurring cannabinoid, delta-9-tetrahydrocannabinol, on brain activity associated with cutaneous pain and hyperalgesia temporarily induced using capsaicin. The analgesic effect on hyperalgesia was linked to diminished activity in the cingulate cortex, whilst the drug-induced reduction in “unpleasantness” was correlated with activity in the right amygdala, known to be primed by pain.
Generally used for moderate to severe pain, opioids include morphine, hydromorphone, oxycodone, hydrocodone, codeine, fentanyl, and methadone. These are the mainstay of the World Health Organization three-step analgesic “ladder,” first proposed in 1986, which provides some 71%–100% of patients with adequate analgesia when used appropriately.9 Various other guidelines are in place.20 However, up to 30% of patients do not respond to morphine, or experience excessive side effects, or both.21 In the multicenter European Pharmacogenetic Opioid Study, opioids were evaluated in pain management of 22 mesothelioma and 373 lung cancer patients receiving 30–960 mg and 10–5,072 mg, respectively.22 However, only 18% (four) of mesothelioma and 10% (seven) of lung cancer patients reported complete pain relief with opioids. Combining administration of methadone with another opioid to patients with moderate to severe pain resulted in improved pain control and seems safe, well tolerated, and practical.23
A novel formulation, fentanyl pectin nasal spray, is showing promise in treatment of breakthrough cancer pain.24,25 The pectin-based delivery helps modulate fentanyl’s transmucosal absorption, and the nasal formulation achieves a greater maximum plasma concentration than oral transmucosal fentanyl products, and at a much quicker rate. Throughout all phases of clinical studies, the fentanyl pectin nasal was shown to be safe and effective at doses between 100 and 800 μg per breakthrough pain episode. For the various oral formulations of fentanyl, a literature review identified relevant studies for a mixed-treatment meta-analysis to indirectly compare fentanyl preparations, morphine, and placebo for the treatment of breakthrough cancer pain.26 While fentanyl preparations provided better pain relief than placebo in the first half-hour after dosing, oral morphine performed little better than placebo.
A transdermal fentanyl matrix patch also exists, and was tested for up to 2 weeks in 474 Chinese patients with moderate-to-severe cancer pain.27 The study authors concluded that the product was safe and effective, and resulted in significant improvements in quality of life.
Denosumab is a monoclonal antibody drug that binds to RANKL, a protein involved in the formation, function, and survival of osteoclasts. Denosumab was evaluated against zoledronic acid in a randomized, double-blind Phase 3 clinical study of pain in 2,046 advanced breast cancer patients with bone metastases.28 The authors concluded that denosumab showed improved pain prevention and similar pain palliation when compared with zoledronic acid; also, fewer individuals receiving denosumab transitioned to the use of strong opioid analgesics.
Adjuvant analgesics (drugs with a primary indication other than pain) include a wide range of drugs and are normally combined with an opioid regimen to treat painful conditions.29 Some adjuvant analgesics that are useful in several painful conditions are termed “multipurpose adjuvant analgesics” (such as antidepressants, corticosteroids, α2-adrenergic agonists and neuroleptics). Others are specific for neuropathic pain (local anesthetics, anticonvulsants, N-methyl-D-aspartate [NMDA] receptor antagonists), bone pain (bisphosphonates, calcitonin, radiopharmaceuticals), musculoskeletal pain (muscle relaxants), or pain from bowel obstruction (anticholinergics, octreotide). Adjuvant medications include steroids, anxiolytics, antidepressants, hypnotics, anticonvulsants, antiepileptic-like gabapentinoids (gabapentin and pregabalin), membrane stabilizers, voltage-gated sodium channel (VGSC) blockers, and NMDA receptor antagonists for the treatment of neuropathic pain.30
Possible impact of orthodox analgesics on cancer progression
Clearly, cancer and pain are intimately associated, and a variety of analgesics and their combinations are used to control cancer pain. The question arises, therefore, whether painkillers prescribed routinely to cancer patients could themselves affect the cancer process. The current evidence is examined below.
NSAIDs
These generally inhibit the enzyme, cyclooxygenase-2, which normally causes swelling in response to inflammation and signals pain. Importantly, NSAIDs have also been associated with inhibition of cancer cell proliferation and metastasis.31,32 In particular, regular use of aspirin has been demonstrated consistently to reduce the incidence of numerous cancers, leading to the possibility of its usefulness as a preventative and/or therapeutic agent for cancer. In several randomized trials, daily use of aspirin was found to reduce the long-term (20-year) risk of developing several cancers and metastatic disease.33–35 A risk reduction of around 10% was found among prostate cancer patients with slow-growing or aggressive tumors; a few case-control studies have indicated a favorable effect of aspirin on bladder cancer, although a modest but nonsignificant increased risk was reported for kidney cancer.36
A large prospective cohort study of 64,839 people aged 50–76 years found that high doses of acetaminophen (more than 4 days per week for 4 or more years) was associated with an almost twofold increase in the risk of hematological malignancies other than chronic lymphocytic leukemia and small lymphocytic lymphoma.37 There was no association of increasing use of aspirin, nonaspirin NSAIDs, or ibuprofen with raised risk of incident hematological malignancies.
A recent meta-analysis by Choueiri et al38 concluded that acetaminophen and nonaspirin NSAIDs were associated with a significant risk of developing kidney cancer. The authors evaluated 20 studies (14 for acetaminophen, 13 for aspirin, and five for other NSAIDs) done in six countries and including 8,420 cases of kidney cancer. Use of acetaminophen and nonaspirin NSAIDs were associated with an increased risk of kidney cancer. For aspirin use, no overall increased risk was identified.
In conclusion, although there is a general trend for long-term NSAID use to reduce cancer incidence, adverse effects can also occur.
GABAergic drugs and GABA “mimetics”
There is some evidence that deregulation of the GABA system can cause or contribute to the onset of cancer.39 Indeed, some tumor characteristics, such as uncontrolled proliferation, can be induced by GABA itself.40 For example, Azuma et al41 and Takehara et al42 reported that GABA promoted prostate and pancreatic cancer. Azuma et al studied expression of GABA, glutamate decarboxylase, and matrix metalloproteinase (MMP) in the prostates of individuals with cancer or benign prostatic hypertrophy using immunohistochemical approaches. They concluded that raised expression of GABA may be involved in metastasis by promoting production of MMP in cancer cells, and that the GABAB receptor pathway may be involved in the process. Takehara et al42 used the genome-wide cDNA microarray analysis of pancreatic ductal adenocarcinoma (PDAC) cells, along with reverse transcription-polymerase chain reaction and Northern blot analyses, to detect overexpression of GABA receptor pi subunit (GABRP) in PDAC cells. The authors suggest that GABA and GABRP could have a key role in PDAC development and progression, a pathway that might hold promise as a molecular target for new therapies.
In contrast, Ortega43 proposed a generally inhibitory role of GABA on tumor cell migration. Overexpression of the ionotropic GABAA receptor in PDAC cells stimulated proliferative activity, dependent upon the π subunit.44 Similarly, GABA induced proliferation of undifferentiated gastric carcinoma (KATO III) cells and increased their invasiveness.45 On the other hand, Zafrakas et al46 found that the GABA π subunit downregulated tumor cell progression in breast cancer but this effect was thought to be independent of GABAA receptor function.
Al-Wadei et al47 suggest that targeted inhibition of stress-induced pathways may hold promise as an intervention to improve prognosis in PDAC. The authors noted that the poor prognosis of PDAC could be linked to psychological distress, promoted by beta-adrenergic signaling and that GABA inhibits these responses in in-vitro and in-vivo mouse models of PDAC. Thus, according to Al-Wadei et al,47 epinephrine-induced increase in proliferation and migration of PDAC cells was inhibited in a concentration-dependent way by the cyclooxygenase-2 inhibitor, celecoxib, and this effect was significantly increased by co-treatment with GABA.
There are several analgesic and mood-adjusting drugs that act upon GABAA receptors, especially benzodiazepines and barbiturates,48 and promote GABAergic signaling. Some studies point to an antiproliferative effect of benzodiazepines, while others have demonstrated a link to neoplastic progression.49 However, the generally accepted clinical view is that benzodiazepine use is not associated with an overall cancer risk.50 Although barbiturates are not normally administered to cancer patients for pain, they can be used against seizure disorders and as anesthetics.As with benzodiazepines, there is some controversy in the literature over the possible cancer risk of barbiturates. Some studies have shown a weak positive association of barbiturate use with cancer, while others have revealed antiproliferative and potential anticancer effects.51
As regards cell behaviors related to cancer, one study found gabapentin to be a low-level mitogen on isolated pancreatic cells of rats (Figure 2), although the effect was harder to detect in vivo.52 Also, gabapentin-lactam, a derivative of gabapentin (but not gabapentin itself) induced dendritic filopodia and increased motility in cultured hippocampal neurons.53 In contrast, gabapentin did not affect the proliferative activity of pancreatic tumor (AR42J) cells (Figure 2). Gabapentin induced pancreatic acinar cell neoplasia in male rats after a 2-year exposure, but this was not found be the case in mice.54 These results indicate that the effects of gabapentin may be gender-, species-, and dose-specific. In a case-control screening study of the possible association of gabapentin with 55 cancers, the only cancer that met the screening criteria for a possible increase was renal (including renal pelvis) cancer.55
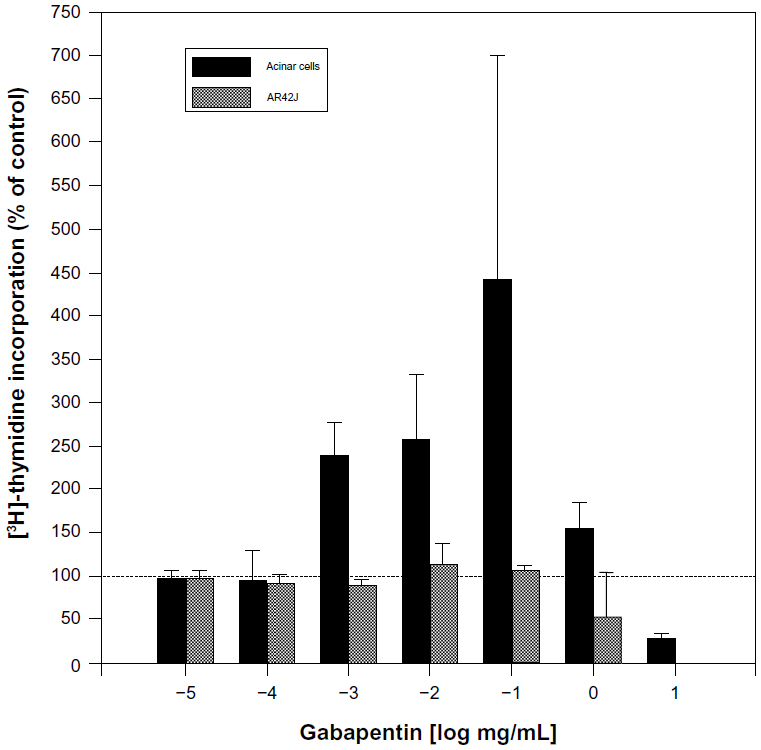 | Figure 2 Dose-dependent effects of gabapentin on pancreatic cell proliferation measured by uptake of 3H-thymidine. Compared with untreated cells, gabapentin induced proliferation of normal pancreatic acinar cells by 140%–220% after 18-hour incubations. The effect was statistically significant for 1 and 10 μg/mL. In contrast, gabapentin had no effect on proliferation of pancreatic cancer AR42J cells. Similar to the normal acinar cells, thymidine incorporation in AR42J cells decreased below control levels at gabapentin concentrations of 1 mg/mL and above, probably due to cytotoxicity at the high concentrations. |
For pregabalin, Criswell et al56 found that this drug induced proliferation in mammalian endothelial cells in a species-specific manner. Also, other studies have shown that pregabalin increased incidence of hemangiosarcomas in mice but, again, that this was species-specific and, at clinical doses, there was no proliferative effect on various mammalian cells.57–59
In conclusion, although it would appear that GABAergic drugs and GABA “mimetics” are generally safe to use against cancer pain, the possibility of carcinogenicity, albeit small, cannot completely be ruled out at present, and should be carefully investigated considering their broad usage. In particular, for gabapentin, a commonly prescribed analgesic against cancer pain, it would seem essential to (1) perform further in-vitro mechanistic and in-vivo studies on human cancer cells to elucidate its possible cellular and molecular effects; and (2) monitor possible adverse effects of its long-term use by cancer patients.
Cannabinoids and opioids
Several publications have suggested a relationship between cannabis use and certain types of cancer.60 In users under 40 years of age, cannabis has been suspected of increasing the risk of squamous cell carcinoma of tongue and larynx and possibly of lung, in dose-dependent fashion. Promotion of nonlymphoblastic acute leukemia and astrocytoma have also been suspected. An increased risk of head and neck cancer was observed in long-term (years) regular (more than once a day) smokers of cannabis. Epidemiological studies are needed to further clarify the apparent effects of cannabinoids on cancer.
A study in a mouse model suggested that cannabinoid receptor 2 agonists have potential as a novel treatment for breast cancer-induced bone pain, potentially achieving disease modifications including reduced bone loss, suppressed cancer growth, attenuation of severe bone pain, and increased survival – apparently without the major side effects of current therapeutic options such as opiates.61 The inhibition of breast cancer proliferation was attributed to cytokine/chemokine suppression. In vitro, a cannabinoid receptor 2 agonist, JWH015, was found to reduce cancer cell proliferation and lower levels of inflammatory mediators that have been shown to promote pain, bone loss, and proliferation.
Following an initial report showing that morphine stimulates human microvascular endothelial cell proliferation and angiogenesis in vitro and in vivo,62 much work has been done to determine the potential impact of morphine on cancer. A recent paper by Qin et al63 highlighted the “double-edged sword” of morphine’s impacts on cancer. These authors noted that although morphine may have potential to induce angiogenesis, when human gastric carcinoma MCG-803 cells were incubated with morphine, growth and proliferation of the cells were inhibited. Furthermore, the cells underwent morphological changes consistent with induction of apoptosis. A recent paper by Gong et al64 hypothesized that morphine might induce cancer recurrence by disturbing the behavior of regulatory T cells, possibly through vascular endothelial growth factor receptor 2 and opioid receptors. These authors proposed that morphine affected neoplastic tissues by modulating immune responses and promoting angiogenesis, and that morphine might affect regulatory T cells by modulating the function of other immune cells or cytokines, such as tumor growth factor-β and interleukin-2.
The opioid, fentanyl, which is used to control cancer pain, could also affect tumor growth in many cell lines.65 These authors examined the impact of fentanyl on growth of human gastric carcinoma MGC-803 cells and expression of apoptosis-related genes such as nuclear factor-kappa-B and PTEN. It was found that fentanyl inhibited cell growth and proliferation, and led to arrest of the cell cycle at the G2/M phase. The cells also had a higher rate of apoptosis and appeared less motile.
Opium is also an emerging risk factor for gastric adenocarcinoma according to Shakeri et al.66 The authors found that long-term use of opium was linked to a higher risk of cancers of the esophagus, bladder, larynx, and lung. Their study enrolled 309 cases of gastric adenocarcinoma in Iran, and 613 matched controls. The effect was dose dependent.
In conclusion, it is impossible to conclude from the existing data whether the morphine-induced changes in tumor cell proliferation and invasion, inflammation, angiogenesis, and immune response might result in harm or benefit and both seem possible.67,68 This is likely to be due to the complexity of cellular pathways and physiological responses elicited by opioids (endogenous and exogenous) in vitro and in vivo (Figure 3). Among the factors influencing the effect of morphine on metastasis are dose, duration, the route of administration, possible receptor desensitization/withdrawal effects, central versus peripheral actions, and the models employed, which can differ markedly in their opioid receptor expression, characteristics, and metabolism. Clearly, further work is required to elucidate the possible impacts of morphine in cancer patients.
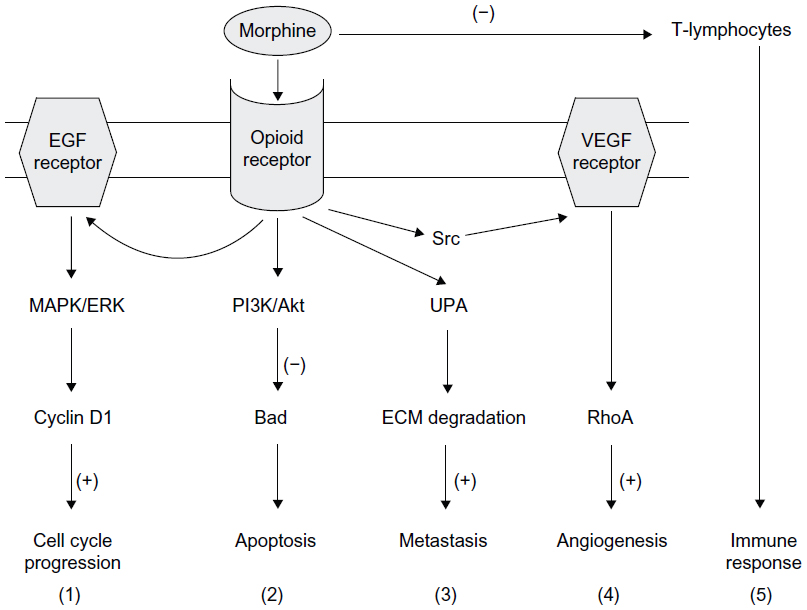 | Figure 3 Possible mechanisms of opioid receptor-mediated influence of morphine on tumor growth. Morphine binds to the μ-opioid receptor and stimulates a range of signaling pathways, and can thus promote different aspects of the cancer process as follows: (1) MAPK/ERK pathway, promoting cell cycle progression; (2) PI3K/Akt pathway, mediating anti-apoptotic effects; (3) uPA expression upregulation and secretion, promoting metastasis; (4) transactivation of VEGF receptors which induces angiogenesis; and (5) suppression of the function of T lymphocytes, leading to immunosuppression. (+) and (−) denote stimulation and inhibition, respectively. |
Novel areas of research
The pharmacology of pain remains a very active field of research and development, and new analgesics and targets are being evaluated all the time in three complementary areas. First, there is continuing interest in the role of ion channels in analgesia research.69 Currently, ion channels, including VGCCs (especially Cav2.2 and Cav3.2) and VGSCs, eg, Nav1.7 and Nav1.8, are undergoing scrutiny as novel analgesic targets.70,71 Venom peptides from marine cone snails can target VGCC subtypes specifically, and a conotoxin-based drug (ziconotide), selectively inhibiting Cav2.2 and inducing analgesia in chronic pain states, has reached the market. However, its mode of administration (intrathecal) and adverse side-effects have restricted its clinical use. Novel Cav2.2 inhibitors are in development and may overcome the existing limitations. Importantly, both VGCC and VGSC signaling have also been implicated in the cancer process.72,73 In particular, functional upregulation of VGSCs occurs in carcinoma progression, VGSC activity promotes metastatic cell behaviors, and VGSCs have been proposed as viable targets for controlling metastasis.74 Accordingly, possible future use of VGSC blockers against cancer pain may produce significant additional benefit by suppressing metastasis. A variety of transient receptor potential (TRP) channels are also involved in both pain signaling75 and cancer.76 Second, G-protein coupled receptors, including purinergic P2X and P2Y receptors, represent novel targets for analgesics.77,78 These receptors also are involved in cancer.79,80 Third, growth factors can also signal pain, modulate ion channel expression at a hierarchy of levels and can play a significant role in the cancer process itself. A particular analgesic focus is nerve growth factor and its receptors.81,82 Interestingly, also, use of local anesthetics, such as lidocaine, during cancer surgery can subsequently lead to lesser pain, reduced dependence on morphine, and even a reduced chance of the cancer reoccurring due to the suppression of the “showering” of tumor cells during the surgery.83,84
Conclusion
In overall conclusion, cancer pain, originating from a variety of sources in the body, continues to be a major problem in reducing the quality of life of cancer patients and survivors. Although there are many orthodox analgesics in use for treatment of pain, most of these are only partially effective (less than 30% in placebo-controlled trials) and many have unpleasant side-effects (eg, gastrointestinal, and cognitive) and abuse potential.85 New pharmacological approaches to pain management are being developed based on our evolving understanding of the mechanisms involved in pain. Finally, importantly, the potential of common analgesics (in current or future use) to impact upon cancer development and progression, both positively and negatively, is not just intriguing but needs further investigation and continued clinical evaluation.
Disclosure
The authors report no conflicts of interest in this work.
References
Bray F, Jemal A, Grey N, Ferlay J, Forman D. Global cancer transitions according to the Human Development Index (2008–2030): a population-based study. Lancet Oncol. 2012;13(8):790–801. | |
Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19(8):1893–1907. | |
Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. | |
Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2011;4:448–456. | |
Williams AC de C, Gessler S. Empathy in cancer pain. In: Pace JA, Bell RF, Kalso EA, Soyannwo OA, editors. Cancer Pain: From Molecules to Suffering. Washington, DC: International Association for the Study of Pain; 2010: chapter 16. | |
Ripamonti C, Longo M. Cancer pain syndromes and pharmacotherapy of cancer pain. Arch Oncol. 2004;12(3):168–170. | |
Paice JA, Ferrell B. The management of cancer pain. CA Cancer J Clin. 2011;61(3):157–182. | |
Sabiston CM, Brunet J, Burke S. Pain, movement, and mind: does physical activity mediate the relationship between pain and mental health among survivors of breast cancer? Clin J Pain. 2012;28(6):489–495. | |
Anand U. Mechanisms and management of cancer pain. In: Alison MR, editor. The Cancer Handbook. 2nd ed. Chichester, UK: John Wiley & Sons; 2007. | |
Theriault RL, Theriault RL. Biology of bone metastases. Cancer Control. 2012;19(2):92–101. | |
Greenspan JD, Craft RM, LeResche L, et al; the Consensus Working Group of the Sex, Gender, and Pain SIG of the IASP. Studying sex and gender differences in pain and analgesia: a consensus report. Pain. 2007;132(Suppl 1):S26–S45. | |
Fallon M. When morphine does not work. Support Care Cancer. 2008;16:771–775. | |
Portenoy RK, Ahmed E, Keilson YY. Cancer pain management: use of acetaminophen and nonsteroidal anti-inflammatory drugs. UpToDate. May 1, 2012. 18. | |
Costa E, Guidotti A, Mao CC, Suria A. New concepts on the mechanism of action of benzodiazepines. Life Sci. 1975;17:167–185. | |
Dworkin RH, Backonja M, Rowbotham MC, et al. Advances in neuropathic pain: diagnosis, mechanisms, and treatment recommendations. Arch Neurol. 2003;60:1524–1534. | |
Hendrich J, Van Minh AT, Heblich F, et al. Pharmacological disruption of calcium channel trafficking by the α2δ ligand gabapentin. Proc Natl Acad Sci U S A. 2008;105:3628–3633. | |
Gilron I. Gabapentin and pregabalin for chronic neuropathic and early postsurgical pain: current evidence and future directions. Curr Opin Anaesthesiol. 2007;20:456–472. | |
Portenoy RK, Ganae-Motan ED, Allende S, et al. Nabiximols for opioid-treated cancer patients with poorly-controlled chronic pain: a randomized, placebo-controlled, graded-dose trial. J Pain. 2012;13(5):438–449. | |
Lee MC, Ploner M, Wiech K, et al. Amygdala activity contributes to the dissociative effect of cannabis on pain perception. Pain. 2013;154(1):124–134. | |
National Institute for Health and Clinical Excellence. Opioids in Palliative Care: Safe and Effective Prescribing of Strong Opioids for Pain in Palliative Care of Adults. Clinical Guideline 140. Available from: http://publications.nice.org.uk/opioids-in-palliative-care-safe-and-effective-prescribing-of-strong-opioids-for-pain-in-palliative-cg140. Accessed August 23, 2013. | |
Cherny N, et al. (2001). Strategies to manage the adverse effects of oral morphine: an evidence based report. Journal of Clinical Oncology, 19, 2542–2554. Cited in Uma Anand. Mechanisms and Management of Cancer Pain. The Cancer Handbook, 2nd Edition. Edited by Malcolm R Alison. 2007 John Wiley & Sons, Ltd. | |
Salminen EK, Silvoniemi M, Syrjänen K, Kaasa S, Kloke M, Klepstad P. Opioids in pain management of mesothelioma and lung cancer patients. Acta Oncol. 2013;52(1):30–37. | |
Wallace E, Ridley J, Bryson J, Mak E, Zimmermann C. Addition of methadone to another opioid in the management of moderate to severe cancer pain: a case series. J Palliat Med. 2013;16(3):305–309. | |
Bulloch MN, Hutchison AM. Fentanyl pectin nasal spray: a novel intranasal delivery method for the treatment of breakthrough cancer pain. Expert Rev Clin Pharmacol. 2013;6(1):9–22. | |
Escobar Y, Mañas A, Juliá J, et al. Optimal management of breakthrough cancer pain (BCP). Clin Transl Oncol. Epub December 21, 2012. | |
Jandhyala R, Fullarton JR, Bennett MI. Efficacy of rapid-onset oral fentanyl formulations vs oral morphine for cancer-related breakthrough pain: a meta-analysis of comparative trials. J Pain Symptom Manage. February 1, 2013. pii: S0885–3924(12)00815–00819. | |
Zhu YL, Song GH, Liu DQ, et al. Multicenter clinical study for evaluation of efficacy and safety of transdermal fentanyl matrix patch in treatment of moderate to severe cancer pain in 474 Chinese cancer patients. Chin J Cancer Res. 2011;23(4):317–322. | |
Cleeland CS, Body JJ, Stopeck A, et al. Pain outcomes in patients with advanced breast cancer and bone metastases: results from a randomized, double-blind study of denosumab and zoledronic acid. Cancer. 2013;119(4):832–838. | |
Lussier D, Huskey AG, Portenoy RK. Adjuvant analgesics in cancer pain management. Oncologist. 2004;9:571–591. | |
Vargas-Shaffer G. Is the WHO analgesic ladder still valid? Twenty-four years of experience. Can Fam Physician. 2010;56(6):514–517. | |
Grösch S, Maier TJ, Schiffmann S, Geisslinger G. Review: Cyclooxygenase-2 (COX-2) – independent anticarcinogenic effects of selective COX-2 inhibitors. J Nat Cancer Inst. 2006;98(11):736–747. | |
Garcia Rodriguez LA, Cea-Soriano L, Tacconelli S, Patrignani P. Coxibs: pharmacology, toxicity and efficacy in cancer clinical trials. Recent Results Cancer Res. 2013;191:67–93. | |
Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012;13(5):518–527. | |
Rothwell PM, Wilson M, Price JF, Belch JFF, Meades TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591–1601. | |
Rothwell PM, Price JF, Fowkes FGR, et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet Oncol. 2012;379:1602–1612. | |
Bosetti C, Rosato V, Gallus S, La Vecchia C. Aspirin and urologic cancer risk: an update. Nat Rev Urol. 2012;9(2):102–110. | |
Walter RB, Milano F, Brasky TM, White E. Long-term use of acetaminophen, aspirin, and other nonsteroidal anti-inflammatory drugs and risk of hematologic malignancies: results from the Prospective Vitamins and Lifestyle (VITAL) study. J Clin Oncol. 2011;29(17):2424–2431. | |
Choueiri TK, Je Y, Cho E. Analgesic use and the risk of kidney cancer: a meta-analysis of epidemiologic studies. Int J Cancer. Epub February 7, 2013. | |
Roberts SS, Mori M, Pattee P, et al. GABAergic system gene expression predicts clinical outcome in patients with neuroblastoma. J Clin Oncol. 2004;22:4127–4134. | |
Takehara A, Hosokawa M, Eguchi H, et al. Gamma-aminobutyric acid (GABA) stimulates pancreatic cancer growth through overexpressing GABAA receptor P subunit. Cancer Res. 2007;67:9704–9712. | |
Azuma H, Inamoto T, Sakamoto T, et al. Gamma-aminobutyric acid as a promoting factor of cancer metastasis; induction of matrix metalloproteinase production is potentially its underlying mechanism. Cancer Res. 2003;63:8090–8096. | |
Takehara A, Hosokawa M, Eguchi H, et al. γ-aminobutyric acid (GABA) stimulates pancreatic cancer growth through overexpressing GABAA receptor π subunit. Cancer Res. 2007;67:9704–9712. | |
Ortega A. A new role for GABA: inhibition of tumor cell migration. Trends Pharmacol Sci. 2003;24:151–154. | |
Johnson SK, Dennis RA, Barone GW, Lamps LW, Haun RS. Differential expression of insulin-like growth factor binding protein-5 in pancreatic adenocarcinomas: identification using DNA microarray. Mol Carcinog. 2006;45(11):814–827. | |
Maemura K, Shiraishi N, Sakagami K, et al. Proliferative effects of γ-aminobutyric acid on the gastric cancer cell line are associated with extracellular signal-regulated kinase 1/2 activation. J Gastroenterol Hepatol. 2009;24;688–696. | |
Zafrakas M, Chorovicer M, Klaman I, et al. Systematic characterisation of GABRP expression in sporadic breast cancer and normal breast tissue. Int J Cancer. 2006;118(6):1453–1459. | |
Al-Wadei HA, Al-Wadei MH, Ullah MF, Schuller HM. Celecoxib and GABA cooperatively prevent the progression of pancreatic cancer in vitro and in xenograft models of stress-free and stress-exposed mice. PLoS One. 2012;7(8):e43376. | |
Löscher W, Rogawski MA. How theories evolved concerning the mechanism of action of barbiturates. Epilepsia. 2012;53 Suppl 8:12–25. | |
Miyawaki I, Moriyasu M, Funabashi H, Yasuba M, Matsuoka N. Mechanism of clobazam-induced thyroidal oncogenesis in male rats. Toxicol Lett. 2003;145:291–301. | |
Pottegård A, Friis S, Andersen M, Hallas J. Use of benzodiazepines or benzodiazepine related drugs and the risk of cancer: a population-based case-control study. Br J Clin Pharmacol. 2013;75(5):1356–1364. | |
Xie J, Li Y, Huang Y, et al. Anesthetic pentobarbital inhibits proliferation and migration of malignant glioma cells. Cancer Lett. 2009;282(1):35–42. | |
Dethloff L, Barr B, Bestervelt L, Bulera S, Sigler R, LaGattuta M. Gabapentin-induced mitogenic activity in rat pancreatic acinar cells. Toxicol Sci. 2000;55:52–59. | |
Henle F, Leemhuis J, Fischer C, et al. Gabapentin-lactam induces dendritic filopodia and motility in cultured hippocampal neurons. J Pharmacol Exp Ther. 2006;319:181–191. | |
Sigler RE, Gough AW, De La Iglesia FA. Pancreatic acinar cell neoplasia in male Wistar rats following 2 years of gabapentin exposure. Toxicology. 1995;98(1–3):73–82. | |
Friedman GD, Udaltsova N, Chan J, Quesenberry CP Jr, Habel LA. Screening pharmaceuticals for possible carcinogenic effects: initial positive results for drugs not previously screened. Cancer Causes Control. 2009;20(10):1821–1835. | |
Criswell KA, Cook JC, Morse D, et al. Pregabalin induces hepatic hypoxia and increases endothelial cell proliferation in mice, a process inhibited by dietary vitamin E supplementation. Toxicol Sci. 201;128:42–56. | |
Pegg D, Bleavins M, Herman J, et al. Hemangiosarcoma in mice administered pregabalin: analysis of genotoxicity, tumor incidence, and tumor genetics. Toxicol Sci. 2012;128:9–21. | |
Criswell KA, Wojcinski Z, Pegg D, et al. Key components of the mode of action for hemangiosarcoma induction in pregabalin-treated mice: evidence of increased bicarbonate, dysregulated erythropoiesis, macrophage activation, and increased angiogenic growth factors in mice but not in rats. Toxicol Sci. 2012;128:22–41. | |
Criswell KA, Cook JC, Wojcinski Z, et al. Mode of action associated with development of hemangiosarcoma in mice given pregabalin and assessment of human relevance. Toxicol Sci. 2012;128:57–71. | |
Carriot F, Sasco AJ. Cannabis and cancer. Rev Epidemiol Sante Publique. 2000;48:473–483. | |
Lozano-Ondoua AN, Hanlon KE, Symons-Liguori AM, et al. Disease modification of breast cancer-induced bone remodeling by cannabinoid 2 receptor agonists. J Bone Miner Res. 2013;28(1):92–107. | |
Gupta K, Kshirsagar S, Chang L, et al. Morphine stimulates angiogenesis by activating proangiogenic and survival-promoting signaling and promotes breast tumor growth. Cancer Res. 2002;62:4491–4498. | |
Qin Y, Chen J, Li L, et al. Exogenous morphine inhibits human gastric cancer MGC-803 cell growth by cell cycle arrest and apoptosis induction. Asian Pac J Cancer Prev. 2012;13(4):1377–1382. | |
Gong L, Dong C, Ouyang W, Qin Q. Regulatory T cells: a possible promising approach to cancer recurrence induced by morphine. Med Hypotheses. 2013;80(3):308–310. | |
Qin Y, Li L, Chen J, et al. Fentanyl inhibits progression of human gastric cancer MGC-803 cells by NF-kappaB downregulation and PTEN upregulation in vitro. Oncol Res. 2012;20(2–3):61–69. | |
Shakeri R, Malekzadeh R, Etemadi A, et al. Opium; an emerging risk factor for gastric adenocarcinoma. Int J Cancer. 2013;133(2):455–461. | |
Gach K, Wyrębska A, Fichna J, Janecka A. The role of morphine in regulation of cancer cell growth. Naunyn Schmiedebergs Arch Pharmacol. 2011;384(3):221–230. | |
Afsharimani B, Cabot P, Parat MO. Morphine and tumor growth and metastasis. Cancer Metastasis Rev. 2011;30:225–238. | |
Wood JN. Ion channels in analgesia research. Handb Exp Pharmacol. 2007;(177):329–358. | |
Vink S, Alewood PF. Targeting voltage-gated calcium channels: developments in peptide and small molecule inhibitors for the treatment of neuropathic pain. Br J Pharmacol. 2012;167:970–989. | |
Liu M, Wood JN. The roles of sodium channels in nociception: implications for mechanisms of neuropathic pain. Pain Med. 2011;12(Suppl 3):S93–S99. | |
Panner A, Wurster RD. T-type calcium channels and tumor proliferation. Cell Calcium. 2006;40:253–259. | |
Fraser SP, Diss JK, Chioni AM, et al. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin Cancer Res. 2005;11:5381–5389. | |
Onkal R, Djamgoz MB. Molecular pharmacology of voltage-gated sodium channel expression in metastatic disease: clinical potential of neonatal Nav1.5 in breast cancer. Eur J Pharmacol. 2009;625:206–219. | |
Premkumar LS, Abooj M. TRP channels and analgesia. Life Sci. 2013;92(8–9):415–424. | |
Ouadid-Ahidouch H, Dhennin-Duthille I, Gautier M, Sevestre H, Ahidouch A. TRP channels: diagnostic markers and therapeutic targets for breast cancer? Trends Mol Med. 2013;19(2):117–124. | |
Magni G, Ceruti S. P2Y purinergic receptors: new targets for analgesic and antimigraine drugs. Biochem Pharmacol. 2013;85(4):466–477. | |
North RA, Jarvis MF. P2X Receptors as drug targets. Mol Pharmacol. 2013;83(4):759–769. | |
Kłopocka W, Korczyński J, Pomorski P. Cytoskeleton and nucleotide signaling in glioma C6 cells. Adv Exp Med Biol. 2013;986:103–119. | |
Braganhol E, Wink MR, Lenz G, Battastini AM. Purinergic signaling in glioma progression. Adv Exp Med Biol. 2013;986:81–102. | |
Watson JJ, Allen SJ, Dawbarn D. Targeting nerve growth factor in pain: what is the therapeutic potential? BioDrugs. 2008;22:349–359. | |
Brackenbury WJ, Djamgoz MB. Nerve growth factor enhances voltage-gated Na+ channel activity and Transwell migration in Mat-LyLu rat prostate cancer cell line. J Cell Physiol. 2007;210:602–608. | |
Vigneault L, Turgeon AF, Côté D, et al. Perioperative intravenous lidocaine infusion for postoperative pain control: a meta-analysis of randomized controlled trials. Can J Anaesth. 2011;58(1):22–37. | |
Snyder GL, Greenberg S. Effect of anaesthetic technique and other perioperative factors on cancer recurrence. Br J Anaesth. 2010;105(2):106–115. | |
Borsook D. A future without chronic pain: neuroscience and clinical research. Cerebrum. May 2012;2012:7. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.