")
Back to Journals » Cancer Management and Research » Volume 11
Malignant Triton Tumor in a Child: Case Report and Literature Review
Authors Zhao A, Ding D, Li X, Wang J
Received 27 June 2019
Accepted for publication 14 November 2019
Published 24 December 2019 Volume 2019:11 Pages 10759—10766
DOI https://doi.org/10.2147/CMAR.S221110
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Harikrishna Nakshatri
Ailing Zhao,1–3,* Daling Ding,4,* Xueqin Li,1–3 Jiangtao Wang1–3
1Department of Infant Ward, Children’s Hospital Affiliated of Zhengzhou University, Zhengzhou, Henan 450018, People’s Republic of China; 2Department of Infant Ward, Henan Children’s Hospital, Zhengzhou, Henan 450018, People’s Republic of China; 3Department of Infant Ward, Zhengzhou Children’s Hospital, Zhengzhou, Henan 450018, People’s Republic of China; 4Department of Neurosurgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450052, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jiangtao Wang
Department of Infant Ward, Children’s Hospital Affiliated of Zhengzhou University, Zhengzhou, Henan 450018, People’s Republic of China
Email [email protected]
Objective: Malignant triton tumor (MTT) is a rare and devastating malignant peripheral nerve-sheath tumor, which shows rapid growth and poor clinical outcomes. Here, we reported a 2-year-old girl who was diagnosed as MTT, an overview of the literature was conducted to discuss the clinical features and optimal treatment strategies of MTT.
Methods: We conducted an analysis of 42 patients from the PubMed, Medline, Embase and Web of Science databases for relevant articles published between 1938 and 2018.
Results: A 2-year-old girl died of tumor recurrence. Forty-two eligible cases of MTT in children (birth to 18 years; mean age, 8.3 years), the highest frequency of occurrence was in 12–16 years; and the male-to-female ratio was 1.7:1. Only 33 provided complete treatment details: 11 patients received treatment by surgery alone; 2 received both surgery and chemotherapy; 4 received both surgery and radiation therapy; 14 received surgery, chemotherapy, and radiation therapy; 1 case received chemotherapy and radiation therapy; and only 1 received supportive care. In the 33 cases, the average OS and 5-year OS probability were 23.9 months (range, 0.3–156 months) and 27.5 ± 4.3%. There were significant differences between radiation therapy and patient survival (p<0.05), postoperative chemotherapy/radiation therapy and patient prognosis (p<0.05).
Conclusion: The clinical and histopathological features and therapeutic options for MTT are discussed in the light of published data. Further studies are needed to improve survival in children with this rare malignant tumor.
Keywords: malignant triton tumor, malignant peripheral nerve-sheath tumors, children, treatment strategies, prognosis
Introduction
Malignant triton tumor (MTT) is a histological subtype of malignant peripheral nerve-sheath tumors (MPNST) that contains rhabdomyoblasts. It is an extraordinarily rare and highly malignant tumor, to date approximately 170 cases of MTT have been reported.1–3 And, data on clinical features and optimal treatment strategies for MTT are rarely, so the tumor is associated with poor clinical outcome.4
The average age of patients with MTT is 31.7 years, and the morbidity rates are approximately equal among men and women. The tumor usually occurs in the head, neck, and trunk. Thus far, the lack of information on the incidence of MTT in the literature has made the tumor unfamiliar to most pediatricians and surgeons. Here, we report an unusual case of MTT in a child, and discuss the clinical features of this tumor as well as its treatment and prognosis in children.
Case Report
Patient Case
A 2-year-old girl presented with a large mass in the right shoulder, gradually increasing in size over 2 months. Upon physical examination, the patient was found to be normal. There were no signs of neurofibromatosis (NF), such as café au lait spots or subcutaneous nodules, or a family history thereof. Physical examination revealed an obvious, firm, subcutaneous, nontender, incompressible, and immobile mass measuring approximately 4.0 × 3.0 × 3.0 cm located on the medial side of the right shoulder. There was no palpable axillary lymphadenopathy. The findings of enhanced computed tomography revealed a large posterior mediastinal mass; the lesion was unevenly enhanced, and there was no clear boundary between the lesion and surrounding tissue. There was another nodular soft-tissue mass located at the tip and middle of the right lung, near the diaphragm (Figure 1A–D). A differential diagnosis of neurogenic tumor was made on the basis of imaging findings. The patient underwent a wide range of laboratory investigations, the results of which were all negative.
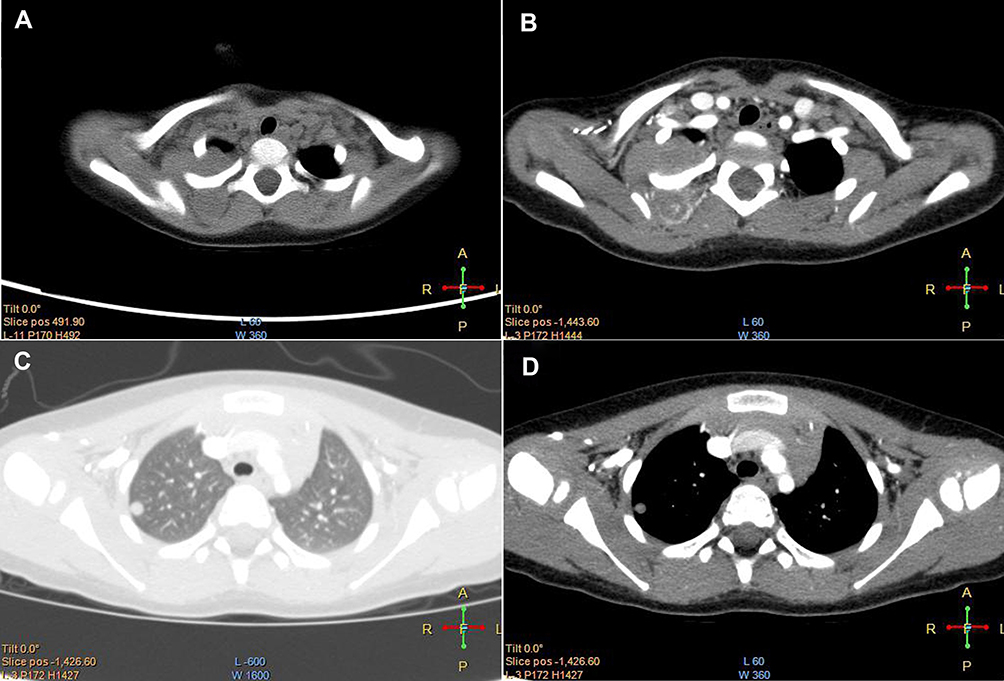 |
Figure 1 Preoperative image. (A) Axial unenhanced CT scan revealed a large posterior mediastinal mass. (B) The lesion was uneven enhanced. (C and D) There was another nodular soft tissue located in the right lung. Abbreviation: CT, computed tomography. |
The patient underwent surgery for tumor resection both for alleviating the symptoms and confirming the diagnosis. During surgery, the tumor was found to be located in the supraclavicular fossa; it was solid, oval in shape, and lacked a complete capsule. However, because a part of the tumor was wrapped around a nerve, the solid mass was only partially resected.
The resected tumor was hard and light red in color, with moderate blood supply. Histological findings of multiple sections revealed spindle cells with wavy nuclei and scattered or focal large cells that were round or elongated and contained abundant and deep eosinophilic cytoplasm. A few cells exhibited cross striations, and mitotic figures were scarce (Figure 2A).
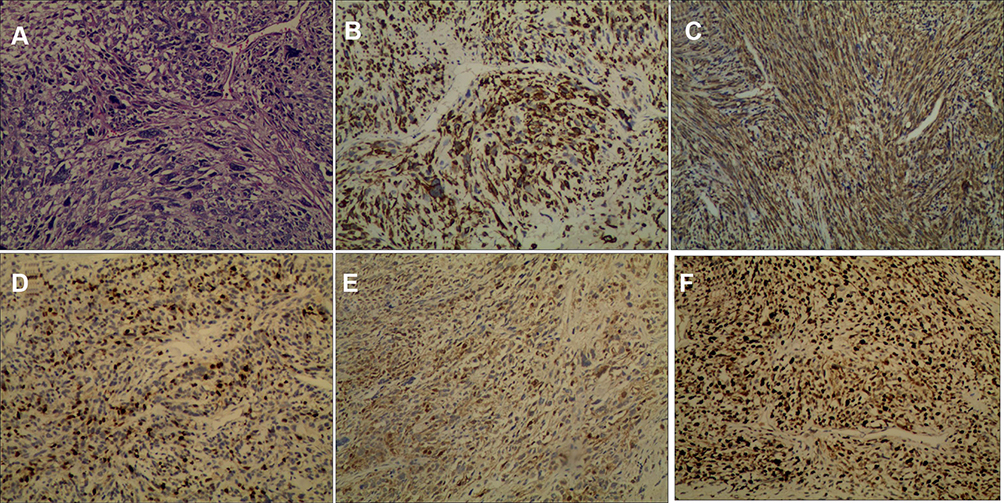 |
Figure 2 Immunostaining of the resected lesion. (A) Under the microscope, the tissue illustrated a remarkably uniform histological feature (hematoxylin and eosin, ×100). Immunohistochemical staining showed the tumor was strong positive for Desmin (×100) (B), Myoglobin (×100) (C), Myogenin (×100) (D), S-100 (×100) (E), and Ki-67 (×100) (F), which the Ki-67 index was 30%. The tumor was negative for other markers. |
Upon immunohistochemical staining, the spindle cells showed focal positivity for S-100 protein, while the large cells were positive for vimentin, myoglobin, and myogenin, with a Ki-67 proliferation index of 30%. Both cell types showed negative staining for glial fibrillary acidic protein, neuron-specific enolase, cytokeratin, epithelial membrane antigen, alpha-fetoprotein, CD30, SM-actin, and CD34 (Figure 2B–F). Therefore, on the basis of existing evidence and diagnostic criteria, a diagnosis of malignant peripheral nerve-sheath tumor with rhabdomyoblastic differentiation, i.e., MTT-was made. The diagnosis of MTT was mainly based on the histopathological and immunohistochemical characteristics of the tumor.
Although it is a highly malignant tumor, there is no standardized chemotherapy strategy for MTT currently. Therefore, we used the generally accepted multiagent MTT chemotherapy regimen, including vincristine, actinomycin, and cyclophosphamide, for treatment in the present case. The patient tolerated chemotherapy well. Although radiotherapy was also considered, her parents did not consent to it. Therefore, the patient was recommended continued chemotherapy. However, the patient died 14 months after surgery.
Ethical Requirements
This study protocol was approved by the Ethical Committee of the Zhengzhou University. Written informed consents were collected from all participants involved in the study. The patient’s parents or legal guardians provided written informed consent for the case details and images to be published.
Review of the Literature
We searched the PubMed, Medline, Embase and Web of Science databases for relevant articles published between 1938 and 2017, using the following terms: “triton tumor”, “rhabdomyosarcoma”, and “malignant peripheral nerve sheath tumor”. There was no exclusion for languages, details regarding patient characteristics was extracted from the articles. According to existing reports, Kaplan–Meier analysis was used and the log rank test was performed for comparison. The chi-square test was used to determine statistical significance, with P<0.05 being considered statistically significant. All statistical analyses were performed using the SPSS statistical software.
Result
Only 42 eligible cases of MTT in children (birth to 18 years; mean age, 8.3 years) were retrieved from available data, details regarding patient characteristics are summarized in Table 1.4–19 Among these 42 cases of MTT, the highest frequency of occurrence was in 12–16 years, and the male-to-female ratio was 1.7:1.
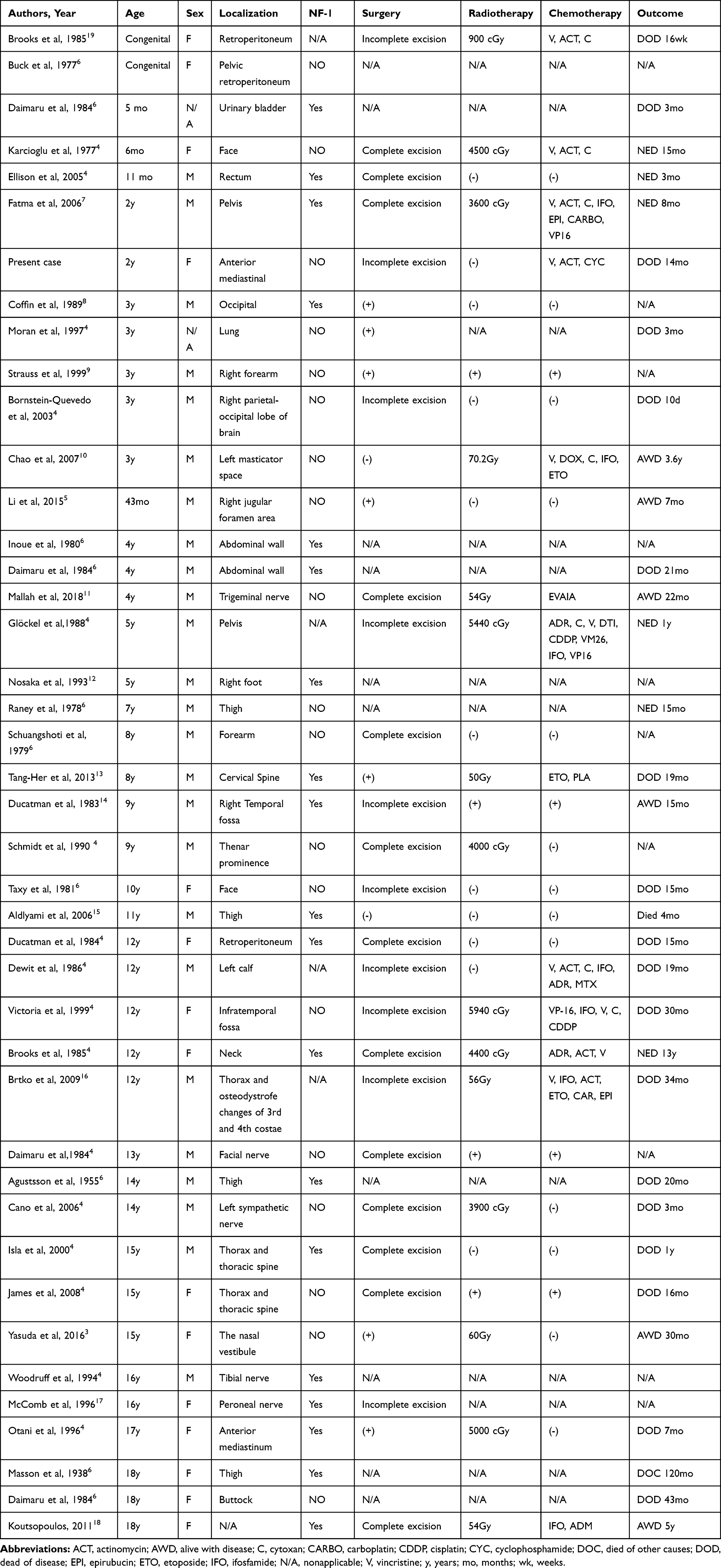 |
Table 1 Characteristics, Treatment, and Outcome of Pediatric Patients with MTT |
The treatment protocols were varied, and some patients received more than one type of therapy. Although there were 42 reported cases of MTT in children, only 33 (including the present report) provided complete treatment details (Table 1). In these 33 cases, 11 patients received treatment by surgery alone; 2 received both surgery and chemotherapy; 4 received both surgery and radiation therapy; 14 received surgery, chemotherapy, and radiation therapy; 1 case received chemotherapy and radiation therapy; and 1 received supportive care only.
In the 33 cases that provided data on prognosis, the average OS and 5-year OS probability were 23.9 months (range, 0.3–156 months) and 27.5 ± 4.3%, respectively (Figure 3A). Postoperative radiotherapy had been administered in 16 cases, patients who received radiotherapy most often received radiation doses of 40–60 Gy. Furthermore, there was a significant difference in survival between patients whose treatment protocols did and did not include radiation therapy (P =0.001, Figure 3B).
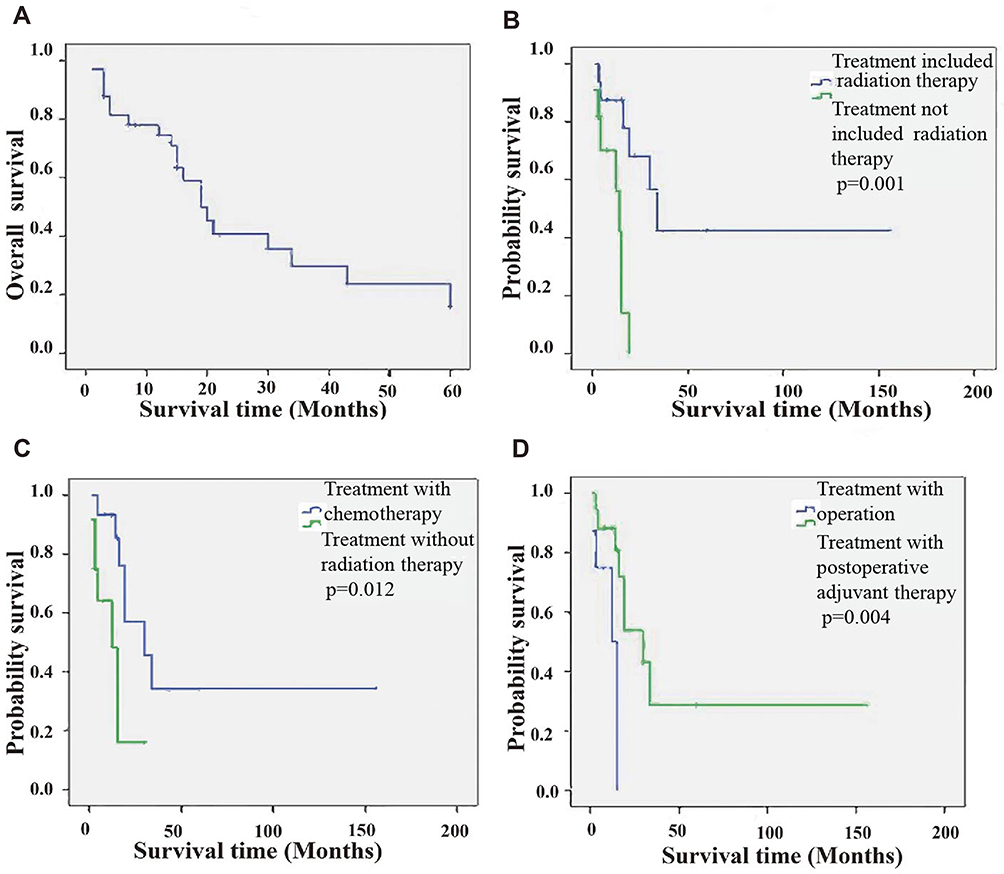 |
Figure 3 KaplanMeier survival analysis of the available data. (A) Overall survival. (B) Comparison of survival for patients who received radiation versus no radiation, p=0.001. (C) Comparison of survival for patients who received chemotherapy versus no chemotherapy, p=0.012. (D) Comparison of survival for patients who received operation only versus postoperative adjuvant therapy, p=0.004. |
Patients described in these studies were often treated with postoperative chemotherapy alone or in combination with radiotherapy. Postoperative chemotherapy had been administered in 15 cases, and the chemotherapy regimens varied but often included V, ACT, C, ADR, IFO, etc. There was a statistically significant association between treatment protocols that did and did not include chemotherapy (P=0.012, Figure 3C).
Patients in these studies often received treatment by surgery alone or in combination with postoperative adjuvant therapy (including chemotherapy and/or radiation therapy). In eight cases, postoperative first-line treatment involved surgery. One of the patients, who had received postoperative chemotherapy and radiation therapy, was still alive 13 years after treatment.19 Patients who received postoperative chemotherapy and/or radiation therapy had a significant association with prognosis than those who received treatment by surgery alone (P =0.004, Figure 3D).
Discussion
Malignant triton tumor is a subgroup of MPNSTs which have been reported to exhibit rhabdomyosarcomatous differentiation and follow a particularly aggressive disease course. The diagnosis of MTT was mainly based on the histopathological and immunohistochemical characteristics of the tumor. It exhibits the growth characteristics of Schwann cells; rhabdomyoblasts can be demonstrated within the peripheral nerve tumor, but they do not originate from extension or metastasis of an extrinsic rhabdomyosarcoma.19 Daimaru et al6 later broadened the definition of MTT: 1) Tumors without NF-1 that are microscopically compatible with a malignant schwannoma and contain focal rhabdomyoblasts and 2) tumors consisting predominantly of rhabdomyoblastic differentiation with focal Schwann cell elements occurring within a nerve or in patients with NF-1. However, the cell type of origin of MTT remains unclear. Immunohistochemical staining helps demonstrate the origin of cells. There is a consensus that MTT can be diagnosed on the basis of morphological findings, supported by positive staining results for S-100 protein.16
Including the present case, only 42 such cases of MTT in children have been reported (Table 1). In this study, we have reported a case of MTT in a 2-year-old girl and reviewed the relevant literature. Comparison of clinical data among relevant studies is possible only to a limited degree, because MTT is a very rare tumor and has been reported either in single case reports, frequently with no or only scant data on treatment and follow-up, or as part of larger reports on MPNSTs. Moreover, the outcomes are not usually elaborated upon separately.
As the rarity of this entity and the lack of prospective trials, there is no standard treatment regimen for this rare tumor. The more popular treatments include radical excision only, radical excision followed by postoperative irradiation and chemotherapy, and excision in conjunction with radiotherapy. Data collected in the present literature review demonstrate that patients in previous reports received various types of therapy and had varying prognoses. We also found that postoperative radiotherapy, chemotherapy, and combined chemotherapy and radiotherapy could improve survival (Figure 3B–D).
Analysis of published reports on MTT revealed a trend towards better outcomes among patients undergoing complete tumor resection. Some authors have reported that complete tumor resection with or without adjuvant therapy tends to portend better survival than incomplete resection with or without adjuvant therapy.4 One of the patients, who had received postoperative chemotherapy and radiation therapy, was still alive 13 years after treatment.
The Oncology Consensus Group recommends postoperative radiotherapy as part of a uniform treatment policy for MTT. For patients who received radiotherapy for MTT in previous studies, the radiation doses varied considerably but were mostly between 40 and 60 Gy, depending on the clinical context.3,12,15 It is worth noting that radiotherapy, as a part of primary treatment for MTT, seemed to have a positive effect on survival (P=0.001, Figure 3B).
The value of chemotherapy for MTT remains questionable since published accounts of clinical experience are limited. Proposed chemotherapy regimens include CEI (cisplatin, etoposide, and ifosfamide) as first-line treatment and IA (ifosfamide and adriamycin) or MDID (mesna, doxorubicin, ifosfamide, and dacarbazine) as second-line treatment. In studies included in the present review, we also found a statistically significant association between treatment included chemotherapy and treatment did not include chemotherapy (P=0.012, Figure 3C). Thus, neoadjuvant chemotherapy potentially plays an important role in a multimodal treatment approach for a subset of patients with high-grade, metastatic, primarily unresectable MTT.5
Conclusions
Children with MTT have extremely poor prognosis, and optimal treatment strategies have yet to be established. When possible, complete tumor resection with adjuvant radiotherapy and/or chemotherapy should be recommended. We believe that the present findings will help attain a greater understanding of this rare tumor, but this study is limited due to the small sample size. Further research is required on the clinical behavior, tumor biology, and potential prognostic factors of MTT. Additionally, effective risk-adapted treatment recommendations and optimization of the balance between treatment intensity and adverse effects are needed.
Abbreviations
MTT, malignant triton tumor; MPNST, malignant peripheral nerve-sheath tumors; NF, neurofibromatosis.
Acknowledgements
This study was supported by grants from the Youth Foundation of The First Affiliated Hospital of Zhengzhou University and Henan Province’s Natural Science Foundation (grant no.162300410311).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Ram R, Gardner J, Alapati S, et al. Malignant triton tumor (malignant peripheral nerve sheath tumor with rhabdomyoblastic differentiation) occurring in a vascularized free flap reconstruction graft. Int J Surg Pathol. 2017;25(5):462–467. doi:10.1177/1066896917700725
2. Elleuch W, Briki S, Mnif H, Abdelmoula M. Malignant triton tumour of the maxilla: a case report. Eur Ann Otorhinolaryngol Head Neck Dis. 2018;135(4):297–298. doi:10.1016/j.anorl.2018.05.005
3. Yasuda M, Muto Y, Kuremoto T, et al. A case of recurrent malignant triton tumor successfully treated with radiotherapy. Auris Nasus Larynx. 2016;43(6):710–714. doi:10.1016/j.anl.2016.04.003
4. McConnell YJ, Giacomantonio CA. Malignant triton tumors-complete surgical resection and adjuvant radiotherapy associated with improved survival. J Surg Oncol. 2012;106(1):51–56. doi:10.1002/jso.v106.1
5. Li G, Liu C, Liu Y, et al. Analysis of clinical features and prognosis of malignant triton tumor: a report of two cases and literature review. Oncol Lett. 2015;10(6):3551–3556. doi:10.3892/ol.2015.3762
6. Daimaru Y, Hashimoto H, Enjoji M. Malignant “triton” tumors: a clinicopathological and immunohistochemical study of nine cases. Hum Pathol. 1984;15(8):768–778. doi:10.1016/S0046-8177(84)80169-0
7. Fatma VO, Aynur O, Ceyda K, Caglar C, Pelin B, Oznur B. Malignant triton tumor of the pelvis in a 2-year-old boy. J Pediatr Hematol Oncol. 2006;28(3):173–176. doi:10.1097/01.mph.0000201419.05182.29
8. Coffin CM, Dehner LP. Peripheral neurogenic tumors of the soft tissues in children and adolescents: a clinicopathologic study of 139 cases. Pediatr Pathol. 1989;9(4):387–407.
9. Strauss BL, Gutmann DH, Dehner LP, Liapis H. Molecular analysis of malignant triton tumors. Hum Pathol. 1999;30(8):984–988. doi:10.1016/S0046-8177(99)90255-1
10. Chao MM, Levine JE, Ruiz RE, et al. Malignant triton tumor in a patient with Li-Fraumeni syndrome and a novel TP53 mutation. Pediatr Blood Cancer. 2007;49(7):1000–1004. doi:10.1002/pbc.20700
11. Mallah FA, Tanrıkulu B, Çorapçıoğlu F, Özek MM. Malignant triton tumor of trigeminal nerve-case report. Childs Nerv Syst. 2018;34(5):983–986.
12. Nosaka S, Kao SC. MRI of malignant triton tumor in a child. Clin Imaging. 1993;17(1):53–55. doi:10.1016/0899-7071(93)90014-E.
13. Jaing TH, Chuang CC, Jung SM, Wu CT, Tseng CK, Chen CS. Malignant triton tumor of the cervical spine: report of one case and review of the literature. Pediatr Neonatol. 2015;56(1):58–61. doi:10.1016/j.pedneo.2013.01.013
14. Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, llstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathological study of 120 cases. Cancer. 1986;57(10):2006–2021. doi:10.1002/1097-0142(19860515)57:10<2006::AID-CNCR2820571022>3.0.CO;2-6
15. Aldlyami E, Dramis A, Grimer RJ, Abudu A, Carter SR, Tillman RM. Malignant triton tumor of the thigh- a retrospective analysis of nine cases. Eur J Surg Oncol. 2006;32(7):808–810. doi:10.1016/j.ejso.2006.04.008
16. Brtko J, Sejnová D, Ondková S, Macejová D. Malignant triton tumour exhibits a complete expression pattern of nuclear retinoid and rexinoid receptor subtypes. Gen Physiol Biophys. 2009;28(4):425–427.
17. McComb EN, McComb RD, DeBoer JM, Neff JR, Bridge JA. Cytogenetic analysis of a malignant triton tumor and a malignant peripheral nerve sheath tumor and a review of the literature. Cancer Genet Cytogenet. 1996;91(1):8–12. doi:10.1016/S0165-4608(96)00125-2
18. Koutsopoulos AV, Mantadakis E, Katzilakis N, et al. Long-term survival of a patient with a neurofibromatosis Type 1 associated retroperitoneal malignant triton tumor after multi-modality treatment. Clin Neuropathol. 2011;30(6):333–335. doi:10.5414/NP300393
19. Brooks JS, Freeman M, Enterline HT. Malignant “triton” tumors. Natural history and immunohistochemistry of nine new cases with literature review. Cancer. 1985;55(11):2543–2549. doi:10.1002/1097-0142(19850601)55:11<2543::AID-CNCR2820551105>3.0.CO;2-4
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.