")
Back to Journals » Cancer Management and Research » Volume 6
Malignant gliomas: current perspectives in diagnosis, treatment, and early response assessment using advanced quantitative imaging methods
Authors Ahmed R, Oborski M, Hwang M, Lieberman F, Mountz J
Received 19 September 2013
Accepted for publication 14 December 2013
Published 24 March 2014 Volume 2014:6 Pages 149—170
DOI https://doi.org/10.2147/CMAR.S54726
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Rafay Ahmed,1 Matthew J Oborski,2 Misun Hwang,1 Frank S Lieberman,3 James M Mountz1
1Department of Radiology, 2Department of Bioengineering, University of Pittsburgh, Pittsburgh, PA, USA; 3Department of Neurology and Department of Medicine, Division of Hematology/Oncology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Abstract: Malignant gliomas consist of glioblastomas, anaplastic astrocytomas, anaplastic oligodendrogliomas and anaplastic oligoastrocytomas, and some less common tumors such as anaplastic ependymomas and anaplastic gangliogliomas. Malignant gliomas have high morbidity and mortality. Even with optimal treatment, median survival is only 12–15 months for glioblastomas and 2–5 years for anaplastic gliomas. However, recent advances in imaging and quantitative analysis of image data have led to earlier diagnosis of tumors and tumor response to therapy, providing oncologists with a greater time window for therapy management. In addition, improved understanding of tumor biology, genetics, and resistance mechanisms has enhanced surgical techniques, chemotherapy methods, and radiotherapy administration. After proper diagnosis and institution of appropriate therapy, there is now a vital need for quantitative methods that can sensitively detect malignant glioma response to therapy at early follow-up times, when changes in management of nonresponders can have its greatest effect. Currently, response is largely evaluated by measuring magnetic resonance contrast and size change, but this approach does not take into account the key biologic steps that precede tumor size reduction. Molecular imaging is ideally suited to measuring early response by quantifying cellular metabolism, proliferation, and apoptosis, activities altered early in treatment. We expect that successful integration of quantitative imaging biomarker assessment into the early phase of clinical trials could provide a novel approach for testing new therapies, and importantly, for facilitating patient management, sparing patients from weeks or months of toxicity and ineffective treatment. This review will present an overview of epidemiology, molecular pathogenesis and current advances in diagnoses, and management of malignant gliomas.
Keywords: glioblastoma multiforme, malignant gliomas, MRI, PET, FLT, early therapy response assessment, quantitative molecular imaging
Epidemiology and classification of brain tumors
The estimated number of new cases (adjusted for age) using the world standard population of primary malignant brain and central nervous system cancer in 2008, was 3.8 per 100,000 in males and 3.1 per 100,000 in females. The incidence rates were higher in more developed countries (males: 5.8 per 100,000; females: 4.4 per 100,000) than in less developed countries (males: 3.2 per 100,000; females: 2.8 per 100,000).1 In the US, the annual incidence of primary malignant gliomas is approximately five cases per 100,000 people.2,3 Every year, about 22,500 new cases of malignant primary brain tumor are diagnosed in adults in the US, out of which 70% are malignant gliomas.2,3 Glioblastomas account for approximately 60% to 70% of malignant gliomas, anaplastic astrocytomas for 10% to 15%, and anaplastic oligodendrogliomas and anaplastic oligoastrocytomas for 10%; less common tumors, such as anaplastic ependymomas and anaplastic gangliogliomas, account for the rest.2,3
The World Health Organization (WHO) classifies astrocytomas based on histologic type,2,4 with grading based on the most malignant region of the tumors. Tumor grade depends upon the degree of nuclear atypia, mitotic activity, microvascular proliferation, and necrosis, with increased anaplasia corresponding to higher tumor grade. Grades include low-grade, or WHO grade I (pilocytic astrocytoma) and grade II (diffuse astrocytoma); and high-grade, or WHO grade III (anaplastic astrocytoma) and grade IV (glioblastoma multiforme, GBM). Grade III and IV tumors are considered malignant gliomas. The median age at the time of diagnosis is 64 years for glioblastomas and 45 years in the case of anaplastic gliomas.5
Apart from primary brain tumors, brain metastases from common solid tumors that spread to the brain primarily include those of lung, breast, and melanoma. However, a recent increase in the incidence of brain metastases from other cancer types, such as renal, prostate, and colorectal cancers, has been observed.6,7
Molecular pathology
Molecular pathology of primary brain tumors
In the past 2 decades, the application of molecular pathology in diagnosis and classification has transformed the management of malignant gliomas.8 Molecular biomarkers have been able to differentiate oligodendroglial tumors from astrocytomas, resolve controversies regarding classification of mixed oligoastrocytic tumors, and identify clinically significant subgroups of anaplastic astrocytoma and glioblastoma.9,10 Recent clinical pathologic correlations between outcome and molecular biomarkers have also validated predictive markers for oligodendrogliomas and identified subgroups of glioblastoma susceptible to epidermal growth factor receptor (EGFR) signal transduction inhibitors.11–13
The European Organization for Research and Treatment of Cancer has identified six different types of anaplastic oligodendrogliomas using microarray unsupervised gene expression analysis of the tumor specimens obtained as part of the European Organization for Research and Treatment of Cancer trial (EORTC 26951).13 These intrinsic molecular subtypes had prognostic significance for progression-free survival (PFS) independent of the previously recognized prognostic factors, including 1p/19q deletion, isocitrate dehydrogenase gene (IDH1) mutation, and O6-methylguanine-DNA-methyltransferase (MGMT) promoter methylation status. One subgroup, with a 1p/19q deletion and IDH1 mutation, especially benefitted from the addition of chemotherapy to external beam radiation, demonstrating an overall survival (OS) of 12.8 years with adjuvant chemotherapy contrasted with 5.5 years for those patients treated with radiation alone.
It is now recognized that patients with oligodendroglial tumors with 1p/19q deletions have a consistently better prognosis for survival than those with tumors of equivalent grade and similar histologic appearance that lack the deletions.14,15 In two recently reported prospective randomized trials of fractionated external beam radiotherapy (EBRT) with or without alkylator-based chemotherapy for newly diagnosed anaplastic astrocytoma, the presence of 1p deletions was a predictive marker for the cohort of patients in which the addition of chemotherapy led to prolonged OS.11
The identification of mutations in isocitrate dehydrogenase (IDH) isoenzymes 1 and 2 in a high percentage of low grade gliomas and in subsets of anaplastic astrocytoma, oligodendroglioma, and glioblastoma has further refined the delineation of prognosis. IDH1 is a good prognostic marker for anaplastic astrocytoma and glioblastoma.9,16 For anaplastic astrocytoma, lack of an IDH1 mutation appears to identify a subgroup of histologically indistinguishable tumors with a prognosis similar to glioblastoma.17 The oncogenic mechanism appears to be the production of a metabolite, 2-hydroxyglutarate (2HG), which inhibits ketoglutarate-dependent dioxygenases, leading to aberrant histone and DNA methylation.16
In clinical trials of alkylator-based chemotherapy regimens for glioblastoma, anaplastic astrocytoma, and oligodendroglioma,9,18 the MGMT promoter methylation status has proven to be a prognostic, though not a specific predictive biomarker. Hegi et al demonstrated that promoter methylation silencing of the MGMT gene correlates strongly with long-term survival in patients receiving chemotherapy.19 At the same time, Brandes et al showed that for patients receiving chemoradiation for newly diagnosed GBM, MGMT promoter methylation silencing correlates with increased frequency of vascular permeability of vessels in the radiation treatment field.20 This may produce a transient increase in the volume of contrast taken up by the lesion, known as “pseudoprogression”.20
GBMs that arise de novo appear to be different genetically from those that arise from prior low-grade astrocytomas.9 IDH and p53 mutations are rare in primary GBM. In contrast, primary GBMs are characterized by EGFR amplification and mutation, loss of heterozygosity on chromosome 10q, and inactivation of the phosphatase and tensin homolog (PTEN) gene.21 Secondary GBMs are characterized by tumor protein p53 (TP53) mutations and platelet-derived growth factor receptor activation.21 A poor prognosis subgroup of secondary GBM in older adults, in which relapse occurs in the first year after treatment, appears to be characterized by lack of IDH1 mutations, similar to primary GBM’s molecular signature.22
Microarray-based unsupervised genome-wide analysis of gene expression in glioblastomas has identified at least four subgroups differentiable by molecular profile.23 Phillips et al examined 107 grade III and IV astrocytomas, and using a set of 35 signature genes, segregated into three subtypes: proneural, proliferative, and mesenchymal.24 In this study, the proneural subset had a better prognosis than the proliferative and mesenchymal subsets, which had worse prognoses.
The investigators of The Cancer Genome Atlas (TCGA) pilot project25 proposed a four-subgroup classification based on analysis of 202 GBMs. The subtypes include proneural, neural, classical, and mesenchymal. In the context of the cancer genome atlas, Noushmehr et al profiled promoter DNA methylation alterations in 272 glioblastomas (43 low and intermediate grade gliomas and 57 additional primary GBMs).26 They reported a distinct subset of tumors with increased DNA methylation at large number of loci, indicating the existence of a glioma–CpG island methylator phenotype (G–CIMP).26 Within the GBM cohort, the G–CIMP phenotype correlates with IDH1 mutation, younger age, proneural genotype, and a better prognosis.
The EGFR gene is the most frequently amplified gene in primary GBM and is seen in 94% of the TCGA classical type,25 and in the proliferative and mesenchymal subtypes in the Phillips classification.24 A specific in-frame deletion of exons 2–7 is present in 20%–30% of GBM overall and 50%–60% of GBM with EGFR gene amplification.27 The protein product of this truncated mRNA is the EGFRvIII mutant protein. This protein is the target antigen for immunotherapy strategies, including vaccines. Although the small molecule EGFR tyrosine kinase inhibitors trials for patients with GBM and anaplastic astrocytoma demonstrated low response rates and no benefit in PFS, a small subset of patients had durable responses.28 A specific genotype correlated with response in which EGFRvIII mutation was present in the context of intact AKT pathway function, with wild-type PTEN.29
BRAF (an oncogene located on chromosome 7) encodes a serine threonine kinase involved in cell signaling, and also involved in mitogen-activated protein kinases/extracellular signal regulated kinases pathway activation, and cell growth is most commonly associated with low-grade pediatric gliomas, but is commonly seen in high-grade diffuse gliomas as well. The most common BRAF abnormalities involve gene duplication with fusions leading to a mutant protein with a constitutively active kinase domain.9 Mutation in p53 and BRAF appear to be mutually exclusive.30 The presence of activating BRAF mutations may identify a therapeutic target in the high-grade gliomas in which it is expressed. BRAF inhibitor vemurafenib is US Food and Drug Administration (FDA)-approved as treatment for melanoma with BRAFV600E mutation.31
Molecular markers are also useful to predict a response to chemotherapy in three settings: 1p and 19q loss, MGMT methylation, and possibly the EGFR–PI3 kinase pathways in response of glioblastomas to specific EGFR inhibitors.
1p and 19q deletions
Allelic loss of chromosomes 1p and 19q is a powerful predictor of chemotherapeutic response and longer PFS and OS following chemotherapy with either temozolomide or procarbazine, lomustine, and vincristine (PCV) in patients with anaplastic oligodendrogliomas. Those tumors with 1p and 19q loss in the setting of polysomy of chromosomes 1 and 19 have intermediate prognoses. Allelic loss of 1p alone is also predictive of response to temozolomide in patients with grade II oligodendroglial tumors.32 Thus, testing for 1p and 19q status is now widespread and is used to influence therapeutic decisions.
MGMT promoter methylation
In the course of tumor development, the MGMT gene may be silenced by methylation of its promoter, thereby preventing repair of DNA damage and increasing the potential effectiveness of chemotherapy. Several clinical studies have indicated that such promoter methylation is associated with an improved survival in patients receiving adjuvant alkylating agent chemotherapy.33
EGFR-PI3 kinase pathways
Two studies evaluated patients with glioblastomas treated with the EGFR inhibitors, erlotinib or gefitinib34,35 and found that, in contrast to other studies that did not report objective responses,36 patients with recurrent glioblastoma responded to these two agents. Furthermore, the studies showed associations between response and activation of EGFR itself (one report implicating the wild-type receptor34 and the other implicating the vIII mutant EGFR35), as well as between response and whether the PI3 kinase pathway was functionally intact (one report measuring phosphorylated AKT and the other measuring PTEN expression). If responses continue to be documented with these agents, immunohistochemical testing for EGFR and the PI3K pathway may prove useful.
Molecular pathology of brain metastases
The pathophysiology of brain metastasis is complex and distinct from primary brain tumors. It is dependent upon both oncogenic processes and host organ responses. Some of the multiple mechanisms that ultimately determine the development of a brain metastasis include, but are not limited to, the phenotype of the brain-trophic tumor cells, tumor cell survival in the vasculature and extravasation of those cells from the bloodstream and into a host organ, and the structure and function of the blood–brain barrier (BBB).
Since the brain does not contain lymphatics, circulating tumor cells reach the brain parenchyma only via a hematogenous route. Invading metastatic cancer cells interact with all cell types, including endothelium, pericytes, and astrocytes, to breach the BBB and gain access to brain parenchyma.37 Once tumor cells enter the brain parenchyma, a number of factors are released by both the tumor cells and the underlying brain. In co-culture experiments, lung-cancer-derived cells release tumor-associated factors, including macrophage migration inhibitory factor, interleukin-8, and plasminogen activator inhibitor-1, which stimulate astrocytes. In turn, the activated astrocytes release interleukin-6, tumor necrosis factor-α, and interleukin-1β, which induce tumor cell proliferation.38,39
Receptor biomarkers indicating an enhanced potential for the development of central nervous system metastases may be identified in the primary tumor cell and thereby define future therapeutic targets. For example, overexpression of human epidermal growth factor receptor 2 (HER2/neu) is predictive of a three-fold increase in metastases to the lungs, liver, and brain as compared with HER2/neu-negative breast carcinomas.40–42 In lung adenocarcinoma, genetic alterations in homeobox protein Hox-B9 and lymphoid enhancer-binding factor 1 lead to hyperactivity of the Wnt/T Cell Factor (Wnt/TCF) pathway, which has been implicated in the growth of cancer stem cells and enhanced competence to metastasize to the bone and brain.43,44
Diagnosis
Clinical signs and symptoms
Although the symptoms and signs produced by malignant gliomas will vary with the location of the tumor, a unifying characteristic of the clinical presentation is relentless progression. For tumors that are located in or subjacent to cortical regions with specific functions, the symptoms and signs will relate to the functions of the brain regions affected. Patients may present with progressive motor or sensory disturbances, language dysfunction, visual field abnormalities, or focal seizures. Tumors arising in the brain stem may cause rapidly progressing cranial neuropathies as well as motor and sensory deficits. Neurologic deficits with less localizing features may include headache, confusion, memory loss, and personality changes.
As the size of tumor increases, the edema surrounding the tumor increases, resulting in increased intracranial pressure and subsequent headaches. The headaches associated with increased intracranial pressure are typically worse when the patient is recumbent. When intracranial pressure rises to a critical threshold, changes in blood pressure due to dysfunctional autonomic reflexes may produce a syndrome of position-evoked crescendo headache, visual obscurations, lightheadedness, and exacerbation of focal symptoms. This cluster of symptoms is associated with intracranial pressure waves and is usually associated with papilledema.
Neuroimaging
Neuroimaging plays a crucial role in diagnosing and assessing the location, extent, and biologic activity of the tumor before, during, and after treatment. Its role in low-grade tumors lies in the monitoring of possible recurrent disease or anaplastic transformation into high-grade tumors. In high-grade tumors, neuroimaging is much needed for differentiating recurrent tumor from treatment-induced changes such as radiation necrosis.
Gliomas are often characterized by diffuse infiltration of white matter tracts,45 and stereotactic biopsy studies have demonstrated that these regions appear normal on conventional contrast-enhanced computed tomography (CT) and magnetic resonance imaging (MRI).46 Since complete resection of infiltrative high-grade neoplasms is not an option,47 the development of improved posttreatment imaging to detect residual tumor is pivotal in clinical outcome.
MRI serves as the current gold standard in tumor treatment response monitoring; however, prognostic information cannot be obtained until weeks after the initiation of treatment.48 Determination of recurrence versus treatment effects on CT or MRI cannot be accurately evaluated.49–51 Functional imaging can distinguish cerebral necrosis from viable brain tumor, and determine viability grade.52–54
The realization that the MacDonald criteria1 for response assessment in clinical trials of treatments for high-grade gliomas failed to account for nonenhancing progression has led to the development of a new paradigm, the Response Assessment for Neuro-Oncology (RANO) criteria.55 Differentiating tumor response related to cytotoxicity from physiologic modifications of BBB function is a major focus of translational imaging research. MRI techniques that interrogate the vascular density and permeability of tumor vasculature as well as positron emission tomography (PET) techniques56 are being evaluated as imaging biomarkers of tumor response in treatment trials of anti-angiogenic therapy.57
CT
Most of the time, CT is the first imaging modality for evaluating symptoms of gliomas. Contrast-enhanced CT scans can delineate disruptions in the BBB, but CT sensitivity is much lower than that of MRI. The attenuation difference can offer limited information on tumor biology. For instance, slightly increased tissue density during tumor monitoring may indicate increase in tissue cellularity, or tumor growth. On the other hand, decreased attenuation in the treated region indicates low tumor cellularity or edema. However, the exact delineation of tumor borders or the extent of treatment-related changes is not feasible using this modality.
MRI
The clinical gold standard for brain tumor imaging, MRI, utilizes T1- and T2-weighted sequences, fluid-attenuated inversion recovery sequences, and contrast-enhanced T1-weighted imaging for tumor monitoring. Glioblastoma is classically hypointense to isointense, with a ring-pattern of enhancement on gadolinium-enhanced T1-weighted images, and is hyperintense on both T2-weighted and FLAIR (fluid-attenuated inversion recovery) images (Figure 1).58,59 It can be focal, multifocal, or diffuse (gliomatosis cerebri).
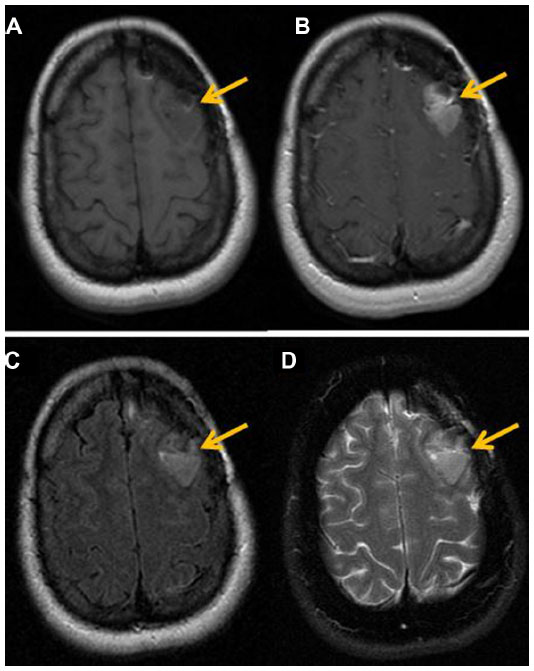 | Figure 1 Magnetic resonance findings in GBM. |
MRI provides excellent anatomic detail; however, it cannot reliably differentiate between radiation necrosis and recurrence posttreatment (Figure 2).60,61 This is of critical importance in monitoring tumor response to chemoradiation and stereotactic radiosurgery, both of which are associated with high prevalence of post-therapy necrosis.
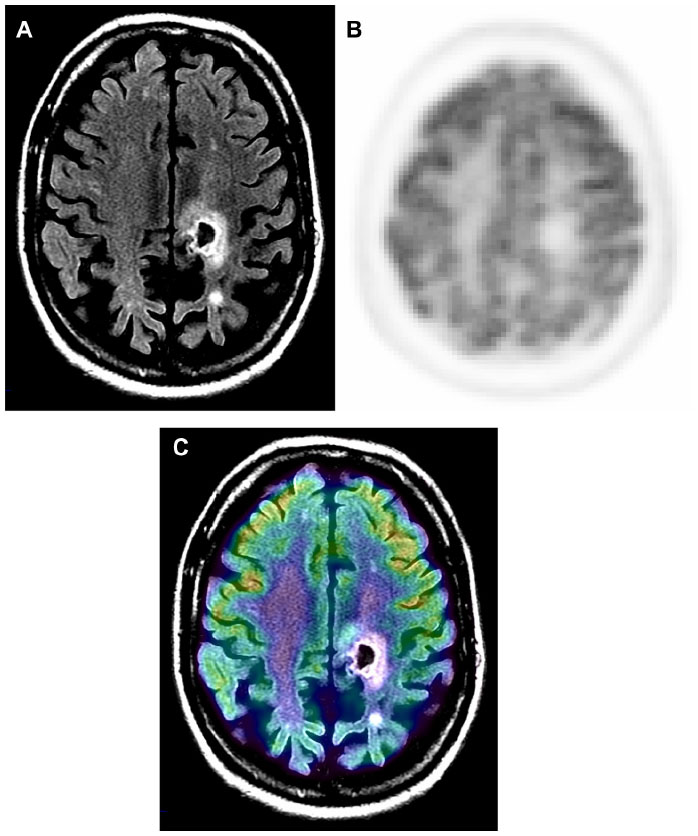 | Figure 2 Radiation necrosis versus viable tumor on MRI. |
Although BBB destruction with subsequent leakage of contrast medium is commonly seen in most high-grade tumors, such as glioblastomas, it is not a reliable distinguishing feature of tumor grade.62 In fact, approximately one-third of nonenhancing gliomas are malignant.63 Moreover, glioblastoma may initially present as a nonenhancing lesion, especially in older patients. In addition, contrast enhancement cannot always be used to assess response since therapy may result in BBB disruption without a corresponding change in tumor status.64,65
After therapy, physiologic MRI can provide insights into changes in tumor environment related to metabolism (magnetic resonance spectroscopy [MRS]), perfusion (perfusion-weighted imaging), and microstructure (diffusion-weighted imaging [DWI]). Indeed, apparent diffusion coefficient (ADC) measurements,66–68 DWI values,69 and fluid-attenuation inversion recovery images70 correlate with the probability of response to therapy.
1H MRS
The magnetic resonance spectrum from 1H MRS contains peaks representative of different (hydrogen-containing) metabolites. The relative concentration of each metabolite is determined from the area under the corresponding peak. Whereas single-voxel spectroscopy yields a single spectrum from a defined tissue area, two- and three-dimensional chemical shift imaging depict one or more tissue slices with several voxels in each slice to better account for tissue inhomogeneities.
In the case of tumor monitoring, tumor metabolite data are compared to those of the contralateral healthy side. The most commonly examined metabolites include lactate as a product of anaerobic glycolysis,71 N-acetylaspartate as a sign of neuronal viability and density,72,73 choline as an indicator of high membrane turnover and thus cell proliferation,74,75 and creatine as a signature of cell energy expenditure used for an internal reference value.76 Increasing choline/creatine ratios and lactate concentrations,75 and decreasing N-acetylaspartate77 correlate with tumor progression, and can also be seen in tumor recurrence (Figure 3). Whereas elevated creatine values (normalized to normal brain) correlate with a shorter time-to-progression in WHO grade II and III astrocytomas,78,79 no correlation was identified between tumor grading and choline/creatine ratio.80
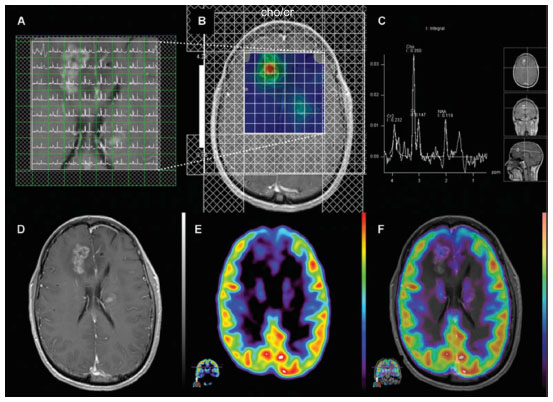 | Figure 3 Magnetic resonance spectroscopy. |
A study by Imani et al compared the accuracy of high-field proton MRS (1H MRS) and 18F 2-fluorodeoxyglucose PET (18F-FDG PET) for identification of viable tumor recurrence in 12 grade II and III glioma patients and showed that 1H MRS imaging was more accurate in low-grade glioma and 18F-FDG PET provided better accuracy in high-grade gliomas.80 The study also suggested that the combination of 1H MRS data and 18F-FDG PET imaging can enhance detection of glioma progression. While the sensitivity of 18F-FDG PET in detecting glioma progression was very high (100%), its specificity in differentiating post-therapy inflammation from true tumor progression was low (71%), leading to a high false positive rate (29%) in post-radiation therapy patients.
Studies have looked into the significance of IDH mutational status in the diagnosis and classification of gliomas and the identification of an oncometabolite, 2HG, which accumulates in IDH mutant tumors.9,16 Recent investigations using ultrahigh field strength MRI suggest that the presence of IDH mutations in a tumor can be noninvasively detected by spectroscopic measurement of 2HG.81 Recently, investigators in the US and Europe have demonstrated that MRS can differentiate 2HG from neighboring metabolites, such as gamma amino butyric acid, glutamine, and glutamate. Kalinina et al82 analyzed brain tumor specimens to show the feasibility of using MRS to quantitate 2HG for the classification of IDH mutant tumors. Subsequently, Pope et al83 demonstrated detection of 2HG by MRS in glioma patients prior to resection, with analysis of IDH1 status by DNA sequencing, and measurement of concentrations of 2HG and other metabolites by liquid chromatography–mass spectroscopy. In tumors with IDH1 mutations, 2HG levels, as measured in vivo using water suppressed proton (1H) MRS, correlate with measured amounts in the resected tumor specimens. Tumors with IDH1 mutations have elevated choline and decreased glutathione levels. Elkhaled et al81 also demonstrated that levels of 2HG correlate with levels of choline, lactate, and glutathione, as well as with histopathologic grade. While it appears that MRS can provide a noninvasive measure of 2HG in human gliomas, further studies are needed to validate the utility of 2HG quantitation and the relevance of other alterations in metabolites as prognostic biomarkers.
DWI-MRI
DWI relies on the microscopic motion of water molecules within tissue. The process is influenced by temperature and tissue architecture84 and is commonly quantified by the ADC. Tumor infiltration alters tissue architecture and thus water diffusion. ADC decreases with an increase in viscosity, cellular density, and reduction of extracellular space. Low values in ADC maps in solid gliomas are associated with higher-grade tumors.85 Complicating the interpretation is coexistent posttreatment edema which may alter ADC values. The recently introduced higher-order diffusion technique, diffusion kurtosis imaging,86 is being studied to characterize microstructural changes, and initial findings appear promising in the differential diagnosis of brain tumors.87
Perfusion-weighted MRI
Perfusion-weighted imaging involves the quantification of cerebral blood volume (CBV) after contrast administration with a dynamic MRI sequence sensitive to T2* effects. A graph of contrast enhancement is generated to calculate the area under the signal curve as an estimate of relative CBV (rCBV). High-grade gliomas, in particular, are associated with disruption of the BBB, which causes more contrast extravasation and consequent adjustments to rCBV calculations with sophisticated mathematical models.88 Preloading of contrast medium has been applied to minimize the effects of leakage.89,90 Increased angiogenesis in high-grade gliomas is also correlated with higher CBV relative to contralateral normal white matter rCBV and tumor aggressiveness.91–93 Quantitative analysis found a threshold of rCBV =1.75 for determining a high-grade glioma91 and a higher rCBV ratio of about 2.14 for oligodendrogliomas.94 It has also been shown that an increase in rCBV occurs up to 12 months prior to malignant transformation as assessed by new contrast enhancement.95
PET
Imaging glucose metabolism – 18F-FDG
18F-FDG PET has allowed monitoring of therapeutic response in brain tumors with a greater specificity than CT or MRI. 18F-FDG, a glucose analog, is taken up by high-glucose-using cells, including normal brain and cancer cells. FDG is actively transported across the BBB into the cell and the 18F-FDG-6-phosphate formed when 18F-FDG enters the cell and prevents its further metabolism. As a result, the distribution of 18F-FDG is a good reflection of the distribution of glucose uptake and utilization by cells in the body.
Since most cancer cells, including gliomas, demonstrate a high rate of glycolysis,96 18F-FDG helps in differentiation between tumor and normal brain tissue. It should be noted, however, that the correlation between 18F-FDG uptake and glucose metabolism in tumors may differ from that in normal tissue.97 In untreated tumor, the degree of 18F-FDG uptake has been correlated with tumor grade: high-grade tumors demonstrate increased tracer uptake, and high uptake in a previously categorized low-grade tumor confirms anaplastic transformation of the tumor.98,99 Quantitatively, ratios of 18F-FDG uptake in tumors to that of white matter (>1.5) or gray matter (>0.6) were able to distinguish low-grade (grades I and II) from high-grade tumors (grades III and IV).100 Based on a preliminary finding, delayed imaging at 3–8 hours after injection can further distinguish tumor and normal gray matter due to the faster tracer excretion in normal brain than in tumor.101 However, after therapy the degree of tracer uptake does not necessarily correlate with tumor grade in that high-grade tumors may have uptake similar to or slightly above that of white matter.102
18F-FDG PET also plays a role in differentiating between recurrent or residual tumor and radiation necrosis (Figures 4 and 5). However, due to the 18F-FDG uptake in normal brain, the sensitivity of detecting recurrent or residual tumor is low.103,104 The specificity is also low in the initial few weeks post-therapy due to radiation necrosis. A study showed a sensitivity of 81%–86% and a specificity of 40%–94% for distinguishing between radiation necrosis and tumor.105 It is thus recommended that 18F-FDG PET should not be performed before 6 weeks after the completion of radiation treatment.
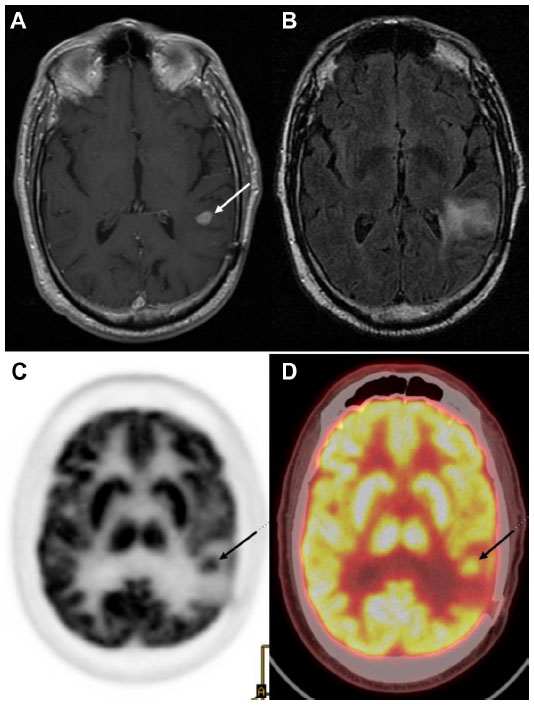 | Figure 4 Tumor recurrence versus radiation induced changes: images of a 77-year-old male who was originally diagnosed with glioblastoma multiforme, treated with external beam radiation and adjuvant chemotherapy with temozolomide. |
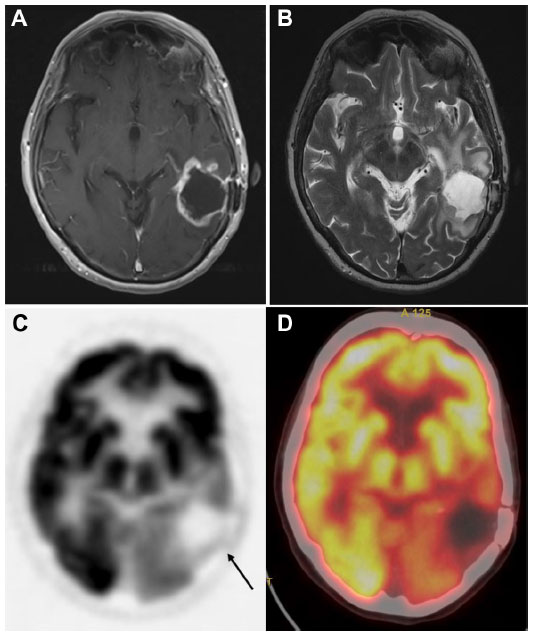 | Figure 5 18F-FDG PET for tumor recurrence: 71-year-old male patient with history of glioblastoma multiforme, status post-resection presents for evaluation of recurrence. |
Recently, new issues have emerged regarding the evaluation of disease response, and also with the identification of patterns such as pseudoprogression, frequently indistinguishable from real disease progression,106 and pseudoresponse. The Macdonald criteria,107 widely used clinically as a guideline for evaluating therapeutic response in high-grade gliomas, uses contrast-enhanced CT and MRI, and defines progression as greater than a 25% increase in size of enhancing tumor. Enhancement of brain tumors, however, primarily reflects a disturbed BBB.
By definition, pseudoprogression of gliomas is a treatment-related reaction of the tumor with an increase in enhancement and/or edema on MRI, suggestive of tumor progression, but without increased tumor activity (Figure 6). Typically, the absence of true tumor progression is shown by a stabilization or decrease in size of the lesion during further follow-up and without new treatment. Pseudoprogression occurs frequently after combined chemo-irradiation with temozolomide, the current standard of care for glioblastomas.20,65
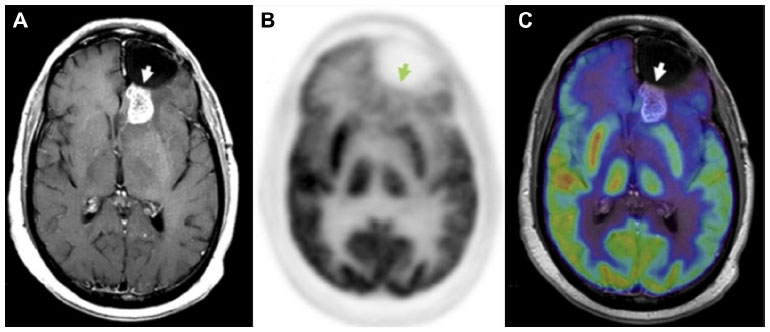 | Figure 6 18F-FDG PET diagnosis of pseudoprogression. |
In an effort to identify patients likely to exhibit pseudoprogression, some studies have attempted to correlate MGMT promoter methylation status with pseudoprogression.20 Studies have demonstrated that MGMT methylation status is an important biomarker for assessing primary brain tumors, as MGMT status has been shown to correlate with both therapy response and OS in GBM when therapy includes alkylating agents.19,108 However, similar studies of MGMT promoter methylation in anaplastic oligodendrogliomas were unable to find a correlation between MGMT methylation status and either response rate, time-to-progression, or OS, suggesting that MGMT promoter methylation patterns may be dependent on cell type.109
Another phenomenon, pseudoresponse, is the decrease in contrast-enhancement and/or edema of brain tumors on MRI without a true antitumor effect. It occurs after treatment with agents that induce a rapid normalization of abnormally permeable blood vessels or regional cerebral blood flow.110 Recent trials on high-grade gliomas with agents that modify the signaling pathways of vascular endothelial growth factor (VEGF), formerly also known as the vascular permeability factor111,112 (eg, bevacizumab, cediranib), have shown a rapid decrease in contrast enhancement with high response rate and 6-month PFS (PFS-6), but with rather modest effects on OS.111–113
These two opposing phenomena emphasize that enhancement by itself is not a measure of tumor activity, but only reflects a disturbed BBB. A recent case report by our group emphasizes the value of 18F-FDG PET when pseudoprogression is strongly suspected by the referring physician.56 Currently, 18F-FDG PET is not a clinically standard method for evaluating therapeutic response in high-grade gliomas, as it is only used for initial staging and to confirm suspected recurrence observed on gadolinium MRI (Gd-MRI). However, a central advantage of 18F-FDG PET is that it can be used to determine the metabolic state of tumor cells, in contrast to Gd-MRI, which is limited to evaluating changes in size of contrast enhancement. This is an important distinction in comparing 18F-FDG PET and Gd-MRI results, as changes in contrast enhancement are generally a conglomeration of many effects, such as local vascularity, changes in both normal and tumor cell density, necrosis, apoptosis, and BBB breakdown. All of these morphological changes are presumably preceded by changes in tumor metabolism, suggesting that, in many cases, 18F-FDG PET may allow for comparatively faster discrimination of pseudoprogression from true progression and pseudoresponse from true response.
Recent efforts have focused on the coregistration of PET and MRI images, which has increased sensitivity over using either modality alone.114,115 The simultaneous PET–MRI scan, which offers better MRI-based motion correction of PET data, is also being studied in more centers.116,117
Amino acid PET tracers
Amino acid and amino acid analog PET tracers are better suited than 18F-FDG for quantitative monitoring of tumor response due to higher tumor-to-normal-tissue contrast.118–122 The use of amino acids for tumor imaging is based on the observation that amino acid transport is upregulated in malignant transformation.123,124 Response after chemotherapy can be detected by amino acid PET early in the course of treatment,125–127 suggesting that deactivation of amino acid transport is an early sign of response to chemotherapy. Amino acids are transported across the cell via a carrier-mediated mechanism.128 For example, transport of the 18F amino acid analog 3-O-methyl-6-18F-fluoro-L-DOPA via sodium-independent, high-capacity amino acid transport systems has been demonstrated in tumor cell lines.129 In gliomas, increased amino acid uptake is mediated by type L amino acid carriers, which are upregulated in tumor vasculature.124,130 This is in part attributed to the increased metabolic demand of tumor cells. Several amino acid tracers are available, though they are not FDA-approved in the US; eg, O-(2-18F-fluoroethyl)-L-tyrosine (FET), 3,4-dihydroxy-6-18F-fluoro-L-phenylalanine, and of 11C methionine (MET).131,132
MET: The best-studied PET amino acid isotope has been l-[methyl-11C] methionine (11C-MET),133 which is able to differentiate tumor recurrence from radiation necrosis.134 However, due to the relatively short 11C half-life of 20 minutes, it requires a nearby cyclotron. The extent of tracer uptake is greater than the degree of contrast enhancement indicative of better delineation of tumor margins.135 In low-grade gliomas, the uptake is increased in the absence of BBB breakdown, which is a significant advantage over CT, conventional MRI, and 18F-FDG PET.136,137 The tracer uptake has been shown to correlate with prognosis and survival in low-grade gliomas.138,139 In high-grade gliomas, 11C-MET uptake is greater than in low-grade tumors,140–142 establishing its potential for use in monitoring anaplastic transformation. In fact, recent findings show that increased 11C-MET uptake during tumor growth parallels an upregulation of angiogenic markers such as VEGF.143 Moreover, the addition of 11C-MET PET changed patient management in half the cases.144
18F-FET (fluoro-3′-deoxy-3′-l-fluorothymidine) is another PET tracer studied for its potential role in the differentiation of radiation necrosis and residual tumor. Indeed, the absence of 18F-FET uptake in a case of radiation necrosis was shown,131 but further systematic studies are necessary to confirm this finding. In contrast to 18F-FDG, 18F-FET uptake was absent from macrophages, a common inflammatory mediator.145 In another study, the ratio of 18F-FET uptake in radiation necrosis to that in normal cortex was much lower than the corresponding ratios for 18F-FDG and 18F choline, supportive of its potential for differentiating radiation necrosis from tumor recurrence.146
In the last decade, studies on combined 18F-FET and MRI have shown improved identification of tumor tissue as compared with either modality alone.147,148 The specificity of distinguishing gliomas from normal tissue could be increased from 68% with the use of MRI alone to 97% with the use of MRI in conjunction with 18F-FET PET and MRI spectroscopy.149
Nucleic acid analogs – 18F-FLT
The pyrimidine analog, 18F-FLT, is a PET radiotracer specifically used for noninvasive in vivo evaluation of the cell proliferation rate. 18F-FLT reflects the activity of thymidine kinase-1 during phase S of DNA synthesis.150 18F-FLT, introduced by Shields et al for PET imaging of tumor proliferation in animals and humans,151 has been used in both preclinical and clinical studies.152,153 Transport of 18F-FLT is mediated by both passive diffusion and Na+-dependent carriers. The tracer is subsequently phosphorylated by thymidine kinase 1 (TK1) into 18F-FLT-monophosphate, where TK1 is a principal enzyme in the salvage pathway of DNA synthesis. Whereas the TK1 activity is virtually absent in quiescent cells, its activity reaches the maximum in the late G1 and S phases of the cell cycle in proliferating cells.154 The phosphorylation of the tracer by TK1, therefore, makes 18F-FLT a good marker for tumor proliferation.
Recent findings suggest that 18F-FLT is a promising biomarker for differentiating between radiation necrosis and tumor recurrence (Figure 7).155,156 A study by Hatakeyama et al155 showed its superiority over 11C-MET in tumor grading. Chen et al demonstrated 18F-FLT PET as a promising imaging biomarker that seems to be predictive of OS in bevacizumab and irinotecan treatment of recurrent gliomas in which both early and later 18F-FLT PET responses were more significant predictors of OS compared with the MRI responses.157 In addition, a recent prospective study by Schwarzenberg et al158 showed that 18F-FLT uptake was highly predictive of PFS and OS in patients with recurrent gliomas on bevacizumab therapy (Avastin®; Genentec, South San Francisco, CA, USA; a recombinant humanized monoclonal antibody targeting VEGF, a protein released by tumor cells to recruit novel blood vessels to support tumor growth),159,160 and that 18F-FLT PET seems to be more predictive than MRI for early treatment response.
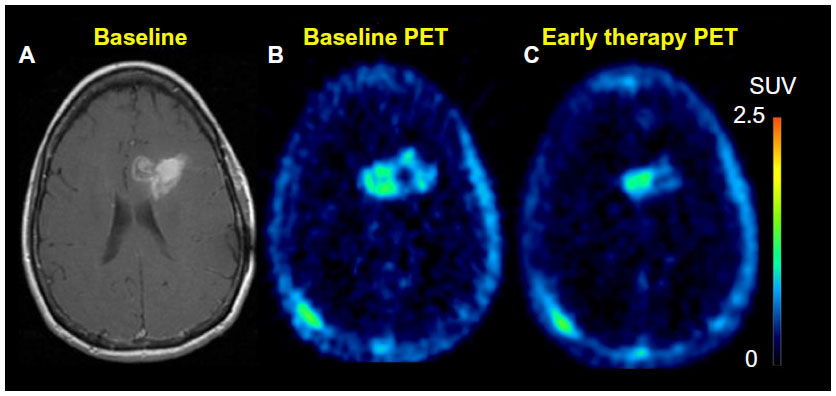 | Figure 7 18F-FLT PET. |
Hypoxia imaging – 18F-fluoromisonidazole
18F-Fluoromisonidazole is a nitroimidazole derivative PET agent used to image hypoxia,161 a physiologic marker for tumor progression and resistance to radiotherapy (RT).162 Its preferential uptake in high-grade rather than low-grade gliomas,163 a significant relationship with upregulation of angiogenic markers such as VEGF receptor 1,164 and correlation to progression and survival after RT,165 suggest its potential role in monitoring response to therapy targeting hypoxic tissue.
Biopsy
A tissue diagnosis can be obtained at the time of surgical resection or through stereotactic biopsy. Biopsy alone is used in situations where the lesion is not amenable to resection, or when a meaningful amount of tumor tissue cannot be resected, or the patient’s overall clinical condition will not permit invasive surgery.
Stereotactic image-guided brain biopsy is an accurate and safe diagnostic procedure in patients with focal lesions.166,167 The combined use of computerized imaging and stereotactic framing devices allows neurosurgeons to perform deep brain biopsies with continuous and accurate intraoperative tumor localization. Frameless stereotaxy establishes a computerized link between the preoperative three-dimensional tumor volume and the surface landmarks of the patient. This link permits the neurosurgeon to be aware of the three-dimensional position of surgical instruments within the intracranial space during the biopsy based upon the preoperative imaging, with an accuracy of 1 mm within the intracranial space.
Treatment
After decades of minimal incremental advances in outcomes for multimodality treatment of malignant gliomas, the last decade has seen a series of transformative clinical trials establish new standards of care. At the same time, the limitations of these transformative strategies have raised new questions for therapeutic clinical trials. Addressing these questions requires innovative neuroimaging strategies to better assess treatment response. The application of molecular neuropathology, quantitative imaging of tumor response, and systematic evaluation of molecularly targeted therapies, as well as cytotoxic chemotherapy are expected to improve outcomes even further.
Surgery
Surgical resection has been a critical component of the multimodality management of malignant gliomas since the advent of modern neurosurgery and the original case series by Cushing and Dandy.168 The role of neurosurgery has expanded in recent years to include techniques for intratumoral delivery of drugs, monoclonal antibodies, viral gene vectors, and immunotherapeutics. Resection or image-guided techniques for accessing the tumor microenvironment are increasingly critical components of therapeutic clinical trials as they help to show drug delivery to the tumor site and to verify that the anticipated physiologic effects relevant to the mode of action of the drug have occurred.169,170 In the era of molecularly-targeted therapies and personalized therapeutics, determination of the pattern of genetic and epigenetic changes in tumor tissue is critical to understanding the mechanisms of tumor response and resistance.171
For GBM patients, there is compelling, though not level-one evidence, that maximal resection of newly diagnosed tumor improves survival.172–174 For anaplastic astrocytoma and anaplastic oligodendrogliomas, the survival benefit of aggressive surgical resection is less clearly documented, but expert consensus supports similar resection goals as for GBM patients.175 Maximal surgical resection provides the advantages of rapid cytoreduction, relief of symptoms related to mass effect, allows for institution of fractionated radiation therapy and chemotherapy with reduced target volumes, and provides tissue for diagnosis.168
Image-guided resection and the incorporation of functional MRI information as well as intraoperative mapping has allowed for resection of tumors in close proximity to eloquent cortical structures and expanded the indications for resection.176–181 Innovations in MRI design have allowed for intraoperative MRI, in which the neurosurgeon can assess completeness of resection prior to closure of the craniotomy.
Minimally invasive neurosurgical techniques, exemplified by endoscopic resection techniques182 are being applied to resection of malignant gliomas, facilitating more complete resection of deeply located tumors, and intraventricular or periventricular tumors.183 Neurosurgical techniques for intratumoral drug delivery are also being investigated. Stereotactic MRI or CT-guided techniques allow for biopsy and intratumoral delivery of therapeutic agents, though limited capacity for diffusion limits this technique in most settings. Microdialysis catheters placed at the time of tumor resection allow direct measurement of drug pharmacodynamics in clinical trials of systemically administered agents.
RT
Shortly after the initial attempts to control malignant gliomas with aggressive surgical resection, neurosurgeons and oncologists turned to EBRT as the second component of multimodality therapy. Seminal clinical trials by the early brain tumor clinical trial collaborative groups demonstrated that EBRT prolongs survival as compared with surgery alone, for GBM, anaplastic astrocytoma, and anaplastic oligodendrogliomas.184–186 Collaborative group trials established optimal dose and fractionation schema for the different histologies and grades of malignant tumors.
Involved field radiation therapy, which involved delivery of RT only to involved regions of the brain, has become the standard approach for adjuvant RT. The rationale for limiting the RT field is based upon the observation that, following whole brain radiation therapy, recurrent malignant gliomas develop within 2 cm of the original tumor site in 80%–90% of cases, while fewer than 10% are multifocal.187–189 To encompass infiltrating tumor cells, the RT dose of typically 60 Gray is usually delivered to the tumor plus a margin of radiographically apparently normal tissue. If the tumor is defined based upon contrast enhancement, a margin of 2.0 to 3.0 cm is often used, while if the RT field is defined by T2-weighted MRI abnormality, a 1.0 to 2.0 cm margin is used.
Over the past 3 decades, innovations in computer-based three-dimensional treatment planning have led to an increase in conformal radiation therapy. In academic centers of excellence, as well as in the community, these techniques have provided a new approach to treat malignant gliomas using an increased dose with less morbidity. Current three-dimensional conformal radiation therapy utilizes CT-based treatment planning with dosimetric software to create composite treatment plans. The fusion of planning CT with MRI is extremely helpful in assisting with target definition.190,191 The incorporation of PET or MRS data is still largely investigational and most commonly used to define boost volumes rather than primary target volumes. Photons of 6 to 8 MV are most commonly used with three to four angled radiation fields. Radiation oncologists work with medical physicists and dosimetrists to design optimal treatment plans. Optimization requires the consideration of beam energy, field size and shape, beam modifiers, irradiated tissue density and heterogeneity, and radiation tolerance of surrounding normal tissues. No benefit in PFS or OS has been demonstrated, although these techniques help avoid excess RT to normal brain.192,193
In the past several years, intensity-modulated radiation therapy (IMRT), at least for academic radiation oncology centers, has been the technique of choice due to the elegance and precision of the dosimetry, especially if the tumor is in close proximity to radiosensitive structures such as the optic nerve. The IMRT technique uses advanced technology to manipulate beams of radiation to conform to the shape of a tumor. It uses nonuniform small radiation beams of varying intensities to deliver a treatment plan that maximizes the homogenous delivery of radiation to the intended treatment volume, while minimizing irradiation to normal tissue outside the target. The radiation intensity of each beam is controlled, and the beam shape changes throughout each treatment. The goal of IMRT is to bend the radiation dose to avoid or reduce exposure of healthy tissue and limit the side effects of treatment. The application of IMRT in the treatment of malignant gliomas has become increasingly prevalent as it may decrease radiation-related adverse effects.194 IMRT can also be used to escalate doses to the tumor, but there are no proven benefits to delivering doses beyond 60 Gray.195 The most appropriate application of IMRT in the brain will likely be when the radiation target abuts radiation-sensitive structures such as the eyes, optic nerves, optic chiasm, or brainstem. The disadvantages of IMRT include increased radiation scattering to surrounding non-target tissues and the complexity of radiation planning, which requires adaptation of the hardware of linear accelerators, skilled physicist support, and increased delivery time for treatment.
Despite decades of trials investigating permutations of total dose and fractionation schemes, the typical one per day treatment with external beam, 5 days per week, has remained the standard of care. With present technologies and strategies for radioprotection of normal structures, improvements in survival are unlikely to result from modifications in total dose or fraction size.
Proton beam RT is being investigated in the treatment of low-grade gliomas, medulloblastomas and ependymomas, and in malignant gliomas. At present, there is no level-one evidence that proton beam therapy improves survival in either the newly diagnosed or recurrent setting for GBM or anaplastic astrocytoma and oligodendroglioma. As with IMRT, proton techniques may have a specialized role in treatment of targets close to critical radiosensitive structures.
Stereotactic radiosurgery has been used to boost fractionated RT for the treatment of newly diagnosed GBM following either biopsy or resection.196–198 Stereotactic radiosurgery uses three-dimensional planning techniques to precisely deliver narrowly collimated beams of ionizing radiation in a single high-dose fraction to small (<4 cm) intracranial targets. When this approach is divided into several factions it is called stereotactic RT.
In some centers, Gamma Knife radiosurgery is used, in which a hemispherical compartment with an array of cobalt-60 sources is the source of collimated beams. The Gamma Knife uses a fixed frame to stabilize the head relative to the radiation sources.
Frameless linear-accelerator-based stereotactic radiosurgery employs a linear accelerator that moves in multiple arcs around the target volume. The linear accelerator techniques do not employ a fixed frame, and the relationship of the target volume to the radiation source is determined by registration of fiducials.
Radiosurgery has transformed the treatment of brain metastasis and benign tumors such as acoustic schwannoma, but has yet to claim a clear role in the treatment of malignant gliomas.199–202 In the newly diagnosed setting, radiosurgery in conjunction with fractionated EBRT has not improved survival outcomes. However, in the recurrent setting, radiosurgery is an FDA-approved treatment modality, but progression at the margin of the target is a ubiquitous pattern of failure. More recently, radiosurgery has been combined with bevacizumab therapy. Initial institutional Phase II trials of this combination have not demonstrated superior time-to-progression or OS than either treatment alone, but some patients have durable tumor control.203 The nuances in designing treatment fields may be critical in this setting.204
Chemotherapy/drug therapy
The current standard of care for newly diagnosed GBM combines surgical resection, RT and adjuvant temozolomide treatment, leading to an increased median survival time205 of approximately 14.6 months. The EORTC trial206 established that concomitant low-dose temozolomide and external beam fractionated radiation followed by adjuvant temozolomide results in a survival benefit to the chemotherapy arm versus radiation alone. The trial demonstrated a benefit in OS to the group receiving chemotherapy, and a tripling of the percentage of patients alive 2 years after therapy. Subsequent prospective trials showed that a dose-intensive adjuvant temozolomide regimen in which patients received 75 mg/m2 daily for 21 days followed by a 7-day rest was not superior to the shorter monthly courses of temozolomide.
Management of newly diagnosed anaplastic oligodendroglioma is now based on level-one evidence. Two prospective randomized trials comparing external beam radiation alone to radiation therapy plus alkylator-based adjuvant or neoadjuvant chemotherapy were initially reported as showing no survival benefit with the addition of chemotherapy.11,13 However, long-term follow-up demonstrated that for patients with tumors expressing 1p/19q deletions, chemotherapy confers a significant survival advantage.11,13 The predictive value of the 1p deletion status makes this one of the first robust predictive biomarkers for malignant gliomas. In addition to the impact of 1p deletion status on outcome, these anaplastic oligodendroglioma studies also led to the delineation of subgroups of tumors with prognostic significance using microarray genome-wide expression analysis.207 It is clear that future studies of anaplastic oligodendrogliomas and astrocytomas will need to include stratification by prognostic subgroups.
For newly diagnosed anaplastic astrocytoma, the optimal application of radiation and chemotherapy is an active clinical trial question.208 The EORTC and the Radiation Therapy Oncology Group (RTOG) are conducting a randomized prospective trial comparing fractionated radiation therapy alone, to 1) radiation followed by adjuvant temozolomide, to 2) concurrent chemoradiation without subsequent adjuvant therapy, and to 3) the regimen of concurrent chemoradiation followed by adjuvant chemotherapy that is the standard treatment for GBM.209,210 In addition, these prospective randomized trials are stratifying tumors based on MGMT promoter methylation status and molecular biomarkers.208
For patients with recurrent GBM, treatment outcomes are poor; the median time to tumor progression is 9 weeks, and the median survival is 25 weeks.211 PFS is correlated with OS and has become the benchmark for assessing treatment efficacy in patients with recurrent GBM in whom the PFS-6 rate ranges between 9% and 15%.211–213 For recurrent GBM and anaplastic astrocytoma, the transformative trials involve the use of anti-angiogenic drugs.214 The RTOG has completed two prospective randomized trials; one comparing two different adjuvant temozolomide regimens and another evaluating the efficacy of bevacizumab.215,216
Glioblastomas due to expression of a variety of pro-angiogenic factors are among the most vascular tumors. Angiogenesis is a critical process in the progression of gliomas.217 One of the main determinants of angiogenesis is VEGF, which is secreted by glioma cells to induce the tumor vascularization that in turn facilitates growth of the tumor.218 High expression of VEGF is correlated with poor clinical outcome, and it has been demonstrated that inhibition of VEGF decreases the growth of glioma cell lines.219 High-grade gliomas with a high degree of VEGF expression and vessel density respond best to anti-angiogenic therapy.220
Bevacizumab is an anti-angiogenic agent for GBM and received accelerated FDA approval for use in patients with recurrent GBM in 2009.221 Bevacizumab (Avastin) is a humanized monoclonal antibody that binds VEGF, thereby preventing the interaction of VEGF with its receptors VEGF receptor 1 and VEGF receptor 2. Blocking VEGF activity halts angiogenesis. The half-life of bevacizumab is approximately 20 days, so it is administered every 2 weeks and sometimes every 3 weeks. In Phase II studies in previously treated patients with malignant glioma, bevacizumab reduced requirements for steroids and was associated with imaging evidence of tumor response. These results have led to approval of bevacizumab for recurrent malignant glioma as well as investigation of bevacizumab as a component of initial combined modality therapy.222
Bevacizumab has demonstrated significant activity in Phase II trials.221 Bevacizumab alone or in combination with irinotecan resulted in response rates and time-to-progression that were substantially superior to historical controls with a range of cytotoxic regimens, and superior to results with any other molecularly-targeted drug therapy evaluated previously.223 However, the value of bevacizumab in the treatment of recurrent GBM remains uncertain since responses in GBM trials have not been durable. Norden et al224 compared PFS and OS of patients treated with bevacizumab with two contemporaneous trials of cytotoxic chemotherapy testing gimatecan and edotecarin. Median PFS in the bevacizumab cohort was 22 weeks, compared to only 8 weeks for the chemotherapy cohorts, and PFS-6 was 40% versus 11%. However, median OS was only 37 weeks in the bevacizumab cohort versus 39 weeks for the chemotherapy cohorts.224 Bevacizumab appears to have an effect on PFS, but only modest effects on OS.225 When patients progress through bevacizumab, the prognosis is dismal, with PFS of subsequent therapies being 4 weeks and PFS-6 being only 14%.226,227 Current and future trials evaluating combination therapies with molecularly-targeted drugs and bevacizumab have evolved a template structure in which bevacizumab is administered every 2 weeks in 28-day cycles, and the investigational agent is added to the monthly cycles with the scheduling dependent upon the biologic effect of the agent.
Although bevacizumab clearly produces a clinical improvement by decreasing the size of the contrast-enhancing mass lesion as well as ameliorating perilesion edema, the extent to which the drug is modifying the physiology of the BBB rather than killing tumor cells remains complex. When tumors progress after exposure to bevacizumab, subsequent therapies with cytotoxic chemotherapy are uniformly ineffective. In current practice, there is an emerging consensus that bevacizumab should be reserved for patients in whom the tumor is causing neurologic symptoms due to its size and surrounding edema.228 The ability of bevacizumab to suppress the early toxicities of radiation therapy has facilitated re-exploration of reirradiation with fractionated external beam techniques as well as radiosurgery for recurrent malignant gliomas. Several institutional trials229,230 have reported results of combining radiosurgery with Avastin in recurrent GBM and anaplastic astrocytoma. Although bevacizumab clearly reduces the early perilesion edema associated with radiosurgical treatment of recurrent malignant gliomas and produces radiologic responses by RANO criteria, it remains unproven whether the combination of radiosurgery and Avastin produces a more durable response, as measured by OS, than radiosurgery or bevacizumab alone.
Preliminary randomized Phase III trial results do not recommend the routine use of bevacizumab in combination with standard RT and temozolomide in patients with newly diagnosed glioblastoma.231,232 This recommendation is based on the lack of proven survival benefit for bevacizumab when used as part of initial therapy and the increased risk of toxicity associated with combination therapy. Certain subsets of patients may ultimately be shown to benefit from early use of bevacizumab, such as those patients with bulky, nonresectable tumors, but further study is needed. Preliminary results from two Phase III clinical trials231,232 assessing the role of bevacizumab in conjunction with RT plus temozolomide include the AVAglio study, in which 921 patients were randomly assigned to receive bevacizumab or placebo in conjunction with RT and temozolomide.231 After completion of RT, patients were treated with six cycles of monthly temozolomide plus bevacizumab or placebo every 2 weeks, followed by maintenance bevacizumab or placebo every 3 weeks until progression. At the time of the preliminary analysis, 76% of the expected events had occurred. They concluded that median PFS was improved in patients treated with bevacizumab compared with placebo (10.6 versus 6.2 months; hazard ratio 0.64, 95% confidence interval 0.55 to 0.74). However, median OS was not significantly different (hazard ratio 0.89, 95% confidence interval 0.75 to 1.07). As well, there was an increase in the rate of serious adverse events in patients treated with bevacizumab.
In the RTOG 0825 study, 637 patients were randomly assigned to receive bevacizumab or placebo starting at week 4 of standard chemoradiation with temozolomide, followed by six to 12 cycles of maintenance temozolomide plus bevacizumab or placebo.232 The conclusion was that PFS was extended in patients treated with bevacizumab (10.7 versus 7.3 months; P=0.004), but the result did not meet the predefined significance threshold of P<0.002. Median OS did not differ in patients treated with bevacizumab compared with placebo (15.7 versus 16.1 months, P=0.11). Notably, MGMT promoter methylation was strongly associated with improved PFS (14 versus 8 months for methylated versus unmethylated promoter, respectively) and OS (23 versus 14 months, respectively). In the subset of patients whose tumors exhibited both MGMT promoter methylation and a favorable nine-gene signature, there was a trend towards worse survival in patients treated with bevacizumab compared with placebo (15.7 versus 25 months, P=0.08). In addition, there was an increased rate of serious adverse events in patients treated with bevacizumab; primarily neutropenia, hypertension, and thromboembolism.
For recurrent anaplastic astrocytomas, the optimal chemotherapy regimens remain an active clinical trial question. A randomized prospective trial233 for anaplastic astrocytomas at first relapse after fractionated RT alone compared the older regimen PCV to standard (150–200 mg/m2/day for days 1–5 of 28-day cycles) temozolomide and dose-intensive temozolomide (75 mg/m2/day for days 1–21 of 28-day cycles). The day 1–5 regimen was not inferior to PCV, but the more dose-intense regimen was counterintuitively less effective. The optimal regimens for patients relapsing after prior chemoradiation or adjuvant chemotherapy remain to be determined.
For recurrent anaplastic oligodendrogliomas, alkylator-based chemotherapy is the mainstay of treatment, but as for astrocytomas, the optimal regimen and schedules are currently being pursued.12,234–236 A study by Lassman12 of anaplastic oligodendrogliomas suggest that for 1p/19q deleted tumors, the older PCV regimen may be associated with better outcomes. Despite this retrospective data, temozolomide continues to be more widely used in the US.
Despite a quarter century of disappointing results and evidence that the malignant gliomas microenvironment was inhospitable to cytotoxic T cells and natural killer cells, a relentless cadre of investigators has produced Phase II data suggesting that vaccine immunotherapy strategies can produce antitumor immune responses.237 In a study of newly diagnosed GBM tumors expressing the EGFRvIII oncoprotein antigen, an anti-EGFRviii dendritic cell vaccine demonstrated improved time-to-progression and OS as compared with a contemporaneous historical control data set.238 With all the caveats pertaining to historical control analysis and potential differences in distribution of molecular prognostic subgroups, vaccine therapies are demonstrating sufficient evidence of efficacy to warrant Phase III trials. As with clinical trials evaluating anti-angiogenic agents, criteria for determining tumor response and progression must be adapted to account for transient immune-mediated inflammatory responses that might be mistaken for development of tumor progression.239
Summary
In recent times, there has been important progress in our understanding of the molecular pathogenesis of malignant gliomas, leading to the development of targeted chemotherapeutic agents. Additionally, advances in diagnostic imaging have allowed for early diagnosis and treatment of malignant gliomas. As our understanding of the molecular pathogenesis and molecular imaging improves, it may be possible to select the most appropriate therapies on the basis of the patient’s tumor genotype. Furthermore, quantitative imaging biomarker assessment in the early phase of clinical trials could provide a novel approach for testing new therapies, and importantly, for facilitating patient management, sparing patients from weeks or months of toxicity due to ineffective treatment.
Acknowledgments
This work was support by the US National Institutes of Health research grant U01 CA140230, as well as the University of Pittsburgh Cancer Institute shared resources award P30CA047904. We also acknowledge the editorial support from Ms Moira Hitchens, Administrator, Department of Radiology, University of Pittsburgh.
This work was performed at the Department of Radiology, University of Pittsburgh, Pittsburgh, PA, USA.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | |
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. | |
Ostrom QT, Gittleman H, Farah P, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States 2006-2010. Neuro-Oncol 2013;15(sup 2):ii1–ii56. | |
Kleihues P, Louis DN, Scheithauer BW, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61(3):215–225; discussion 226–229. | |
Fisher JL, Schwartzbaum JA, Wrensch M, Wiemels JL. Epidemiology of brain tumors. Neurol Clin. 2007;25(4):867–890, vii. | |
Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol. 2004;22(14):2865–2872. | |
Davis FG, Dolecek TA, McCarthy BJ, Villano JL. Toward determining the lifetime occurrence of metastatic brain tumors estimated from 2007 United States cancer incidence data. Neuro Oncol. 2012;14(9):1171–1177. | |
Gupta K, Salunke P. Molecular markers of glioma: an update on recent progress and perspectives. J Cancer Res Clin Oncol. 2012;138(12):1971–1981. | |
Olar A, Aldape KD. Biomarkers classification and therapeutic decision-making for malignant gliomas. Curr Treat Options Oncol. 2012;13(4):417–436. | |
Clark KH, Villano JL, Nikiforova MN, Hamilton RL, Horbinski C. 1p/19q testing has no significance in the workup of glioblastomas. Neuropathol Appl Neurobiol. 2013;39(6):706–717. | |
Cairncross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013;31(3):337–343. | |
Lassman AB. Success at last: a molecular factor that informs treatment. Curr Oncol Rep. 2013;15(1):47–55. | |
van den Bent MJ, Brandes AA, Taphoorn MJ, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31(3):344–350. | |
Bello MJ, de Campos JM, Kusak ME, et al. Allelic loss at 1p is associated with tumor progression of meningiomas. Genes Chromosomes Cancer. 1994;9(4):296–298. | |
Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W, Collins VP. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol. 1994;145(5):1175–1190. | |
Ichimura K. Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol. 2012;29(3):131–139. | |
Hartmann C, Hentschel B, Wick W, et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. 2010;120(6):707–718. | |
Olson RA, Brastianos PK, Palma DA. Prognostic and predictive value of epigenetic silencing of MGMT in patients with high grade gliomas: a systematic review and meta-analysis. J Neurooncol. 2011;105(2):325–335. | |
Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. | |
Brandes AA, Franceschi E, Tosoni A, et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol. 2008;26(13):2192–2197. | |
Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170(5):1445–1453. | |
Olar A, Raghunathan A, Albarracin CT, et al. Absence of IDH1-R132H mutation predicts rapid progression of nonenhancing diffuse glioma in older adults. Ann Diagn Pathol. 2012;16(3):161–170. | |
Verhaak RG, Hoadley KA, Purdom E, et al; Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. | |
Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. | |
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. | |
Noushmehr H, Weisenberger DJ, Diefes K, et al; Cancer Genome Atlas Research Network. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. | |
Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci. 2009;16(6):748–754. | |
Mellinghoff IK, Schultz N, Mischel PS, Cloughesy TF. Will kinase inhibitors make it as glioblastoma drugs? Curr Top Microbiol Immunol. 2012;355:135–169. | |
Mellinghoff IK, Cloughesy TF, Mischel PS. PTEN-mediated resistance to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res. 2007;13(2 Pt 1):378–381. | |
Kim YH, Nonoguchi N, Paulus W, et al. Frequent BRAF gain in low-grade diffuse gliomas with 1p/19q loss. Brain Pathol. 2012;22(6):834–840. | |
Salama AK, Flaherty KT. BRAF in Melanoma: Current strategies and future directions. Clin Cancer Res. 2013;19(16):4326–4334. | |
Hoang-Xuan K, Capelle L, Kujas M, et al. Temozolomide as initial treatment for adults with low-grade oligodendrogliomas or oligoastrocytomas and correlation with chromosome 1p deletions. J Clin Oncol. 2004;22(15):3133–3138. | |
Hegi ME, Liu L, Herman JG, et al. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol. 2008;26(25):4189–4199. | |
Haas-Kogan DA, Prados MD, Tihan T, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97(12):880–887. | |
Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353(19):2012–2024. | |
Rich JN, Reardon DA, Peery T, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22(1):133–142. | |
Kienast Y, von Baumgarten L, Fuhrmann M, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010;16(1):116–122. | |
Seike T, Fujita K, Yamakawa Y, et al. Interaction between lung cancer cells and astrocytes via specific inflammatory cytokines in the microenvironment of brain metastasis. Clin Exp Metastasis. 2011;28(1):13–25. | |
Stemmler HJ, Kahlert S, Siekiera W, Untch M, Heinrich B, Heinemann V. Characteristics of patients with brain metastases receiving trastuzumab for HER2 overexpressing metastatic breast cancer. Breast. 2006;15(2):219–225. | |
Miller KD, Weathers T, Haney LG, et al. Occult central nervous system involvement in patients with metastatic breast cancer: prevalence, predictive factors and impact on overall survival. Ann Oncol. 2003;14(7):1072–1077. | |
Gabos Z, Sinha R, Hanson J, et al. Prognostic significance of human epidermal growth factor receptor positivity for the development of brain metastasis after newly diagnosed breast cancer. J Clin Oncol. 2006;24(36):5658–5663. | |
Lin NU, Bellon JR, Winer EP. CNS metastases in breast cancer. J Clin Oncol. 2004;22(17):3608–3617. | |
Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–1205. | |
Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–284. | |
Kelly PJ, Daumas-Duport C, Scheithauer BW, Kall BA, Kispert DB. Stereotactic histologic correlations of computed tomography- and magnetic resonance imaging-defined abnormalities in patients with glial neoplasms. Mayo Clin Proc. 1987;62(6):450–459. | |
Earnest F 4th, Kelly PJ, Scheithauer BW, et al. Cerebral astrocytomas: histopathologic correlation of MR and CT contrast enhancement with stereotactic biopsy. Radiology. 1988;166(3):823–827. | |
Buckner JC, Brown PD, O’Neill BP, Meyer FB, Wetmore CJ, Uhm JH. Central nervous system tumors. Mayo Clin Proc. 2007;82(10):1271–1286. | |
Grant R, Liang BC, Slattery J, Greenberg HS, Junck L. Chemotherapy response criteria in malignant glioma. Neurology. 1997;48(5):1336–1340. | |
Wilms G, Marchal G, Demaerel PH, Van Hecke P, Baert AL. Gadolinium-enhanced MRI of intracranial lesions. A review of indications and results. Clin Imaging. 1991;15(3):153–165. | |
Valk PE, Dillon WP. Radiation-Injury of the Brain. Am J Roentgenol. Apr 1991;156(4):689-706. | |
Mountz JM, Deutsch G, Kuzniecky R, Rosenfeld SS. Brain SPECT 1994 update. In: Freeman LM, ed. Nuclear medicine annual 1994. New York: Raven Press, 1994:1–54. | |
Schwartz RB, Holman BL, Polak JF, et al. Dual-isotope single-photon emission computerized tomography scanning in patients with glioblastoma multiforme: association with patient survival and histopathological characteristics of tumor after high-dose radiotherapy. J Neurosurg. 1998;89(1):60–68. | |
Kaplan WD, Takvorian T, Morris JH, Rumbaugh CL, Connolly BT, Atkins HL. Thallium-201 brain tumor imaging: a comparative study with pathologic correlation. J Nucl Med. 1987;28(1):47–52. | |
Black KL, Hawkins RA, Kim KT, Becker DP, Lerner C, Marciano D. Use of thallium-201 SPECT to quantitate malignancy grade of gliomas. J Neurosurg. 1989;71(3):342–346. | |
Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28(11):1963–1972. | |
Oborski MJ, Laymon CM, Lieberman FS, Mountz JM. Distinguishing pseudoprogression from progression in high-grade gliomas: a brief review of current clinical practice and demonstration of the potential value of 18F-FDG PET. Clin Nucl Med. 2013;38(5):381–384. | |
Shah AH, Snelling B, Bregy A, et al. Discriminating radiation necrosis from tumor progression in gliomas: a systematic review what is the best imaging modality? J Neurooncol. 2013;112(2):141–152. | |
Cha S. Update on brain tumor imaging: from anatomy to physiology. AJNR Am J Neuroradiol. 2006;27(3):475–487. | |
Young GS. Advanced MRI of adult brain tumors. Neurol Clin. 2007;25(4):947–973. | |
Levivier M, Becerra A, De Witte O, Brotchi J, Goldman S. Radiation necrosis or recurrence. J Neurosurg. 1996;84(1):148–149. | |
Del Sole A, Falini A, Ravasi L, et al. Anatomical and biochemical investigation of primary brain tumours. Eur J Nucl Med. 2001;28(12):1851–1872. | |
Ginsberg LE, Fuller GN, Hashmi M, Leeds NE, Schomer DF. The significance of lack of MR contrast enhancement of supratentorial brain tumors in adults: histopathological evaluation of a series. Surg Neurol. 1998;49(4):436–440. | |
Scott JN, Brasher PM, Sevick RJ, Rewcastle NB, Forsyth PA. How often are nonenhancing supratentorial gliomas malignant? A population study. Neurology. 2002;59(6):947–949. | |
van den Bent MJ, Vogelbaum MA, Wen PY, Macdonald DR, Chang SM. End point assessment in gliomas: novel treatments limit usefulness of classical Macdonald’s Criteria. J Clin Oncol. 2009;27(18):2905–2908. | |
Taal W, Brandsma D, de Bruin HG, et al. Incidence of early pseudo-progression in a cohort of malignant glioma patients treated with chemoirradiation with temozolomide. Cancer. 2008;113(2):405–410. | |
Hamstra DA, Galbán CJ, Meyer CR, et al. Functional diffusion map as an early imaging biomarker for high-grade glioma: correlation with conventional radiologic response and overall survival. J Clin Oncol. 2008;26(20):3387–3394. | |
Hamstra DA, Chenevert TL, Moffat BA, et al. Evaluation of the functional diffusion map as an early biomarker of time-to-progression and overall survival in high-grade glioma. Proc Natl Acad Sci U S A. 2005;102(46):16759–16764. | |
Nowosielski M, Recheis W, Goebel G, et al. ADC histograms predict response to anti-angiogenic therapy in patients with recurrent high-grade glioma. Neuroradiology. 2011;53(4):291–302. | |
Pope WB, Kim HJ, Huo J, et al. Recurrent glioblastoma multiforme: ADC histogram analysis predicts response to bevacizumab treatment. Radiology. 2009;252(1):182–189. | |
Norden AD, Young GS, Setayesh K, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70(10):779–787. | |
Castillo M, Kwock L, Mukherji SK. Clinical applications of proton MR spectroscopy. AJNR Am J Neuroradiol. 1996;17(1):1–15. | |
Miller BL. A review of chemical issues in 1H NMR spectroscopy: N-acetyl-L-aspartate, creatine and choline. NMR Biomed. 1991;4(2):47–52. | |
Castillo M, Kwock L, Scatliff J, Mukherji SK. Proton MR spectroscopy in neoplastic and non-neoplastic brain disorders. Magn Reson Imaging Clin N Am. 1998;6(1):1–20. | |
Michaelis T, Merboldt KD, Bruhn H, Hänicke W, Frahm J. Absolute concentrations of metabolites in the adult human brain in vivo: quantification of localized proton MR spectra. Radiology. 1993;187(1):219–227. | |
Herminghaus S, Pilatus U, Möller-Hartmann W, et al. Increased choline levels coincide with enhanced proliferative activity of human neuroepithelial brain tumors. NMR Biomed. 2002;15(6):385–392. | |
Kreis R, Ernst T, Ross BD. Development of the human brain: in vivo quantification of metabolite and water content with proton magnetic resonance spectroscopy. Magn Reson Med. 1993;30(4):424–437. | |
Li X, Lu Y, Pirzkall A, McKnight T, Nelson SJ. Analysis of the spatial characteristics of metabolic abnormalities in newly diagnosed glioma patients. J Magn Reson Imaging. 2002;16(3):229–237. | |
Hattingen E, Delic O, Franz K, et al. (1)H MRSI and progression-free survival in patients with WHO grades II and III gliomas. Neurol Res. 2010;32(6):593–602. | |
Hattingen E, Raab P, Franz K, et al. Prognostic value of choline and creatine in WHO grade II gliomas. Neuroradiology. 2008;50(9):759–767. | |
Imani F, Boada FE, Lieberman FS, Davis DK, Deeb EL, Mountz JM. Comparison of proton magnetic resonance spectroscopy with fluorine-18 2-fluoro-deoxyglucose positron emission tomography for assessment of brain tumor progression. J Neuroimaging. 2012;22(2):184–190. | |
Elkhaled A, Jalbert LE, Phillips JJ, et al. Magnetic resonance of 2- hydroxyglutarate in IDH1-mutated low-grade gliomas. Sci Transl Med. 2012;4(116):116ra5. | |
Kalinina J, Carroll A, Wang L, et al. Detection of “oncometabolite” 2- hydroxyglutarate by magnetic resonance analysis as a biomarker of IDH1/2 mutations in glioma. J Mol Med (Berl). 2012;90(10):1161–1171. | |
Pope WB, Prins RM, Albert Thomas M, et al. Non-invasive detection of 2-hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. J Neurooncol. 2012;107(1):197–205. | |
Le Bihan D. Molecular diffusion, tissue microdynamics and microstructure. NMR Biomed. 1995;8(7–8):375–386. | |
Arvinda HR, Kesavadas C, Sarma PS, et al. Glioma grading: sensitivity, specificity, positive and negative predictive values of diffusion and perfusion imaging. J Neurooncol. 2009;94(1):87–96. | |
Jensen JH, Helpern JA. MRI quantification of non-Gaussian water diffusion by kurtosis analysis. NMR Biomed. 2010;23(7):698–710. | |
Raab P, Hattingen E, Franz K, Zanella FE, Lanfermann H. Cerebral gliomas: diffusional kurtosis imaging analysis of microstructural differences. Radiology. 2010;254(3):876–881. | |
Cha S, Knopp EA, Johnson G, Wetzel SG, Litt AW, Zagzag D. Intracranial mass lesions: dynamic contrast-enhanced susceptibility-weighted echo-planar perfusion MR imaging. Radiology. 2002;223(1):11–29. | |
Boxerman JL, Schmainda KM, Weisskoff RM. Relative cerebral blood volume maps corrected for contrast agent extravasation significantly correlate with glioma tumor grade, whereas uncorrected maps do not. AJNR Am J Neuroradiol. 2006;27(4):859–867. | |
Hu LS, Baxter LC, Pinnaduwage DS, et al. Optimized preload leakage-correction methods to improve the diagnostic accuracy of dynamic susceptibility-weighted contrast-enhanced perfusion MR imaging in posttreatment gliomas. AJNR Am J Neuroradiol. 2010;31(1):40–48. | |
Law M, Yang S, Wang H, et al. Glioma grading: sensitivity, specificity, and predictive values of perfusion MR imaging and proton MR spectroscopic imaging compared with conventional MR imaging. AJNR Am J Neuroradiol. 2003;24(10):1989–1998. | |
Provenzale JM, York G, Moya MG, et al. Correlation of relative permeability and relative cerebral blood volume in high-grade cerebral neoplasms. AJR Am J Roentgenol. 2006;187(4):1036–1042. | |
Roberts C, Issa B, Stone A, Jackson A, Waterton JC, Parker GJ. Comparative study into the robustness of compartmental modeling and model-free analysis in DCE-MRI studies. J Magn Reson Imaging. 2006;23(4):554–563. | |
Spampinato MV, Smith JK, Kwock L, et al. Cerebral blood volume measurements and proton MR spectroscopy in grading of oligodendroglial tumors. AJR Am J Roentgenol. 2007;188(1):204–212. | |
Danchaivijitr N, Waldman AD, Tozer DJ, et al. Low-grade gliomas: do changes in rCBV measurements at longitudinal perfusion-weighted MR imaging predict malignant transformation? Radiology. 2008;247(1):170–178. | |
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. | |
Krohn KA, Mankoff DA, Muzi M, Link JM, Spence AM. True tracers: comparing FDG with glucose and FLT with thymidine. Nucl Med Biol. 2005;32(7):663–671. | |
Padma MV, Said S, Jacobs M, et al. Prediction of pathology and survival by FDG PET in gliomas. J Neurooncol. 2003;64(3):227–237. | |
De Witte O, Levivier M, Violon P, et al. Prognostic value positron emission tomography with [18F]fluoro-2-deoxy-D-glucose in the low-grade glioma. Neurosurgery. 1996;39(3):470–476; discussion 476–477. | |
Delbeke D, Meyerowitz C, Lapidus RL, et al. Optimal cutoff levels of F-18 fluorodeoxyglucose uptake in the differentiation of low-grade from high-grade brain tumors with PET. Radiology. 1995;195(1):47–52. | |
Spence AM, Muzi M, Mankoff DA, et al. 18F-FDG PET of gliomas at delayed intervals: improved distinction between tumor and normal gray matter. J Nucl Med. 2004;45(10):1653–1659. | |
Wong TZ, van der Westhuizen GJ, Coleman RE. Positron emission tomography imaging of brain tumors. Neuroimaging Clin N Am. 2002;12(4):615–626. | |
Würker M, Herholz K, Voges J, et al. Glucose consumption and methionine uptake in low-grade gliomas after iodine-125 brachytherapy. Eur J Nucl Med. 1996;23(5):583–586. | |
Ricci PE, Karis JP, Heiserman JE, Fram EK, Bice AN, Drayer BP. Differentiating recurrent tumor from radiation necrosis: time for re-evaluation of positron emission tomography? AJNR Am J Neuroradiol. 1998;19(3):407–413. | |
Langleben DD, Segall GM. PET in differentiation of recurrent brain tumor from radiation injury. J Nucl Med. 2000;41(11):1861–1867. | |
Franceschi E, Tosoni A, Bartolini S, Mazzocchi V, Fioravanti A, Brandes AA. Treatment options for recurrent glioblastoma: pitfalls and future trends. Expert Rev Anticancer Ther. 2009;9(5):613–619. | |
Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8(7):1277–1280. | |
Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–1354. | |
Brandes AA, Tosoni A, Cavallo G, et al; GICNO. Correlations between O6-methylguanine DNA methyltransferase promoter methylation status, 1p and 19q deletions, and response to temozolomide in anaplastic and recurrent oligodendroglioma: a prospective GICNO study. J Clin Oncol. 2006;24(29):4746–4753. | |
Clarke JL, Chang S. Pseudoprogression and pseudoresponse: challenges in brain tumor imaging. Curr Neurol Neurosci Rep. 2009;9(3):241–246. | |
Vredenburgh JJ, Desjardins A, Herndon JE 2nd, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13(4):1253–1259. | |
Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11(1):83–95. | |
Desjardins A, Reardon DA, Herndon JE 2nd, et al. Bevacizumab plus irinotecan in recurrent WHO grade 3 malignant gliomas. Clin Cancer Res. 2008;14(21):7068–7073. | |
Chao ST, Suh JH, Raja S, Lee SY, Barnett G. The sensitivity and specificity of FDG PET in distinguishing recurrent brain tumor from radionecrosis in patients treated with stereotactic radiosurgery. Int J Cancer. 2001;96(3):191–197. | |
Wang SX, Boethius J, Ericson K. FDG-PET on irradiated brain tumor: ten years’ summary. Acta Radiol. 2006;47(1):85–90. | |
Schlemmer HP, Pichler BJ, Schmand M, et al. Simultaneous MR/PET imaging of the human brain: feasibility study. Radiology. 2008;248(3):1028–1035. | |
Boss A, Bisdas S, Kolb A, et al. Hybrid PET/MRI of intracranial masses: initial experiences and comparison to PET/CT. J Nucl Med. 2010;51(8):1198–1205. | |
Tanaka Y, Nariai T, Momose T, et al. Glioma surgery using a multimodal navigation system with integrated metabolic images. J Neurosurg. 2009;110(1):163–172. | |
Grosu AL, Weber WA, Riedel E, et al. L-(methyl-11C) methionine positron emission tomography for target delineation in resected high-grade gliomas before radiotherapy. Int J Radiat Oncol Biol Phys. 2005;63(1):64–74. | |
Weber DC, Zilli T, Buchegger F, et al. [(18)F]Fluoroethyltyrosine- positron emission tomography-guided radiotherapy for high-grade glioma. Radiat Oncol. 2008;3:44. | |
Vees H, Senthamizhchelvan S, Miralbell R, Weber DC, Ratib O, Zaidi H. Assessment of various strategies for 18F-FET PET-guided delineation of target volumes in high-grade glioma patients. Eur J Nucl Med Mol Imaging. 2009;36(2):182–193. | |
Nariai T, Tanaka Y, Wakimoto H, et al. Usefulness of L-[methyl-11C] methionine-positron emission tomography as a biological monitoring tool in the treatment of glioma. J Neurosurg. 2005;103(3):498–507. | |
Busch H, Davis JR, Honig GR, Anderson DC, Nair PV, Nyhan WL. The uptake of a variety of amino acids into nuclear proteins of tumors and other tissues. Cancer Res. 1959;19:1030–1039. | |
Miyagawa T, Oku T, Uehara H, et al. “Facilitated” amino acid transport is upregulated in brain tumors. J Cereb Blood Flow Metab. 1998;18(5):500–509. | |
Galldiks N, Kracht LW, Burghaus L, et al. Use of 11C-methionine PET to monitor the effects of temozolomide chemotherapy in malignant gliomas. Eur J Nucl Med Mol Imaging. 2006;33(5):516–524. | |
Wyss M, Hofer S, Bruehlmeier M, et al. Early metabolic responses in temozolomide treated low-grade glioma patients. J Neurooncol. 2009;95(1):87–93. | |
Galldiks N, Kracht LW, Burghaus L, et al. Patient-tailored, imaging-guided, long-term temozolomide chemotherapy in patients with glioblastoma. Mol Imaging. 2010;9(1):40–46. | |
Jager PL, Vaalburg W, Pruim J, de Vries EG, Langen KJ, Piers DA. Radiolabeled amino acids: basic aspects and clinical applications in oncology. J Nucl Med. 2001;42(3):432–445. | |
Bergmann R, Pietzsch J, Fuechtner F, et al. 3-O-methyl-6-18F-fluoro-L-dopa, a new tumor imaging agent: investigation of transport mechanism in vitro. J Nucl Med. 2004;45(12):2116–2122. | |
Ullrich RT, Kracht LW, Jacobs AH. Neuroimaging in patients with gliomas. Semin Neurol. 2008;28(4):484–494. | |
Weber WA, Wester HJ, Grosu AL, et al. O-(2-[18F]fluoroethyl)-L-tyrosine and L-[methyl-11C]methionine uptake in brain tumours: initial results of a comparative study. Eur J Nucl Med. 2000;27(5):542–549. | |
Becherer A, Karanikas G, Szabó M, et al. Brain tumour imaging with PET: a comparison between [18F]fluorodopa and [11C]methionine. Eur J Nucl Med Mol Imaging. 2003;30(11):1561–1567. | |
Herholz K, Hölzer T, Bauer B, et al. 11C-methionine PET for differential diagnosis of low-grade gliomas. Neurology. 1998;50(5):1316–1322. | |
Hustinx R, Pourdehnad M, Kaschten B, Alavi A. PET imaging for differentiating recurrent brain tumor from radiation necrosis. Radiol Clin North Am. 2005;43(1):35–47. | |
Ericson K, Lilja A, Bergström M, et al. Positron emission tomography with ([11C]methyl)-L-methionine, [11C]D-glucose, and [68Ga]EDTA in supratentorial tumors. J Comput Assist Tomogr. 1985;9(4):683–689. | |
Ogawa T, Inugami A, Hatazawa J, et al. Clinical positron emission tomography for brain tumors: comparison of fludeoxyglucose F 18 and L-methyl-11C-methionine. AJNR Am J Neuroradiol. 1996;17(2):345–353. | |
Chang CC, Kuwana N, Ito S, Yokoyama T, Kanno H, Yamamoto I. Cerebral haemodynamics in patients with hydrocephalus after subarachnoid haemorrhage due to ruptured aneurysm. Eur J Nucl Med Mol Imaging. 2003;30(1):123–126. | |
De Witte O, Goldberg I, Wikler D, et al. Positron emission tomography with injection of methionine as a prognostic factor in glioma. J Neurosurg. 2001;95(5):746–750. | |
Ribom D, Eriksson A, Hartman M, et al. Positron emission tomography (11)C-methionine and survival in patients with low-grade gliomas. Cancer. 2001;92(6):1541–1549. | |
Kaschten B, Stevenaert A, Sadzot B, et al. Preoperative evaluation of 54 gliomas by PET with fluorine-18-fluorodeoxyglucose and/or carbon-11-methionine. J Nucl Med. 1998;39(5):778–785. | |
Sasaki M, Kuwabara Y, Yoshida T, et al. Carbon-11-methionine PET in focal cortical dysplasia: a comparison with fluorine-18-FDG PET and technetium-99m-ECD SPECT. J Nucl Med. 1998;39(6):974–977. | |
Bustany P, Chatel M, Derlon JM, et al. Brain tumor protein synthesis and histological grades: a study by positron emission tomography (PET) with C11-L-Methionine. J Neurooncol. 1986;3(4):397–404. | |
Ullrich RT, Kracht L, Brunn A, et al. Methyl-L-11C-methionine PET as a diagnostic marker for malignant progression in patients with glioma. J Nucl Med. 2009;50(12):1962–1968. | |
Yamane T, Sakamoto S, Senda M. Clinical impact of (11)C-methionine PET on expected management of patients with brain neoplasm. Eur J Nucl Med Mol Imaging. 2010;37(4):685–690. | |
Spaeth N, Wyss MT, Weber B, et al. Uptake of 18F-fluorocholine, 18F-fluoroethyl-L-tyrosine, and 18F-FDG in acute cerebral radiation injury in the rat: implications for separation of radiation necrosis from tumor recurrence. J Nucl Med. 2004;45(11):1931–1938. | |
Chen W, Silverman DH, Delaloye S, et al. 18F-FDOPA PET imaging of brain tumors: comparison study with 18F-FDG PET and evaluation of diagnostic accuracy. J Nucl Med. 2006;47(6):904–911. | |
Pauleit D, Floeth F, Hamacher K, et al. O-(2-[18F]fluoroethyl)-L-tyrosine PET combined with MRI improves the diagnostic assessment of cerebral gliomas. Brain. 2005;128(Pt 3):678–687. | |
Rachinger W, Goetz C, Pöpperl G, et al. Positron emission tomography with O-(2-[18F]fluoroethyl)-l-tyrosine versus magnetic resonance imaging in the diagnosis of recurrent gliomas. Neurosurgery. 2005;57(3):505–511; discussion 505–511. | |
Floeth FW, Pauleit D, Wittsack HJ, et al. Multimodal metabolic imaging of cerebral gliomas: positron emission tomography with [18F]fluoroethyl-L-tyrosine and magnetic resonance spectroscopy. J Neurosurg. 2005;102(2):318–327. | |
Rasey JS, Grierson JR, Wiens LW, Kolb PD, Schwartz JL. Validation of FLT uptake as a measure of thymidine kinase-1 activity in A549 carcinoma cells. J Nucl Med. 2002;43(9):1210–1217. | |
Shields AF, Grierson JR, Dohmen BM, et al. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med. 1998;4(11):1334–1336. | |
Barthel H, Cleij MC, Collingridge DR, et al. 3′-deoxy-3′-[18F]fluorothymidine as a new marker for monitoring tumor response to antiproliferative therapy in vivo with positron emission tomography. Cancer Res. 2003;63(13):3791–3798. | |
Chen W, Cloughesy T, Kamdar N, et al. Imaging proliferation in brain tumors with 18F-FLT PET: comparison with 18F-FDG. J Nucl Med. 2005;46(6):945–952. | |
Munch-Petersen B, Cloos L, Jensen HK, Tyrsted G. Human thymidine kinase 1. Regulation in normal and malignant cells. Adv Enzyme Regul. 1995;35:69–89. | |
Hatakeyama T, Kawai N, Nishiyama Y, et al. 11C-methionine (MET) and 18F-fluorothymidine (FLT) PET in patients with newly diagnosed glioma. Eur J Nucl Med Mol Imaging. 2008;35(11):2009–2017. | |
Jacobs AH, Thomas A, Kracht LW, et al. 18F-fluoro-L-thymidine and 11C-methylmethionine as markers of increased transport and proliferation in brain tumors. J Nucl Med. 2005;46(12):1948–1958. | |
Chen W, Delaloye S, Silverman DH, et al. Predicting treatment response of malignant gliomas to bevacizumab and irinotecan by imaging proliferation with [18F] fluorothymidine positron emission tomography: a pilot study. J Clin Oncol. 2007;25(30):4714–4721. | |
Schwarzenberg J, Czernin J, Cloughesy TF, et al. 3′-deoxy-3′-18F-fluorothymidine PET and MRI for early survival predictions in patients with recurrent malignant glioma treated with bevacizumab. J Nucl Med. 2012;53(1):29–36. | |
Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23(5):1011–1027. | |
Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333(2):328–335. | |
Rasey JS, Koh WJ, Evans ML, et al. Quantifying regional hypoxia in human tumors with positron emission tomography of [18F]fluoromisonidazole: a pretherapy study of 37 patients. Int J Radiat Oncol Biol Phys. 1996;36(2):417–428. | |
Brown JM. Therapeutic targets in radiotherapy. Int J Radiat Oncol Biol Phys. 2001;49(2):319–326. | |
Szeto MD, Chakraborty G, Hadley J, et al. Quantitative metrics of net proliferation and invasion link biological aggressiveness assessed by MRI with hypoxia assessed by FMISO-PET in newly diagnosed glioblastomas. Cancer Res. 2009;69(10):4502–4509. | |
Cher LM, Murone C, Lawrentschuk N, et al. Correlation of hypoxic cell fraction and angiogenesis with glucose metabolic rate in gliomas using 18F-fluoromisonidazole, 18F-FDG PET, and immunohistochemical studies. J Nucl Med. 2006;47(3):410–418. | |
Spence AM, Muzi M, Swanson KR, et al. Regional hypoxia in glioblastoma multiforme quantified with [18F]fluoromisonidazole positron emission tomography before radiotherapy: correlation with time to progression and survival. Clin Cancer Res. 2008;14(9):2623–2630. | |
Paleologos TS, Dorward NL, Wadley JP, Thomas DG. Clinical validation of true frameless stereotactic biopsy: analysis of the first 125 consecutive cases. Neurosurgery. 2001;49(4):830–835; discussion 835–837. | |
Robbins PD, Yu LL, Lee M, et al. Stereotactic biopsy of 100 intracerebral lesions at Sir Charles Gairdner Hospital. Pathology. 1994;26(4):410–413. | |
Sanai N. Emerging operative strategies in neurosurgical oncology. Curr Opin Neurol. 2012;25(6):756–766. | |
Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J. Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol. 2013;10(1):14–26. | |
Gilbert MR. Recurrent glioblastoma: a fresh look at current therapies and emerging novel approaches. Semin Oncol. 2011;38 Suppl 4:S21–S33. | |
Weller M, Wick W, Hegi ME, Stupp R, Tabatabai G. Should biomarkers be used to design personalized medicine for the treatment of glioblastoma? Future Oncol. 2010;6(9):1407–1414. | |
Keles GE, Anderson B, Berger MS. The effect of extent of resection on time to tumor progression and survival in patients with glioblastoma multiforme of the cerebral hemisphere. Surg Neurol. 1999;52(4):371–379. | |
Pope WB, Sayre J, Perlina A, Villablanca JP, Mischel PS, Cloughesy TF. MR imaging correlates of survival in patients with high-grade gliomas. AJNR Am J Neuroradiol. 2005;26(10):2466–2474. | |
Lacroix M, Abi-Said D, Fourney DR, et al. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg. 2001;95(2):190–198. | |
Keles GE, Chang EF, Lamborn KR, et al. Volumetric extent of resection and residual contrast enhancement on initial surgery as predictors of outcome in adult patients with hemispheric anaplastic astrocytoma. J Neurosurg. 2006;105(1):34–40. | |
Duffau H, Capelle L, Sichez J, et al. Intra-operative direct electrical stimulations of the central nervous system: the Salpetriere experience with 60 patients. Acta Neurochir (Wien). 1999;141(11):1157–1167. | |
Duffau H, Capelle L, Sichez N, et al. Intraoperative mapping of the subcortical language pathways using direct stimulations. An anatomo-functional study. Brain. 2002;125(Pt 1):199–214. | |
Duffau H, Capelle L, Denvil D, et al. Usefulness of intraoperative electrical subcortical mapping during surgery for low-grade gliomas located within eloquent brain regions: functional results in a consecutive series of 103 patients. J Neurosurg. 2003;98(4):764–778. | |
Lee SK. Diffusion tensor and perfusion imaging of brain tumors in high-field MR imaging. Neuroimaging Clin N Am. 2012;22(2):123–134, ix. | |
Price SJ, Gillard JH. Imaging biomarkers of brain tumour margin and tumour invasion. Br J Radiol. 2011;84 Spec No 2:S159–S167. | |
Sanai N, Mirzadeh Z, Berger MS. Functional outcome after language mapping for glioma resection. N Engl J Med. 2008;358(1):18–27. | |
Snyderman CH, Pant H, Carrau RL, Prevedello D, Gardner P, Kassam AB. What are the limits of endoscopic sinus surgery?: the expanded endonasal approach to the skull base. Keio J Med. 2009;58(3):152–160. | |
McLaughlin N, Prevedello DM, Engh J, Kelly DF, Kassam AB. Endoneurosurgical resection of intraventricular and intraparenchymal lesions using the port technique. World Neurosurg. 2013; 79(Suppl 2):S18.e1–S18.e8. | |
Walker MD, Alexander E Jr, Hunt WE, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg. 1978;49(3):333–343. | |
Walker MD, Strike TA, Sheline GE. An analysis of dose-effect relationship in the radiotherapy of malignant gliomas. Int J Radiat Oncol Biol Phys. 1979;5(10):1725–1731. | |
Sheline GE. Radiotherapy for high grade gliomas. Int J Radiat Oncol Biol Phys. 1990;18(4):793–803. | |
Hochberg FH, Pruitt A. Assumptions in the radiotherapy of glioblastoma. Neurology. 1980;30(9):907–911. | |
Wallner KE, Galicich JH, Krol G, Arbit E, Malkin MG. Patterns of failure following treatment for glioblastoma multiforme and anaplastic astrocytoma. Int J Radiat Oncol Biol Phys. 1989;16(6):1405–1409. | |
Choucair AK, Levin VA, Gutin PH, et al. Development of multiple lesions during radiation therapy and chemotherapy in patients with gliomas. J Neurosurg. 1986;65(5):654–658. | |
Ten Haken RK, Thornton AF Jr, Sandler HM, et al. A quantitative assessment of the addition of MRI to CT-based, 3-D treatment planning of brain tumors. Radiother Oncol. 1992;25(2):121–133. | |
Glatstein E, Lichter AS, Fraass BA, Kelly BA, van de Geijn J. The imaging revolution and radiation oncology: use of CT, ultrasound, and NMR for localization, treatment planning and treatment delivery. Int J Radiat Oncol Biol Phys. 1985;11(2):299–314. | |
Gross MW, Weber WA, Feldmann HJ, Bartenstein P, Schwaiger M, Molls M. The value of F-18-fluorodeoxyglucose PET for the 3-D radiation treatment planning of malignant gliomas. Int J Radiat Oncol Biol Phys. 1998;41(5):989–995. | |
Douglas JG, Stelzer KJ, Mankoff DA, et al. [F-18]-fluorodeoxyglucose positron emission tomography for targeting radiation dose escalation for patients with glioblastoma multiforme: clinical outcomes and patterns of failure. Int J Radiat Oncol Biol Phys. 2006;64(3):886–891. | |
Narayana A, Yamada J, Berry S, et al. Intensity-modulated radiotherapy in high-grade gliomas: clinical and dosimetric results. Int J Radiat Oncol Biol Phys. 2006;64(3):892–897. | |
Chan JL, Lee SW, Fraass BA, et al. Survival and failure patterns of high-grade gliomas after three-dimensional conformal radiotherapy. J Clin Oncol. 2002;20(6):1635–1642. | |
Shrieve DC, Alexander E 3rd, Black PM, et al. Treatment of patients with primary glioblastoma multiforme with standard postoperative radiotherapy and radiosurgical boost: prognostic factors and long-term outcome. J Neurosurg. 1999;90(1):72–77. | |
Prisco FE, Weltman E, de Hanriot RM, Brandt RA. Radiosurgical boost for primary high-grade gliomas. J Neurooncol. 2002;57(2):151–160. | |
Nwokedi EC, DiBiase SJ, Jabbour S, Herman J, Amin P, Chin LS. Gamma knife stereotactic radiosurgery for patients with glioblastoma multiforme. Neurosurgery. 2002;50(1):41–46; discussion 46–47. | |
Mehta MP, Masciopinto J, Rozental J, et al. Stereotactic radiosurgery for glioblastoma multiforme: report of a prospective study evaluating prognostic factors and analyzing long-term survival advantage. Int J Radiat Oncol Biol Phys. 1994;30(3):541–549. | |
Koga T, Maruyama K, Tanaka M, et al. Extended field stereotactic radiosurgery for recurrent glioblastoma. Cancer. 2012;118(17):4193–4200. | |
Kondziolka D, Flickinger JC, Bissonette DJ, Bozik M, Lunsford LD. Survival benefit of stereotactic radiosurgery for patients with malignant glial neoplasms. Neurosurgery. Oct 1997;41(4):776–783; discussion 783–775. | |
Souhami L, Seiferheld W, Brachman D, et al. Randomized comparison of stereotactic radiosurgery followed by conventional radiotherapy with carmustine to conventional radiotherapy with carmustine for patients with glioblastoma multiforme: report of Radiation Therapy Oncology Group 93-05 protocol. Int J Radiat Oncol Biol Phys. 2004;60(3):853–860. | |
Park KJ, Kano H, Iyer A, et al. Salvage gamma knife stereotactic radiosurgery followed by bevacizumab for recurrent glioblastoma multiforme: a case-control study. J Neurooncol. 2012;107(2):323–333. | |
Koga T, Shin M, Saito N. Role of gamma knife radiosurgery in neurosurgery: past and future perspectives. Neurol Med Chir (Tokyo). 2010;50(9):737–748. | |
Stupp R, Mason WP, van den Bent MJ, et al; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | |
Stupp R, Hegi ME, Mason WP, et al; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. | |
Erdem-Eraslan L, Gravendeel LA, de Rooi J, et al. Intrinsic molecular subtypes of glioma are prognostic and predict benefit from adjuvant procarbazine, lomustine, and vincristine chemotherapy in combination with other prognostic factors in anaplastic oligodendroglial brain tumors: a report from EORTC study 26951. J Clin Oncol. 2013;31(3):328–336. | |
Wang Y, Jiang T. Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. Cancer Lett. 2013;331(2):139–146. | |
Alliance for Clinical Trials in Oncology. Radiation Therapy With Concomitant and Adjuvant Temozolomide or Radiation Therapy With Adjuvant PCV or Temozolomide Alone in Treating Patients With Anaplastic Glioma. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2009 [updated November 7, 2013]. Available from: http://clinicaltrials.gov/ct2/show/study/NCT00887146. NLM identifier: NCT00887146. Accessed February 5, 2014. | |
European Organisation for Research and Treatment of Cancer – EORTC. Radiation Therapy With or Without Temozolomide in Treating Patients With Anaplastic Glioma. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2008 [updated September 16, 2011]. Available from: http://clinicaltrials.gov/show/NCT00626990. NLM identifier: NCT00626990. Accessed February 5, 2014. | |
Wong ET, Hess KR, Gleason MJ, et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol. 1999;17(8):2572–2578. | |
Lamborn KR, Yung WK, Chang SM, et al; North American Brain Tumor Consortium. Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol. 2008;10(2):162–170. | |
Ballman KV, Buckner JC, Brown PD, et al. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro Oncol. 2007;9(1):29–38. | |
Gerstner ER, Batchelor TT. Antiangiogenic therapy for glioblastoma. Cancer J. 2012;18(1):45–50. | |
Vredenburgh JJ, Desjardins A, Reardon DA, et al. The addition of bevacizumab to standard radiation therapy and temozolomide followed by bevacizumab, temozolomide, and irinotecan for newly diagnosed glioblastoma. Clin Cancer Res. 2011;17(12):4119–4124. | |
Haslinghuis LM, Krenning EP, De Herder WW, Reijs AE, Kwekkeboom DJ. Somatostatin receptor scintigraphy in the follow-up of patients with differentiated thyroid cancer. J Endocrinol Invest. 2001;24(6):415–422. | |
Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8(8):610–622. | |
Gupta K, Radotra BD, Banerjee AK, Nijhawan R. Quantitation of angiogenesis and its correlation with vascular endothelial growth factor expression in astrocytic tumors. Anal Quant Cytol Histol. 2004;26(4):223–229. | |
Nam DH, Park K, Suh YL, Kim JH. Expression of VEGF and brain specific angiogenesis inhibitor-1 in glioblastoma: prognostic significance. Oncol Rep. 2004;11(4):863–869. | |
Takano S, Tsuboi K, Matsumura A, Nose T. Anti-vascular endothelial growth factor antibody and nimustine as combined therapy: effects on tumour growth and angiogenesis in human glioblastoma xenografts. Neuro Oncol. 2003;5(1):1–7. | |
Cohen MH, Shen YL, Keegan P, Pazdur R. FDA Drug Approval Summary: Bevacizumab (Avastin (R)) as Treatment of Recurrent Glioblastoma Multiforme. Oncologist. 2009;14(11):1131–1138. | |
Lai A, Tran A, Nghiemphu PL, et al. Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. J Clin Oncol. 2011;29(2):142–148. | |
Reardon DA, Turner S, Peters KB, et al. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J Natl Compr Canc Netw. 2011;9(4):414–427. | |
Norden AD, Drappatz J, Muzikansky A, et al. An exploratory survival analysis of anti-angiogenic therapy for recurrent malignant glioma. J Neurooncol. 2009;92(2):149–155. | |
Beal K, Abrey LE, Gutin PH. Antiangiogenic agents in the treatment of recurrent or newly diagnosed glioblastoma: analysis of single-agent and combined modality approaches. Radiat Oncol. 2011;6:2. | |
Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27(5):740–745. | |
Khasraw M, Lassman AB. Advances in the treatment of malignant gliomas. Curr Oncol Rep. 2010;12(1):26–33. | |
Kunnakkat S, Narayana A. Bevacizumab in the treatment of high-grade gliomas: an overview. Angiogenesis. 2011;14(4):423–430. | |
Memorial Sloan-Kettering Cancer Center. Hypofractionated Stereotactic Radiotherapy With Bevacizumab in the Treatment of Recurrent Malignant Glioma. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2011 [updated December 17, 2013]. Available from http://clinicaltrials.gov/ct2/show/NCT01392209. NLM identifier: NCT01392209. Accessed February 5, 2014. | |
Cabrera AR, Cuneo KC, Vredenburgh JJ, Sampson JH, Kirkpatrick JP. Stereotactic radiosurgery and bevacizumab for recurrent glioblastoma multiforme. J Natl Compr Canc Netw. 2012;10(6):695–699. | |
Cloughsey T, Wick W, Mason W, et al. Phase III trial of bevacizumab added to standard radiotherapy and temozolomide for newly diagnosed glioblastoma: Final progression-free survival and preliminary overall survival results from AVAglio. Neurology. 2013;80(Meeting Abstracts 1): PL02.002. | |
Gilbert MR, Dignam J, Won M, et al. RTOG 0825: Phase III double-blind placebo-controlled trial evaluating bevacizumab (Bev) in patients (Pts) with newly diagnosed glioblastoma (GBM). J Clin Oncol. 2013;31 Suppl:abstr 1. | |
van den Bent MJ, Chinot O, Boogerd W, et al. Second-line chemotherapy with temozolomide in recurrent oligodendroglioma after PCV (procarbazine, lomustine and vincristine) chemotherapy: EORTC Brain Tumor Group phase II study 26972. Ann Oncol. 2003;14(4):599–602. | |
Reardon DA, Galanis E, DeGroot JF, et al. Clinical trial end points for high-grade glioma: the evolving landscape. Neuro Oncol. 2011;13(3):353–361. | |
Abrey LE, Louis DN, Paleologos N, et al; Oligodendroglioma Study Group. Survey of treatment recommendations for anaplastic oligodendroglioma. Neuro Oncol. 2007;9(3):314–318. | |
DeAngelis LM. Anaplastic glioma: how to prognosticate outcome and choose a treatment strategy. [corrected]. J Clin Oncol. 2009;27(35):5861–5862. | |
Murrell J, Board R. The use of systemic therapies for the treatment of brain metastases in metastatic melanoma: Opportunities and unanswered questions. Cancer Treat Rev. 2013;39(8):833–838. | |
Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13(3):324–333. | |
Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412–7420. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.