")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Magnetic resonance imaging of folic acid-coated magnetite nanoparticles reflects tissue biodistribution of long-acting antiretroviral therapy
Authors Li T, Gendelman H, Zhang G , Puligujja P, McMillan J, Bronich T, Edagwa B, Liu X, Boska M
Received 21 February 2015
Accepted for publication 31 March 2015
Published 3 June 2015 Volume 2015:10(1) Pages 3779—3790
DOI https://doi.org/10.2147/IJN.S83279
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Video abstract presented by Michael D Boska.
Views: 510
Tianyuzi Li,1 Howard E Gendelman,1,2 Gang Zhang,1 Pavan Puligujja,1 JoEllyn M McMillan,1 Tatiana K Bronich,2 Benson Edagwa,1 Xin-Ming Liu,1,2 Michael D Boska3
1Department of Pharmacology and Experimental Neuroscience, 2Department of Pharmaceutical Sciences, 3Department of Radiology, University of Nebraska Medical Center, Omaha, NE, USA
Abstract: Regimen adherence, systemic toxicities, and limited drug penetrance to viral reservoirs are obstacles limiting the effectiveness of antiretroviral therapy (ART). Our laboratory’s development of the monocyte-macrophage-targeted long-acting nanoformulated ART (nanoART) carriage provides a novel opportunity to simplify drug-dosing regimens. Progress has nonetheless been slowed by cumbersome, but required, pharmacokinetic (PK), pharmacodynamics, and biodistribution testing. To this end, we developed a small magnetite ART (SMART) nanoparticle platform to assess antiretroviral drug tissue biodistribution and PK using magnetic resonance imaging (MRI) scans. Herein, we have taken this technique a significant step further by determining nanoART PK with folic acid (FA) decorated magnetite (ultrasmall superparamagnetic iron oxide [USPIO]) particles and by using SMART particles. FA nanoparticles enhanced the entry and particle retention to the reticuloendothelial system over nondecorated polymers after systemic administration into mice. These data were seen by MRI testing and validated by comparison with SMART particles and direct evaluation of tissue drug levels after nanoART. The development of alendronate (ALN)-coated magnetite thus serves as a rapid initial screen for the ability of targeting ligands to enhance nanoparticle-antiretroviral drug biodistribution, underscoring the value of decorated magnetite particles as a theranostic tool for improved drug delivery.
Keywords: folic acid, decorated nanoparticles, magnetite, theranostics, magnetic resonance imaging
Introduction
Significant reductions of residual human immunodeficiency virus (HIV) from its cell and tissue reservoirs in the brain, lymph nodes, bone marrow, gut-associated lymphoid tissue, and genital tracts can be achieved by antiretroviral (ARV) drugs or by immune surveillance. However, HIV type one (HIV-1) infected memory effector CD4+ T cells and, in more limited numbers, tissue macrophages remain targets for persistent viral infection.1,2 Both cells are refractory to immune and ARV elimination and are commonly resistant to virus-associated cytopathicity.1 HIV-1 has evolved mechanisms to prolong the life span of infected macrophages.3,4 The virus is, notably, present for long periods of time in gut-associated lymphoid tissue, lymph nodes, and brain reservoirs, thus constituting obstacles for viral eradication.5 One way to improve viral reduction is by viral reservoir-targeted long-acting antiretroviral nanoparticles that engage monocyte-macrophages as drug carriers.6 With this in mind, a number of particle decorations that target circulating monocytes and tissue macrophages are in development. Such decorations include peptides,7 antibodies,8 proteins, and sugars9 that engage cell-specific receptors or facilitate phagocytosis. Outcome measures for improved ARV distribution are required beyond testing of plasma drug levels.10 To such ends, our own works have centered on ways to bridge classic pharmacokinetic (PK) evaluation with magnetic resonance imaging (MRI) assessment of particle distributions.11 We have developed a small magnetite antiretroviral therapy (SMART) nanoparticle platform that employs MRI for determining ARV-magnetite particle biodistribution.11 However, whether SMART technologies could be applied to decorated particles is not yet known. If achieved, magnetic particle biodistribution could be quantitated by conventional MRI and reflect the targeting ability of the ligand-coated particle to bring ARVs to reservoirs of viral infection.12,13 As a secondary step toward realizing this goal, folic acid (FA)-coated SMART (FSMART) particles were developed. FA has emerged as an important targeting moiety capable of specific interaction with cells, notably mononuclear phagocytes (MPs) (monocytes, macrophages, and dendritic cells), expressing the folate receptor (FR). The FR consists of a high-affinity (Kd~10−9–10−10 M) folate binding protein attached to the membrane through a glycosylphosphatidyl-inositol anchor.14 Following binding of FA, endocytosis delivers a fraction of the receptors into the cell.15 Importantly, conjugates of FA with proteins, drugs, or nanoparticles retain FR-binding capabilities.16 This can facilitate the entry of FSMART particles carrying drug by receptor-mediated endocytosis. FR expression is restricted to a few cell types, including kidney epithelial cells (FR1) and MP (FR2).17 Its expression on MP is regulated by cell activation that is initiated by the particle itself. Specific drug targeting can thus parallel tissue sites of infection and consequent inflammation along with the reticuloendothelial system. MP and reticuloendothelial cells would then serve as drug storage depots.18,19 This, taken together, makes FA conjugation suitable for MP targeting of ARV nanoparticles (nanoformulated antiretroviral therapy [nanoART]) for treatment of HIV-1 infection.18,19 In particular, PK and pharmacodynamics (PD) studies have demonstrated that such targeting strategies improve drug biodistribution. However, efficacy determinations have entailed intensive tissue drug chemical analyses.18,20 We posit that testing of targeting ligands can be sped by MRI evaluation of magnetite-labeled ARV particle tissue distributions.
Prior magnetite platforms were developed for purposes such as these.21–23 Among them, polysaccharides are the most widely studied coating materials for magnetite.24–26 However, the potential of in vivo coating disassociation and bare magnetic particle exposure causing cellular toxicity are of significant concern.27 Another limitation of current coating materials for magnetite is the complicated conjugation strategies or limited appreciated functional groups for specific ligand decoration on magnetite. Substantive efforts were made to resolve these issues. Alendronate (ALN) is a bisphosphonate drug that is clinically used to treat osteoporosis. The P–C–P structure of bisphosphonates is known for its ability to bind divalent metal ions, such as Ca2+.28 Taking advantages of this favorable metal binding ability, we conjugated ALN to polymers for stable magnetic particle coating. FA was conjugated into the ALN-associated polymers and then decorated to the exterior coating of magnetite to allow comparisons of magnetite biodistribution with and without particle decoration. Such particles were injected into mice to confirm the usefulness of magnetite nanoparticles to noninvasively estimate FA-targeted nanoART biodistribution. The distribution of FA-targeted magnetite and FA-nanoART techniques were determined by bioimaging and drug quantitation. Both were used to confirm the utility of the new platform. Such a system can provide a relevant screening platform and as such could be used to investigate a broader range of targeting systems for the delivery of nanoART to reservoir sites of viral infection and serve to significantly improve treatment outcomes for infected people.
Materials and methods
Synthesis of magnetite (ultrasmall superparamagnetic iron oxide particles)
The magnetite particles were synthesized with 6 mmol tris(acetylacetonato) iron(III) mixed with 30 mmol 1,2-hexadecanediol, 18 mmol oleic acid (OA), 18 mmol oleylamine, and 60 mL benzyl ether in a three-neck round-bottomed flask equipped with a condenser, magnetic stirrer, thermograph, and heating mantle. The mixture was stirred under nitrogen, slowly heated to 110°C, and maintained at that temperature for 1 hour. This was followed by gradual heating of the mixture to 200°C for 2 hours, followed by slow heating to 298°C. When the mixture was cooled to room temperature, a dark homogeneous colloidal suspension was obtained. The suspension was precipitated in ethanol under a magnetic field. The black precipitate was dissolved in hexane with the presence of OA and oleylamine, and the solution was centrifuged at 3,800× g for 10 minutes to remove any undispersed residue. The black solution was reprecipitated in ethanol and centrifuged at 10,000× g for 30 minutes. Solid products were obtained by drying the precipitate under vacuum and generating dry particles.
Synthesis of ALN conjugated polyethylene glycol
Polyethylene glycol monomethyl ether 5,000 (mPEG5k) (1 g, 0.2 mmol) was dissolved in anhydrous dichloromethane (5 mL) together with 4-dimethylaminopyridine (12 mg, 0.1 mmol) and triethylamine (101 mg, 1 mmol). The mixture was cooled to 0°C, and p-toluenesulfonyl chloride (Tos-Cl) (0.191 g, 1 mmol) was added. Next, the mixture was incubated overnight at room temperature and the mixture precipitated in ether. The precipitate (crude product) was purified using a LH-20 column with methanol as the eluent. The yield of p-toluenesulfonyl-terminated mPEG5k (Tos-PEG) was 65%. Tos-PEG (0.52 g, 0.1 mmol) was dissolved in dimethylformamide (5 mL). Sodium azide (65 mg, 1 mmol) was then added. The reaction proceeded with stirring at 100°C for 1 day. After filtration and solvent removal, the crude product was dissolved in dichloromethane and precipitated into ether. The precipitate (crude product) was further purified using a LH-20 column with methanol as the eluent. The yield of azide-terminated PEG5k (azido-PEG) was 70%. Azido-PEG (0.25 g, 0.05 mmol) and 1-hydroxy-4-pent-4-ynamidobutane-1,1-diyldiphosphonic acid (40 mg, 0.1 mmol) were dissolved in an ethanol-H2O solution (1/1 v/v, 2 mL). Sodium ascorbate (19.8 g, 0.1 mmol) and copper sulfide pentahydrate (2.5 mg, 10 μmol) were then added separately under argon. The reaction mixture was stirred for 3 days at room temperature. After removal of the solvent, the crude product was acidified and purified using a LH-20 column with methanol as the eluent. The yield of the ALN conjugated polyethylene glycol (ALN-PEG) was 70%.
Synthesis of FA conjugated ALN-PEG
Alendronate sodium trihydrate (1 g, 3 mmol) was dissolved in 5 mL water; the pH was adjusted to approximately 8.0 with sodium hydroxide (NaOH). Fluorenylmethoxycarbonyl-PEG5k-N-hydroxysuccinimide (Fmoc-PEG5k-NHS) (0.5 g, ~1 mmol) was then slowly added into the ALN solution. The reaction solution was stirred overnight at room temperature, and then dialyzed to remove excess ALN. After lyophilization, the resulting ALN-PEG-Fmoc powder (0.35 g) was directly dissolved in 20% piperidine in tetrahydrofuran (THF) and reacted at room temperature for 2 hours. The solvent was concentrated using rotavapor under vacuum and precipitated into ether. The resulting precipitate (ALN-PEG-NH2) was further purified by dialysis and lyophilization.
FA (110 mg, 0.25 mmol) was dissolved in 2 mL dimethylsulfoxide (DMSO) plus 50 μL of triethylamine. A 1:1 molar ratio of NHS (31 mg) and N, N′-dicyclohexylcarbodiimide (DCC) (57 mg) was added. The mixture was stirred 4 hours at room temperature in the dark. The by-product, dicyclohexylurea, was removed by filtration. ALN-PEG-NH2 (0.25 g) was then added into the NHS-folate solution for overnight conjugation at room temperature in the dark. The crude product was precipitated with diethylether, and further purified by dialysis. For synthesis of fluorescent CF633-modified ALN-PEG, CF633-NHS (1 μmol), and ALN-PEG-NH2 (10 mg) were dissolved in 0.5 mL DMSO, and then reacted overnight in the dark at room temperature. The resulting CF633-labeled ALN-PEG was purified using a LH-20 column with methanol as the eluent.
Coating of ALN-PEG and ALN-PEG-FA onto magnetite (APM and FAPM)
To synthesize APM, the ALN-PEG polymer (5 mg, 10 mg, 20 mg, and 50 mg) was dissolved in 0.5 mL deionized water. Separately, 5 mg magnetite was dissolved in 10 mL THF. The polymer solution was added slowly to magnetite-THF dispersion with stirring. The mixture was stirred overnight, and THF was removed using a rotary evaporator. The crude product was dissolved in 5 mL deionized water and centrifuged at 500×g for 10 minutes to remove uncoated magnetite. Noncoated polymer was removed by dialysis using 25,000 MW cut off (25 K) dialysis tubing overnight. After dialysis, the dispersion was lyophilized to obtain APM. The mass ratio of magnetite and coating materials was measured using thermal gravimetric analysis (TGA).
To synthesize FAPM, 16 mg ALN-PEG polymer and 4 mg ALN-PEG-folate was dissolved in 0.5 mL deionized water and 5 mg magnetite was dispersed in 10 mL THF. Polymer solution was added slowly to the magnetite-THF dispersion with stirring in the dark. The mixture was stirred overnight, and then THF was removed using a rotary evaporator. The crude product was dissolved in 5 mL deionized water, and then centrifuged at 500×g for 10 minutes to remove uncoated magnetite. Noncoated polymers were removed by dialysis using 25 K dialysis tubing overnight in the dark. After dialysis, the dispersion was lyophilized to get FAPM powder. CF633-labeled APM and FAPM (CF633-APM and CF633-FAPM) were synthesized using the same procedure using 2 mg CF633-labeled ALN-PEG or FA-conjugated ALN-PEG dissolved in the polymer solution.
SMART and FSMART syntheses
One hundred mg of PEG-poly(lactic-co-glycolic acid) (PEG-PLGA) (5 K–20 K), 20 mg of ritonavir (RTV), and 5 mg of magnetite were dissolved in 1 mL dichloromethane. The organic phase was added slowly into the distilled water containing 1% polyvinyl alcohol with constant stirring and then sonicated at 20% amplitude in an ice bath for 6 minutes. Dichloromethane was then removed by rotavapor for 1 hour. A high-speed 10,000× g centrifugation for 20 minutes was used to collect the nanoparticles. After washing twice with distilled water, the nanoparticles were resuspended in phosphate buffered saline (PBS). Next, the particle suspension was centrifuged at 100× g for 5 minutes. The supernatant fluids were collected to remove the aggregated nanoparticles. FSMART was synthesized and purified the same way except for using 100 mg of a polymer mixture of PEG-PLGA (5 K–20 K) and FA-PEG-PLGA (5 K–10 K) with a weight ratio 4:1 instead of only PEG-PLGA (5 K–20 K).
Physicochemical and stability characterization of APM and FAPM
Particle size and size distribution were measured by dynamic light scattering (DLS) using a Malvern Zetasizer Nano-ZS instrument (Malvern Instruments Inc., Westborough, MA, USA) as previously described.6 The pH and concentrations of the particles in the dispersions were fixed before measurements of particle size and size distribution. All measurements were performed at 25°C. The shape and surface morphology of the APM and FAPM particles were determined by transmission electron microscopy (TEM) as previously described.11 Samples were prepared from dilutions in distilled water of particle suspensions and dropped onto stubs. After air-drying, the particles were examined by TEM. The stability of APM was assessed in sodium chloride (NaCl) solutions (1 M and 2 M) and buffer solutions of pH 2, 4, 6, 7.4, 8, 10, and 12, and the size and polydispersity were determined by DLS and TEM. Concentration-dependent relaxivity (r2) (s−1 mL mg−1) was measured by MRI of APM or FAPM dispersion in concentrations ranging from 0 μg/mL to 40 μg/mL.
Biodistribution of APM and FAPM
Biodistribution of APM and FAPM was determined in male Balb/cJ mice (Jackson Labs, Bar Harbor, ME, USA) after 2 weeks on folate-deficient diet (Harlan Teklad TD.00434; Harlan Laboratories, Indianapolis, IN, USA). Twenty-four hours before the administration of magnetite particles, mice were injected with 2 mg/kg lipopolysaccharide (LPS) (Sigma-Aldrich, St Louis, MO, USA) intraperitoneally, or 100 mg/kg P407-coated nanoformulated RTV-boosted atazanavir (ATV) (nanoATV/r) intramuscularly, to activate immune cells. APM or FAPM (2 mg iron/kg body weight) were injected intravenously in a total volume of 100 μL for each mouse.6 The mice were scanned by MRI before and 24 hours after APM or FAPM injection.
Biodistribution of SMART and FSMART
Biodistribution of SMART and FSMART was determined in male Balb/cJ mice after 2 weeks on a folate-deficient diet. SMART and FSMART (2 mg iron/kg) were injected intravenously in a total volume of 100 μL/mouse. The mice were scanned by MRI before and 24 hours after SMART and FSMART injections.
MRI acquisition and analyses
Images were acquired using a 7T/16 cm Bruker PharmaScan MRI scanner (Ettlingen, Germany) and a quadrature birdcage mouse body resonator. Magnetite detection by MRI was done using T2 mapping for quantitation and T2* weighted high-resolution imaging for detection of biodistribution throughout the body. The sequence used for T2 mapping was a Carr–Purcell–Meiboom–Gill (CPMG) phase-cycled multislice multiecho sequence. Forty-one 0.5 mm thick contiguous interleaved coronal images were acquired with an acquisition matrix of 256×192, 40 mm field-of-view (FOV), 12 echoes at 10 milliseconds first echo time and 10 milliseconds echo spacing, a repetition time of 4,680 milliseconds, three averages, for a total acquisition time of 30 minutes. T2* weighted MRI was acquired using a 3D spoiled gradient recalled echo sequence with echo time =3 milliseconds, repetition time =10 milliseconds, 15° pulse angle, 50×40×30 mm FOV, 256×196×128 acquisition matrix, six averages, for a total scan time of 25 minutes.11 T2 maps were reconstructed using custom programs written in Interactive Data Language (IDL) (Exelis Visual Information Solutions, McLean, VA, USA). Preinjection and 24-hour postinjection maps were constructed using the even-echo images from the CPMG phase-cycled imaging data set. Mean tissue T2 was determined using region of interest (ROI) analyses before and after magnetite injection for the 24 hours results. Magnetite concentration was then determined from the change in R2 (916;R2 =1/T2preinjection – 1/T2postinjection) and the per milligram magnetite particle r2 determined as the slope of magnetite concentration versus R2 in phantom studies. The ROI analyses were performed using ImageJ (http://imagej.nih.gov/ij) software.
FA-nanoATV/r biodistribution
NanoATV/r and FA-nanoATV/r were synthesized as previously described.19 Male Balb/cJ mice, 4–6 weeks old, were maintained on folate-deficient diet for 2 weeks and injected with 50 mg/kg of nanoATV/r or FA-conjugated nanoATV/r (FA-nanoATV/r). The mice were sacrificed on day 7. Mice with LPS treatment received intraperitoneal injection of LPS (5 mg/kg) 24 hours before receiving 50 mg/kg of either nanoATV/r or FA-nanoATV/r and were sacrificed on day 14. Tissues were collected and stored at −80°C. Tissue drug levels were determined using ultraperformance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) as described.29
Immunofluorescence
Immunofluorescence staining of magnetite particles was determined in male Balb/cJ mice after 2 weeks on folate-deficient diet. Twenty-four hours before the administration of magnetite particles, mice were injected with 2 mg/kg LPS intraperitoneally or 100 mg/kg nanoATV/r intramuscularly. CF633-APM or CF633-FAPM (2 mg iron/kg) was injected intravenously in a total volume of 100 μL for each mouse. After 24 hours, tissues were collected following the MRI scan for immunofluorescence staining.
Preparations of whole-mount tissues and frozen sections: tissues were collected after perfusion with PBS and fixed in 10% neutral buffered formalin for 24 hours, then incubated in 15% and 30% sucrose for cryoprotection and embedded in optical cutting temperature (OCT) compound (Tissue-Tek) at -20°C. For whole-mount staining, tissues were cut to 20 μm on a cryostat. Tissues were carefully harvested from melting OCT. Immunofluorescence staining of frozen sections: sections were blocked/permeabilized in tris-buffered saline solution containing 10% normal goat serum, and stained with rabbit monoclonal antibody against ionized calcium-binding adaptor molecule 1 (Iba-1) (1:500) (Wako Chemicals, Dallas, Texas, USA) and mouse monoclonal antibody against FR (β) (a generous gift of Dimitri Dimitrov, National Cancer Institute, Bethesda, MD, USA) (1:200) for 1 hour at room temperature. Primary antibodies were labeled with secondary goat anti-mouse and anti-rabbit antibodies conjugated to the fluorescent probes Alexa Fluor 488 and Alexa Fluor 594, and nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI). Images were acquired with a Nikon swept field confocal microscope (Nikon, Tokyo, Japan).
Statistical analyses
The data are presented as mean ± SEM. Statistical significance between groups was assessed using Student’s t-test by Graph-Pad® Prism unless specified. The data were considered statistically significant if P<0.05.
Ethics statement
The acquisition, care, housing, use, and disposition of nonhuman animals used in this research was in full compliance with the ethical standards of the University of Nebraska Medical Center and of the National Institutes of Health that include all applicable laws and regulations for the applied drug trials.
Results and discussion
To integrate classic PK evaluation with MRI assessment of particle distributions, we developed FA-targeted magnetite particles and compared the distribution of these particles to nontargeted magnetite particles using MRI. As an initial step toward securing this end, we manufactured ALN-PEG and ALN-PEG-FA polymers. These were then used to coat magnetite. In order to efficiently conjugate hydrophilic ALN to PEG polymer, the versatile click reaction was used (Figure 1A). mPEG was first tosylated with toluenesulfonyl chloride, and then converted to azido-mPEG under reflux using excess sodium azide. Both steps were achieved by quantitative conversion. To introduce an acetylene group onto ALN, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)/NHS was used to conjugate ALN and pentynoic acid. Finally, these two fragments were successfully conjugated together through copper (I)-catalyzed Huisgen 1,3-dipolar cycloaddition between the azide and alkyne with a high yield at room temperature. From a proton nuclear magnetic resonance (NMR) spectrum, the emergence of the singlet peak of 1,2,3-triazole at 8.32 ppm verified the successful conjugation (Figure 1E). ALN-PEG-FA was synthesized using the amidation strategy (Figure 1B). First, ALN was incorporated into PEG by reaction with Fmoc-PEG-NHS, and the Fmoc was then deprotected with piperidine to synthesize amine-functionalized ALN-PEG (NH2-PEG-ALN) for ligand conjugation, which is one of the most attractive properties of ALN-PEG polymers. Folate was reacted with NH2-PEG-ALN to synthesize the targeted ALN-PEG-FA.
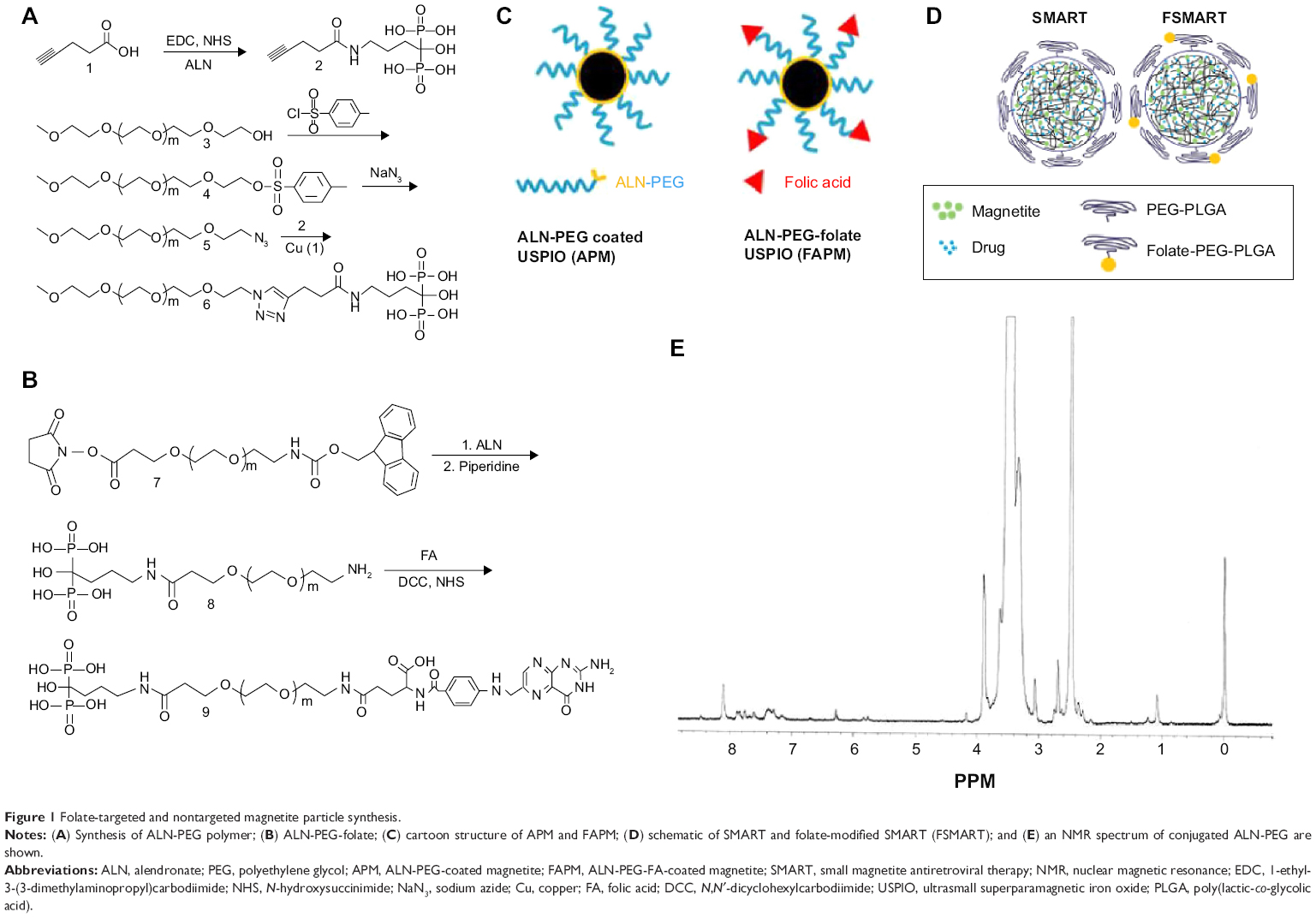 | Figure 1 Folate-targeted and nontargeted magnetite particle synthesis. |
To confirm the strong coating ability of ALN-PEG polymer to magnetite, OA-coated magnetite synthesized by the thermal decomposition method with an average size of 10 nm were selected for replacement coating. The OA coating material is hydrophobic and relatively weakly bound to the core of magnetite. OA-coated magnetite was dispersed into THF, and ALN-PEG was added into this solution for replacement coating overnight at room temperature, turning the magnetite from hydrophobic to hydrophilic. At the end of replacement, THF was evaporated and magnetite particles were redispersed in water and dialyzed. After dialysis, the dispersed solution was lyophilized. The structure of APM is illustrated in Figure 1C. We chose formulation with 20% wt magnetite and 80% wt polymer for further evaluation due to the outstanding stability and ability of this polymer combination to conjugate sufficient number of ligand molecules. The lyophilized particles are easily dispersed in water and the nanoparticles are very stable in biological buffers such as PBS. Under an external magnetic field, APM retained its dispersion stability in PBS while OA-coated magnetite in PBS was colloidally unstable, precipitated, and attracted toward a magnetic field within minutes (Figure 2A).
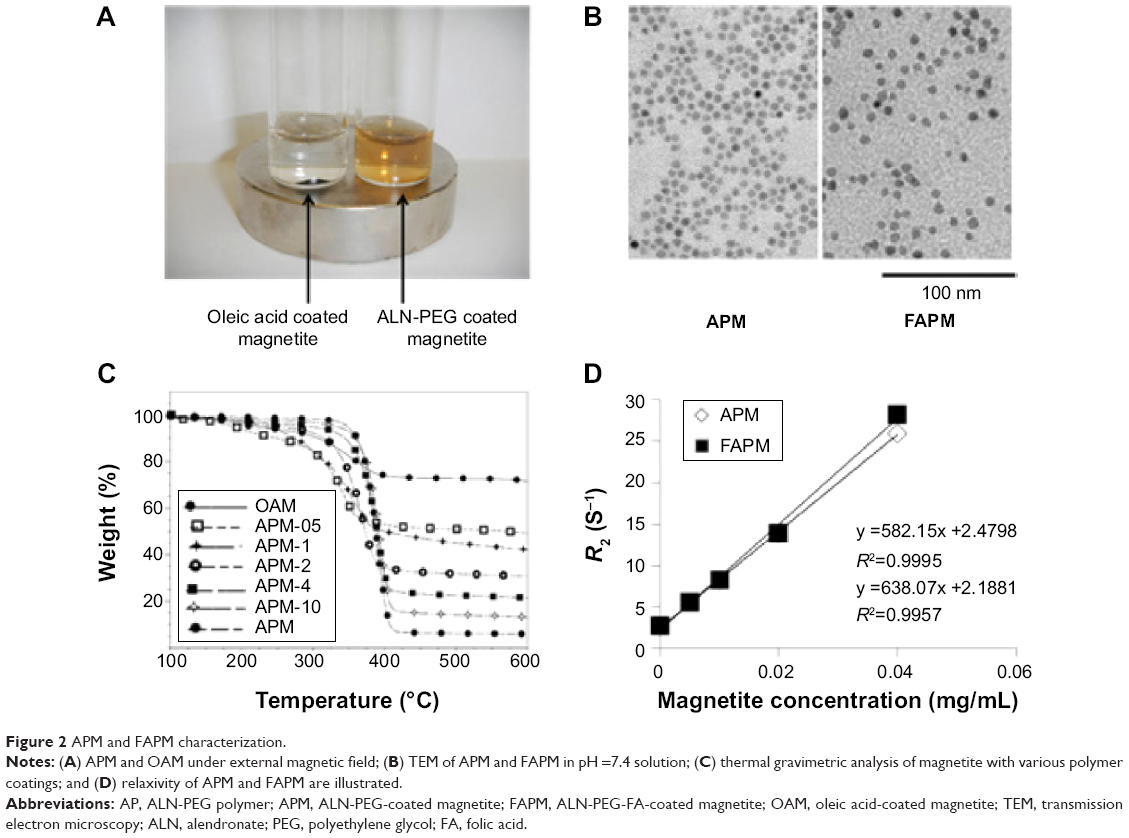 | Figure 2 APM and FAPM characterization. |
To demonstrate the potential of APM as a novel nanomedicine platform for targeting ligand evaluation and optimization, FA was selected to conjugate to the ALN-PEG polymer (ALN-PEG-Folate) using amidation chemistry (Figure 1B). For the preparation of FA-decorated magnetite, OA-coated magnetite was dispersed into THF, and then ALN-PEG and ALN-PEG-FA (weight ratio 4:1) were added into this solution for OA replacement overnight at room temperature. The purification procedures were the same as for APM. The structure of FAPM is demonstrated in Figure 1C. The size of APM and FAPM nanoparticles was assessed by TEM, showing that the iron oxide cores are morphologically indistinguishable from each other, with a mean particle size of 10 nm (Figure 2B). Benefiting from its high coating efficacy, ALN-PEG was coated onto magnetite in a tunable and reproducible manner. Up to 100% of the ALN-PEG was coated onto magnetite as measured by TGA (Figure 2C).
Concentration dependent r2 (s−1 mL mg−1) causing increased relaxivity (R2) (s−1) in tissue as a function of concentration (expressed as mg/mL magnetite) of APM and FAPM is an important parameter. To investigate the MR signal enhancement effects, the R2 of aqueous magnetite particles at different magnetite concentrations was determined using a 7T MRI scanner. The magnetite concentrations in mg/mL were plotted against R2. As shown in Figure 2D, the relaxivity of both APM and FAPM varied linearly with the magnetite concentration. The relaxivity of FAPM was found to be r2=582 s−1 mL mg−1 and of APM r2=638 s−1 mL mg−1 (Figure 2D). The r2 values of these particles were used for noninvasive in vivo quantitation of magnetite concentration using MRI (Figure 4A). Thus, the limit of detection is dominated by the molar relaxivity (r2=600 s−1 mL mg−1) of the magnetite particles and the accuracy of the T2 measurement. In our laboratories, with a 16 echo train and using even numbered echoes for the most accurate measurement from a CPMG phase-cycled refocusing echo train, the accuracy of T2 was found to be within 5% with typical T2 values (preinjection) of 100 milliseconds, providing a minimum level of detection of less than 1 ng/mL for the particles.
To study the stability of APM, we tested its size and polydispersity index (PDI) in acidic, basic, and high ionic strength conditions. APM nanoparticles were very stable in various conditions, and there was no significant increase in size and PDI of APM within the 2 weeks timeframe of the study (Figure 3A). After 1 month of storage on the shelf, APM dispersions remained clear and no nanoparticles precipitated out (Figure 3B). The TEM pictures of APM nanoparticles also confirmed this result (Figure 3C). After 2 months of storage, all APM dispersions remained stable except APM nanoparticles in the 2 M NaCl solution where some precipitation was observed (Figure 3B), which can be reversed with gentle shaking.
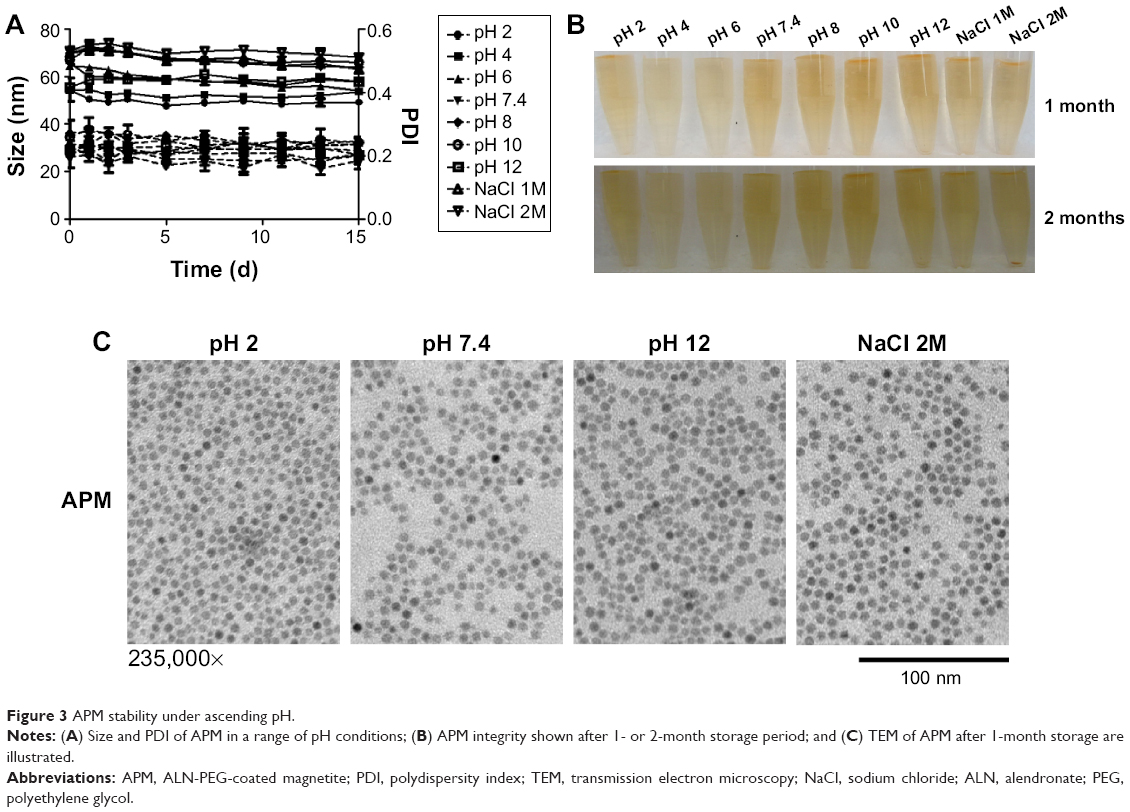 | Figure 3 APM stability under ascending pH. |
Studies have shown that activated macrophages express FR.30–33 Our previous studies demonstrated that FA receptors are operative in differentiated macrophages, as cell lysates were probed with antibodies to FR1 and FR2 and showed that FR2 but not FR1 is expressed and localized to the cell membrane.19 Engagement of the FR receptor in enhancing FA-coated nanoART uptake was demonstrated by the ability of free FA to block enhanced FA-nanoART uptake. Our in vivo studies have demonstrated that the administration of nanoART stimulated the overexpression in the FR receptor in macrophages.19 For the evaluation of the biodistribution of nontargeted APM and macrophage-targeted FAPM, the immune system of mice was first activated with LPS or nanoATV/r, and then APM or FAPM were administered via intravenous injection. The APM and FAPM biodistributions were determined by MRI. Figure 4A displays 0.128 mm thick T2* weighted high-resolution 3D spoiled gradient recalled echo images of the same mouse before and 24 hours after injection of APM or FAPM. The presence of magnetite in tissue causes a reduction of T2*, causing signal loss in proportion to concentration at an echo time (TE) =3 milliseconds in the liver, spleen, and some abdominal regions. The shortening in the T2* value correlates with the decrease in signal intensity as a result of the strong magnetite susceptibility effects. As a result, tissues containing magnetite appear darker in post scans demonstrating a higher accumulation of FAPM systemically in both LPS and nanoATV/r pretreated mice. This method is semi-quantitative; however, it does allow ready identification of the presence of magnetite throughout the body that can be used to guide quantitative ROI analyses using T2 maps.
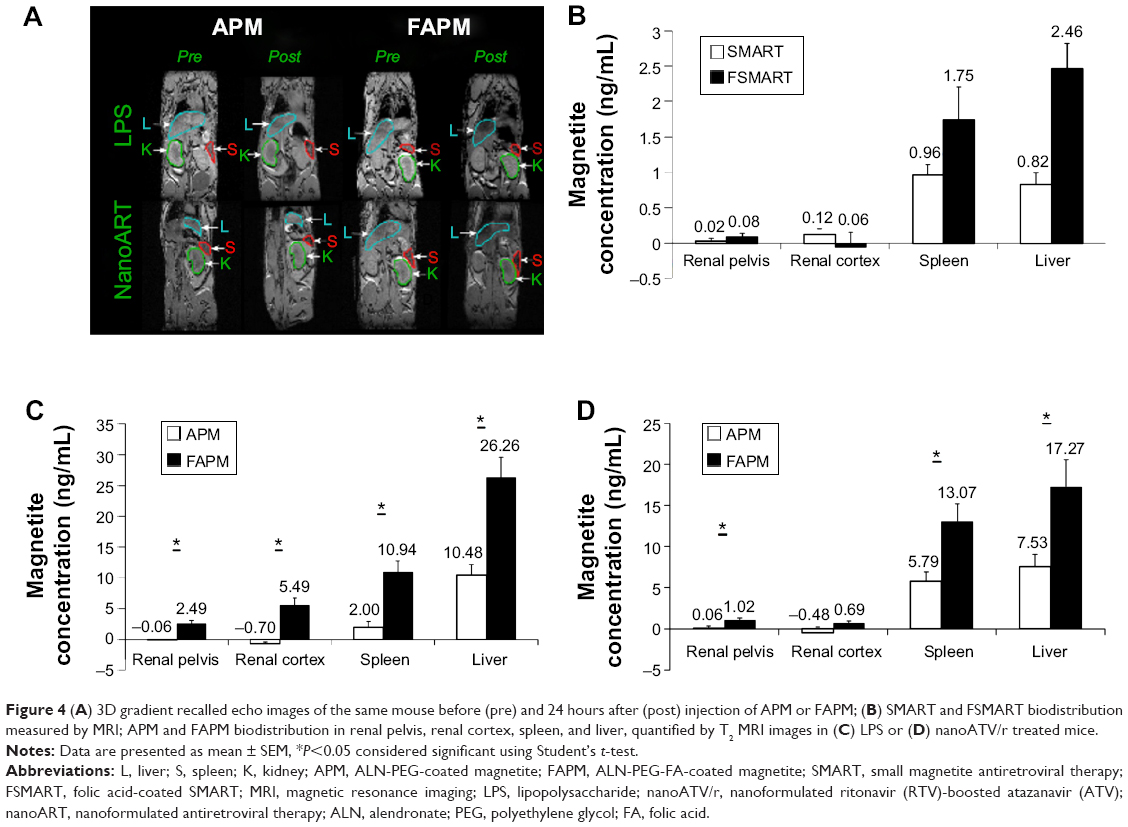 | Figure 4(A) 3D gradient recalled echo images of the same mouse before (pre) and 24 hours after (post) injection of APM or FAPM; (B) SMART and FSMART biodistribution measured by MRI; APM and FAPM biodistribution in renal pelvis, renal cortex, spleen, and liver, quantified by T2 MRI images in (C) LPS or (D) nanoATV/r treated mice. |
APM (n=6) and FAPM (n=6) biodistribution in LPS or nanoATV/r-treated mice were quantified from regions of interest in MRI T2 maps (Figure 4C and D). It can be appreciated that in animals treated with LPS, FAPM showed a higher accumulation in various tissues compared to APM (3 μg/mL versus 0 μg/mL for renal pelvis, 6 μg/mL versus 0 μg/mL for renal cortex, 11 μg/mL versus 2 μg/mL for spleen, and 26 μg/mL versus 11 μg/mL for liver) (Figure 4C). In animals treated with nanoATV/r, FAPM also showed more accumulation in various tissues compared to APM (1 μg/mL versus 0 μg/mL for renal pelvis, 1 μg/mL versus 0 μg/mL for renal cortex, 13 μg/mL versus 6 μg/mL for spleen, and 17 μg/mL versus 8 μg/mL for liver) (Figure 4D). FAPM accumulated more in various tissues, demonstrating its targeting ability to FR on activated tissue macrophages and showing the potential of APM and FAPM for assessing image-guided drug PK and biodistribution.
To further determine the targeting ability of the magnetite nanoparticles, FSMART particles that encapsulate both RTV and magnetite were manufactured to study ARV biodistribution; the structure of these particles is illustrated in Figure 1D. SMART and FSMART particles had similar physicochemical characteristics with a size of 294±2 nm and 354±2 nm, a PDI of 0.1±0.02 and 0.1±0.03, and a zeta potential of −28±1 mV and −19±1 mV, respectively. SMART and FSMART particles were injected into mice at 2 mg/kg of iron, equivalent to the dosage used in APM and FAPM studies. The results showed parallel magnetite distribution to that of APM and FAPM, as such validating the FA targeting ability (Figure 4B).
To confirm that the distribution of FA-targeted magnetite particles accurately reflects the distribution of FA-targeted nanoART, the biodistribution of nanoATV/r or FA-nanoATV/r was evaluated in Balb/cJ mice. Mice were injected with 50 mg/kg of either nanoATV/r or FA-nanoATV/r, and the drug concentrations in different tissues were quantified by UPLC-MS/MS. The tissue ATV concentrations following treatment with FA-nanoATV/r were significantly higher than with nanoATV/r in mice with or without LPS treatment. In LPS-treated mice, FA-nanoATV/r administration provided 2 ng/g and 208 ng/g ATV concentrations in liver and spleen, respectively, compared to 439 ng/g and 145 ng/g in liver and spleen following nanoATV/r administration (Figure 5A). In contrast, the ATV tissue concentrations following FA-nanoATV/r administration were 3 μg/g and 86.3 ng/g compared to 146 ng/g and 2 ng/g for nanoATV/r in liver and spleen, respectively, in mice not treated with LPS (Figure 5B). The differences in drug biodistribution following treatment with targeted versus nontargeted nanoART/r in mice without LPS treatment demonstrated the ability of nanoART itself to activate the immune system. In conclusion, we observed parallel patterns of tissue distribution between nanoATV/r versus FA-nanoATV/r and APM versus FAPM in treated mice. Notably, these comparable differences in distribution of ATV and magnetite concentrations following targeted versus nontargeted nanoparticle administration PK confirmed the ability of our ALN-PEG-coated magnetite system to be used as a targeting ligand approach to predict actual drug PK and tissue biodistribution.
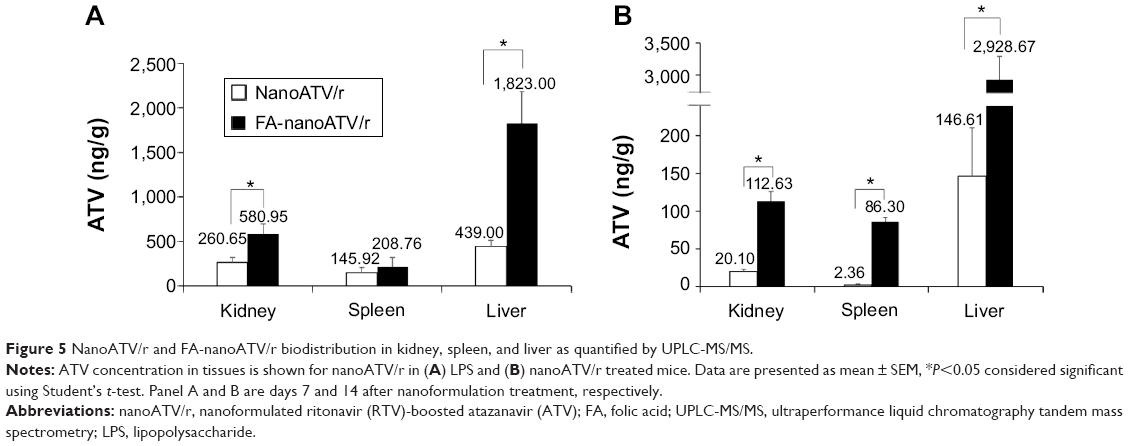 | Figure 5 NanoATV/r and FA-nanoATV/r biodistribution in kidney, spleen, and liver as quantified by UPLC-MS/MS. |
To demonstrate the FR targeting ability of FAPM, the colocalization of fluorescently labeled magnetite and FR2 was studied by administering CF633-APM and CF633-FAPM to mice. Mouse FR2 antibody and Iba-1 antibody were used to illustrate the location of FR2 receptor and macrophages, respectively, and to validate FR targeting. Results obtained by immunofluorescent staining clearly showed colocalization of FR2 and Iba-1 in spleen from mice injected with LPS and nanoATV/r, in agreement with the hypothesis that activated macrophages express FR2. In addition, CF633-labeled FAPM was identified principally in Iba-1+/FR+ cells (Figure 6A–F, white arrows depict merged images). Prussian blue staining of tissue sections was also performed in liver and spleens of FAPM treated animals, but showed discordant results. One potential confounding factor is that the oxidation of magnetite during perfusion and tissue processing could prevent the success of Prussian blue staining. It is also possible that the ultrasmall size of the magnetite results in staining that is undetectable.
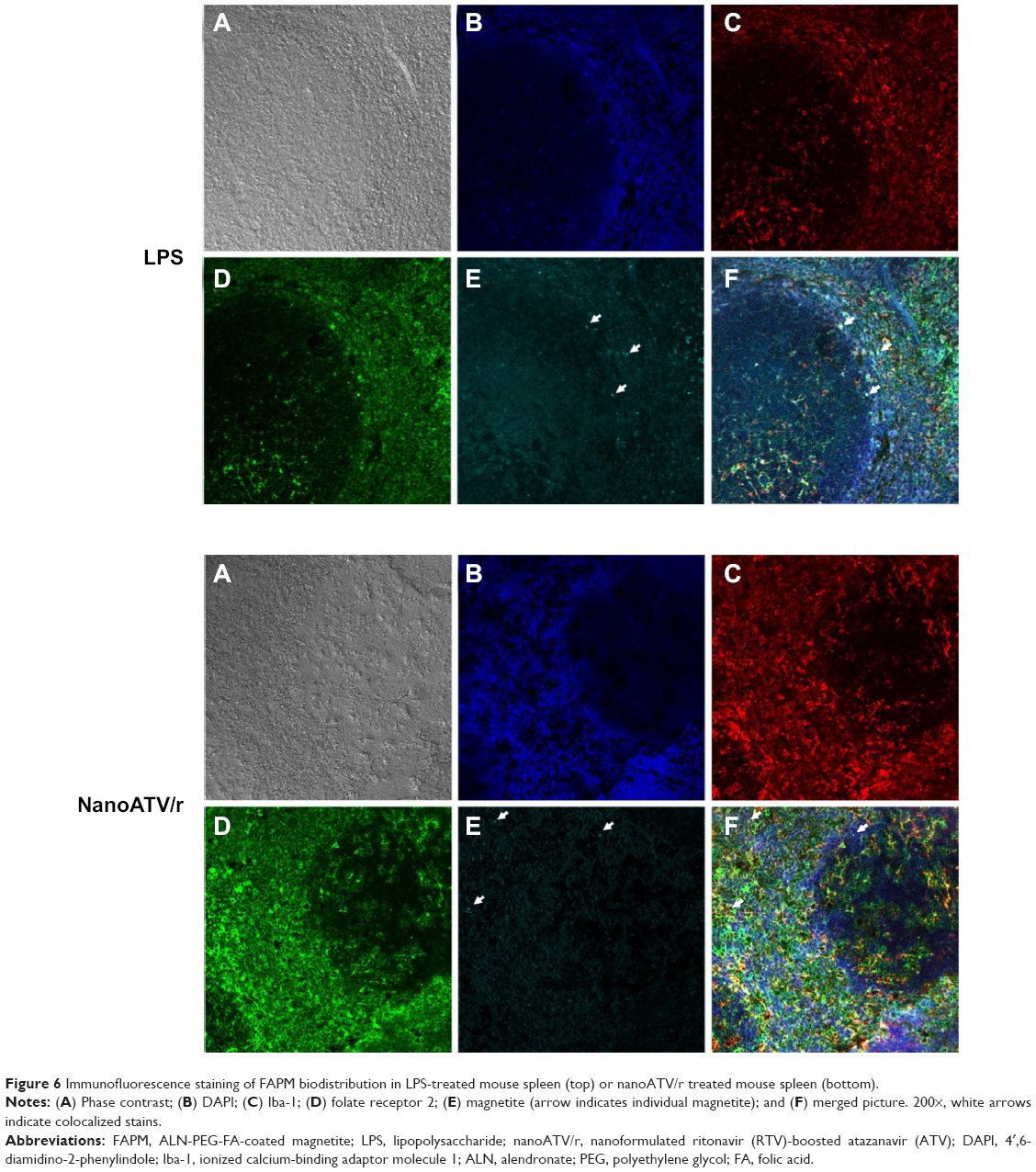 | Figure 6 Immunofluorescence staining of FAPM biodistribution in LPS-treated mouse spleen (top) or nanoATV/r treated mouse spleen (bottom). |
Conclusion
In conclusion, we have successfully developed a stable ALN-polymer platform for tunable magnetite coating. The novel magnetite platform developed with these ALN-polymers has the ability to conjugate various targeting ligands for noninvasive biodistribution evaluation, image-guided drug delivery, and targeting ligand optimizations. The utility of this platform was demonstrated by similar tissue distribution of magnetite, determined by MRI, and nano-ART, determined by UPLC-MS/MS. Rapid uptake by MP of the targeted decorated magnetite provides a noninvasive method of detecting regions of disease-associated infection including virus-induced inflammation. This platform provides a means to rapidly evaluate the efficiency of targeting strategies for HIV reservoirs.
Acknowledgments
This work was supported by the University of Nebraska Foundation that includes individual donations from Dr Carol Swarts and Frances and Louie Blumkin; the Vice Chancellor for Research office, UNMC; ViiV Healthcare; and National Institutes of Health grants P01 MH64570, R01 MH104147, P01 DA028555, R01 NS36126, P01 NS31492, 2R01 NS034239, P01 NS43985, P30 MH062261, and R01 AG043540. We also acknowledge the assistance of the Nanomaterials Core Facility, Nebraska Center for Nanomedicine that is supported by the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103480.
Disclosure
The authors report no conflicts of interest in this work.
References
Igarashi T, Brown CR, Endo Y, et al. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4+ T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): implications for HIV-1 infections of humans. Proc Natl Acad Sci U S A. 2001;98(2):658–663. | ||
Pierson T, McArthur J, Siliciano RF. Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu Rev Immunol. 2000;18:665–708. | ||
Reynoso R, Wieser M, Ojeda D, et al. HIV-1 induces telomerase activity in monocyte-derived macrophages, possibly safeguarding one of its reservoirs. J Virol. 2012;86(19):10327–10337. | ||
Swingler S, Mann AM, Zhou J, Swingler C, Stevenson M. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog. 2007;3(9):1281–1290. | ||
Saksena NK, Wang B, Zhou L, Soedjono M, Ho YS, Conceicao V. HIV reservoirs in vivo and new strategies for possible eradication of HIV from the reservoir sites. HIV AIDS (Auckl). 2010;2:103–122. | ||
Balkundi S, Nowacek AS, Roy U, Martinez-Skinner A, McMillan J, Gendelman HE. Methods development for blood borne macrophage carriage of nanoformulated antiretroviral drugs. J Vis Exp. 2010;46:2460. | ||
Najjar V. Biological effects of tuftsin and its analogs. Drugs Future. 1987;12(2):147–160. | ||
Bestman-Smith J, Gourde P, Desormeaux A, Tremblay MJ, Bergeron MG. Sterically stabilized liposomes bearing anti-HLA-DR antibodies for targeting the primary cellular reservoirs of HIV-1. Biochim Biophys Acta. 2000;1468(1–2):161–174. | ||
Wijagkanalan W, Higuchi Y, Kawakami S, Teshima M, Sasaki H, Hashida M. Enhanced anti-inflammation of inhaled dexamethasone palmitate using mannosylated liposomes in an endotoxin-induced lung inflammation model. Mol Pharmacol. 2008;74(5):1183–1192. | ||
Pretorius E, Klinker H, Rosenkranz B. The role of therapeutic drug monitoring in the management of patients with human immunodeficiency virus infection. Ther Drug Monit. 2011;33(3):265–274. | ||
Guo D, Li T, McMillan J, et al. Small magnetite antiretroviral therapeutic nanoparticle probes for MRI of drug biodistribution. Nanomedicine. 2014;9(9):1341–1352. | ||
Guthi JS, Yang SG, Huang G, et al. MRI-visible micellar nanomedicine for targeted drug delivery to lung cancer cells. Mol Pharm. 2010;7(1):32–40. | ||
Liu W, Dahnke H, Rahmer J, Jordan EK, Frank JA. Ultrashort T2* relaxometry for quantitation of highly concentrated superparamagnetic iron oxide (SPIO) nanoparticle labeled cells. Magn Reson Med. 2009;61(4):761–766. | ||
Antony AC. The biological chemistry of folate receptors. Blood. 1992;79(11):2807–2820. | ||
Kamen BA, Smith AK, Anderson RG. The folate receptor works in tandem with a probenecid-sensitive carrier in MA104 cells in vitro. J Clin Invest. 1991;87(4):1442–1449. | ||
Leamon CP, Low PS. Delivery of macromolecules into living cells: a method that exploits folate receptor endocytosis. Proc Natl Acad Sci U S A. 1991;88(13):5572–5576. | ||
Antony AC. Folate receptors. Annu Rev Nutr. 1996;16:501–521. | ||
Puligujja P, Balkundi SS, Kendrick LM, et al. Pharmacodynamics of long-acting folic acid-receptor targeted ritonavir-boosted atazanavir nanoformulations. Biomaterials. 2015;41:141–150. | ||
Puligujja P, McMillan J, Kendrick L, et al. Macrophage folate receptor-targeted antiretroviral therapy facilitates drug entry, retention, antiretroviral activities and biodistribution for reduction of human immunodeficiency virus infections. Nanomedicine. 2013;9(8):1263–1273. | ||
Gautam N, Puligujja P, Balkundi S, et al. Pharmacokinetics, biodistribution, and toxicity of folic acid-coated antiretroviral nanoformulations. Antimicrob Agents Chemother. 2014;58(12):7510–7519. | ||
Jung CW, Jacobs P. Physical and chemical properties of superparamagnetic iron oxide MR contrast agents: ferumoxides, ferumoxtran, ferumoxsil. Magn Reson Imaging. 1995;13(5):661–674. | ||
Maity D, Chandrasekharan P, Yang CT, et al. Facile synthesis of water-stable magnetite nanoparticles for clinical MRI and magnetic hyperthermia applications. Nanomedicine. 2010;5(10):1571–1584. | ||
Tong S, Hou S, Zheng Z, Zhou J, Bao G. Coating optimization of superparamagnetic iron oxide nanoparticles for high T2 relaxivity. Nano Lett. 2010;10(11):4607–4613. | ||
Huang X, Zhuang J, Chen D, et al. General strategy for designing functionalized magnetic microspheres for different bioapplications. Langmuir. 2009;25(19):11657–11663. | ||
Liang YY, Zhang LM, Jiang W, Li W. Embedding magnetic nanoparticles into polysaccharide-based hydrogels for magnetically assisted bioseparation. Chemphyschem. 2007;8(16):2367–2372. | ||
Zhang L, Yu F, Cole AJ, et al. Gum arabic-coated magnetic nanoparticles for potential application in simultaneous magnetic targeting and tumor imaging. AAPS J. 2009;11(4):693–699. | ||
Sanganeria P, Sachar S, Chandra S, Bahadur D, Ray P, Khanna A. Cellular internalization and detailed toxicity analysis of protein-immobilized iron oxide nanoparticles. J Biomed Mater Res B Appl Biomater. 2015;103(1):125–134. | ||
Rogers M. New insights into the molecular mechanisms of action of bisphosphonates. Curr Pharm Des. 2003;9(32):2643–2658. | ||
Huang J, Gautam N, Bathena SP, et al. UPLC-MS/MS quantification of nanoformulated ritonavir, indinavir, atazanavir, and efavirenz in mouse serum and tissues. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(23):2332–2338. | ||
Kularatne SA, Low PS. Targeting of nanoparticles: folate receptor. Methods Mol Biol. 2010;624:249–265. | ||
Lu J, Li Z, Zink JI, Tamanoi F. In vivo tumor suppression efficacy of mesoporous silica nanoparticles-based drug-delivery system: enhanced efficacy by folate modification. Nanomedicine. 2012;8(2):212–220. | ||
Nukolova NV, Oberoi HS, Cohen SM, Kabanov AV, Bronich TK. Folate-decorated nanogels for targeted therapy of ovarian cancer. Biomaterials. 2011;32(23):5417–5426. | ||
Wang X, Li J, Wang Y, et al. HFT-T, a targeting nanoparticle, enhances specific delivery of paclitaxel to folate receptor-positive tumors. ACS Nano. 2009;3(10):3165–3174. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.