")
Back to Journals » International Journal of Nanomedicine » Volume 17
Magnet-Guided Bionic System with LIFU Responsiveness and Natural Thrombus Tropism for Enhanced Thrombus-Targeting Ability
Authors Fang N, Liu J, Hou J, Zhong Y, Luo Y, Hu L, Zhang W, Wang J, Xu J, Zhou J, Zhang Y , Ran H , Guo D
Received 5 January 2022
Accepted for publication 21 April 2022
Published 4 May 2022 Volume 2022:17 Pages 2019—2039
DOI https://doi.org/10.2147/IJN.S357050
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mian Wang
Ni Fang,1,2,* Jia Liu,1,* Jingxin Hou,1,2 Yixin Zhong,1,2 Ying Luo,1,2 Liu Hu,1,2 Wenli Zhang,1,2 Junrui Wang,1,2 Jie Xu,1 Jun Zhou,1 Yu Zhang,1 Haitao Ran,2 Dajing Guo1
1Department of Radiology, Second Affiliated Hospital of Chongqing Medical University, Chongqing, 400010, People’s Republic of China; 2Chongqing Key Laboratory of Ultrasound Molecular Imaging & Department of Ultrasound, Second Affiliated Hospital of Chongqing Medical University, Chongqing, 400010, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dajing Guo, Email [email protected]
Background: Arterial thrombosis is a serious threat to human health. Recently, many thrombus-targeted nanoparticles (NPs) have been constructed for detecting thrombi or monitoring thrombolysis, but their thrombus-targeting performance is limited. Considering this drawback, we designed a specific bionic system with enhanced thrombus-targeting ability.
Materials and Methods: In the bionic system, gelatin was chosen as a carrier, and Fe3O4 served as a magnetic navigation medium and a magnetic resonance (MR) imaging agent. The CREKA peptide, which targets fibrin, was conjugated to the surface of gelatin to prepare targeted NPs (TNPs), which were then engulfed by macrophages to construct the bionic system. At the targeted site, the bionic system released its interior TNPs under low-intensity focused ultrasound (LIFU) irradiation. Moreover, the targeting performance was further improved by the conjugated CREKA peptide.
Results: In this study, we successfully constructed a bionic system and demonstrated its targeting ability in vitro and in vivo. The results indicated that most TNPs were released from macrophages under LIFU irradiation at 2 W/cm2 for 10 min in vitro. Additionally, the enhanced thrombus-targeting ability, based on the natural tropism of macrophages toward inflammatory thrombi, magnetic navigation and the CREKA peptide, was verified ex vivo and in vivo. Moreover, compared with the bionic system group, the group treated with TNPs had significantly decreased liver and spleen signals in MR images and significantly enhanced liver and spleen signals in fluorescence images, indicating that the bionic system is less likely to be cleared by the reticuloendothelial system (RES) than TNPs, which may promote the accumulation of the bionic system at the site of the thrombus.
Conclusion: These results suggest that the magnet-guided bionic system with LIFU responsiveness is an excellent candidate for targeting thrombi and holds promise as an innovative drug delivery system for thrombolytic therapy.
Keywords: bionic system, low-intensity focused ultrasound, thrombus, magnetic navigation, macrophage
Introduction
Pulmonary embolism, myocardial infarction and acute ischemic stroke caused by thrombus are common life-threatening diseases, and the rate of associated mortality is increasing every year.1 Research on thrombi has been a topic of intense interest, especially in the field of targeted molecular probes. In recent decades, various types of nanocarriers have been developed to target thrombi.2–12 These nanoparticles (NPs), by binding receptors on the thrombus surface or responding to external stimulation (ultrasound or magnetic field), exhibited certain targeting properties. In our previous studies, we tried to conjugate ligands or peptides (RGDs, cRGD, EWVDV, CREKA) on the surface of NPs to target the platelet membrane receptors or fibrin in thrombi, and the results revealed some targeting by these NPs.13–16 In addition, some scholars have found that recombinant antibodies show a remarkable thrombus-targeting effect in vivo.17,18 To further enhance targeting performance, a dual-targeted molecular probe was constructed to simulate the wall attachment function of leukocytes and thereby resist the effects of the high flow velocity and high shear force of arterial blood flow, and the results showed that these dual-targeted molecular probes exhibited enhanced thrombus-targeting binding ability and stability.19 However, there are still problems such as the short circulation time and rapid clearance by the reticuloendothelial system (RES) in vivo.
To enhance the targeting performance of NPs and prevent RES clearance, a bionic system concept was proposed. A bionic strategy to enhance the targeting abilities of molecular probes is reported in Science,20 that is, the construction of a micron-scale NP aggregate that remains intact in blood circulation under physiological conditions. However, under the action of a local high shear force, the aggregate decomposes into smaller NPs. This bionic strategy improves targeting ability and eliminates RES clearance. It is widely believed that, compared with smaller NPs, larger NPs tend not to be removed by the RES and are less likely to accumulate and be cleared by the liver and kidney.21 Therefore, this bionic system is expected to evade RES capture and exhibit an enhanced thrombus-targeting ability. The use of living immune cells for drug delivery is an emerging area of research that involves the ingestion of microsomes and microorganisms by macrophages. Moreover, macrophages are able to sense chemokines and cytokines and thus home to inflammatory tissues, and this property can be used to enhance thrombus-targeting ability.22,23 Therefore, several studies have reported that NP-loaded macrophages displayed excellent drug loading capacity with decreased RES clearance and good biocompatibility in vivo.24,25
However, a study reported that it takes 6 to 12 h for such drug-carrying macrophages to reach inflammatory tissues through chemotaxis.21 Considering the urgency of treating thrombotic diseases, this ability of macrophages to actively home to inflammatory thrombi is far from sufficient. Therefore, we considered combining the active targeting of macrophages with passive targeting guided by magnetic navigation under an external magnetic field to achieve improved targeting performance. Magnetic navigation with permanent magnets was selected to enable macrophages to accurately deliver NPs to vascular embolisms with a reduced time window. Fe3O4 NPs are of great interest in the biomedical field and are currently undergoing preclinical studies, including experimental drug delivery, magnetic hyperthermia, and medical imaging.26,27 Many studies have suggested that magnetic guidance based on Fe3O4 NPs represents the most promising method for drug delivery.28–30 In addition, Fe3O4 NPs have been considered to have a distinct negative contrast effect in magnetic resonance (MR) imaging.31,32 Because of the intrinsic properties of iron oxide, NPs containing Fe3O4 can be used as both a magnetic navigation medium and an MR contrast agent.
The chemotaxis of macrophages and magnetic navigation enable the bionic system to reach the thrombus, and the NPs in the bionic system must be released and aggregate around the thrombus. Low-intensity focused ultrasound (LIFU) has been widely used in clinical nondestructive diagnoses because it can be transmitted in vivo without injurious tissue secondary effects.33 Moreover, the increased temperature, cavitation and mechanical force generated by LIFU can affect the structure and function of biological tissues and cells.34–36 Ikehara showed that mild warming is a promising means of regulating the release of drug molecules in cells.37 Liposomes coated with anticancer drugs and magnetic NPs were injected into the abdominal cavity of mice, and macrophages were loaded with the drugs in vivo. In the presence of alternating magnetic fields, magnetic NPs convert electromagnetic energy into heat energy, which regulates and controls the release of anticancer drugs by macrophages. Therefore, the physical properties of local heating with LIFU are expected to enable the release of NPs from the bionic system.
In summary, we planned to construct a bionic system. First, gelatin was used as a carrier, Fe3O4 was loaded onto the surface, and the CREKA peptides were conjugated to the surface of gelatin to construct Gel-Fe3O4-CREKA NPs. Then, the NPs were phagocytosed by macrophages to construct the bionic system. The tropism of macrophages to naturally home to inflammatory thrombus sites through platelet inflammatory factors, coupled with magnetic navigation under an external magnetic field, allows as much of the bionic system as possible to be delivered near the thrombus and escape surveillance by the RES. In response to LIFU irradiation, NPs are released from macrophages, and the CREKA peptide enables NPs to continue to target fibrin and deposit at the thrombus site, thus enhancing the thrombus-targeting capability. The construction of the bionic system and the process of targeting thrombi are presented in Figure 1. In this paper, the targeting effect of the bionic system guided by magnetic navigation combined with the response to LIFU irradiation was studied in vitro and in vivo. In addition, we tested whether the bionic system is capable of evading RES monitoring and whether it is suitable as an innovative drug carrier for thrombolytic therapy.
 |
Figure 1 Schematic illustration of the construction of the bionic system and the process of targeting thrombi. |
Materials and Methods
Materials
Gelatin (reagent grade, type A, gel strength ~300 g bloom) was purchased from Sigma–Aldrich (St. Louis, MO, USA). Glutaraldehyde solution (50%) and acetone solution were purchased from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). Iron oxide NPs (water-soluble Fe3O4, 10 nm) were obtained from Xi’an Ruixi Biological Technology Co., Ltd. (Xi’an, China). 2-(N-Morpholino) ethanesulfonic acid (MES), 1-ethyl-3-(3-(dimethylamino) propyl) carbodiimide hydrochloride (EDC), and N-hydroxysuccinimide (NHS) were purchased from Sigma–Aldrich (St. Louis, MO, USA). CREKA and CREKA-FITC peptides were synthesized by Chinapeptides Co., Ltd. (Jiangsu, China). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo (Kumamoto Prefecture, Kyushu, Japan). All cell culture reagents were provided by Sigma–Aldrich (St. Louis, MO, USA).
Preparation of NPs
A modified two-step dissolution method was used to prepare gelatin NPs (GNPs).38 First, gelatin (625 mg) was dissolved in 12.5 mL of deionized water at 40°C. Then, 12.5 mL of acetone was added dropwise (6 mL/min) to the gelatin solution and continuously stirred (600 rpm) for 10 min. Afterward, the solution was stratified, and the supernatant was removed. Next, the pH of the residual solution was adjusted to 2.5 by the addition of 1 M HCl. Subsequently, 21 mL of acetone solution was added dropwise (1 mL/min) to the solution under continuous stirring (1000 rpm). Then, 1 mL of acetone containing 30 μL of 50% glutaraldehyde solution was added dropwise (0.05 mL/min) to the solution at 40°C under constant stirring (1000 rpm). After continuous stirring for 16 h, acetone and excess glutaraldehyde were removed by a rotary evaporator. GNPs were obtained after centrifugation (10,000 rpm, 5 min).
To construct the nontargeted NPs (NTNPs), 21 mL of acetone solution was replaced by 21 mL of acetone containing 100 µL of soluble Fe3O4 solution, and the other steps were the same. Finally, NTNPs (Gel-Fe3O4 NPs) were obtained. A carbodiimide-mediated amide bond formation method was used to prepare the targeted NPs (TNPs). Appropriate amounts of EDC and NHS at a molar ratio of 2:1 were added to 2 mL of 0.1 M MES buffer solution (pH = 5), and the resulting solution was added to the NTNPs as described above. The mixture was incubated in a shaker for 3 h. After centrifugation, 2 mL of 0.1 M MES buffer solution (pH = 8) was added to dissolve the mixture. Then, 10 mg of CREKA peptide was added, and the mixture was incubated in a shaker for 12 h. Throughout the process, the NPs were maintained at 40°C. Finally, TNPs (Gel-Fe3O4-CREKA NPs) were obtained after centrifugation and then rinsed three times with double-distilled water.
Characterization of the NPs
First, 100 μg/mL GNPs and TNPs were diluted in double-distilled water to 10 μg/mL, and 1 drop was extracted with a 2 mL syringe and deposited on a 200-mesh copper grid. After 24 h, the morphology and size of the samples were observed by optical microscopy (Olympus CKX41, Olympus Co., Ltd., Tokyo, Japan), transmission electron microscopy (TEM, Hitachi 7500, Hitachi Ltd., Tokyo, Japan) and scanning electron microscopy (SEM, Hitachi S3400N, Japan). GNPs, NTNPs and TNPs diluted to a transparent liquid were dropped into cuvettes and potentiometers. The size, polydispersity index (PDI), and zeta potential of the different NPs were determined at 25°C using a dynamic light scattering (DLS) particle sizer (Zetasizer Nano ZS90, Malvern Instruments, Ltd., Worcestershire, UK). The Fe concentration was analyzed by inductively coupled plasma–mass spectrometry (ICP–MS, Thermo iCAP 6300, Thermo Fisher Scientific, USA). The carrier rate of the CREKA peptide was determined using flow cytometry (FACSVantage SE, Becton Dickinson, San Jose, CA, USA). The NTNPs and TNPs were freeze-dried, and the amide bond was determined by Fourier transform infrared spectroscopy (FTIR, Nicolet iS50, Thermo Scientific Co. Ltd., MA, USA). DiI-stained NTNPs were allowed to react with FITC-labeled CREKA peptide by a carbodiimide method for 12 h, and the binding status was observed by confocal laser scanning microscopy (CLSM, Nikon A1R, Tokyo, Japan). In addition, the stability of the GNPs and TNPs was monitored for 8 consecutive days by DLS. GNPs and TNPs were incubated in solutions of pH 2.5, 3.5, 4.5, 5.5, 7 and 9, and their stability was determined by DLS on day 7.
Cytotoxicity of the NPs
RAW 264.7 cells were obtained from Chongqing Key Laboratory of Ultrasound Molecular Imaging. The cell lines were approved by the Animal Ethics Committee of Chongqing Medical University and conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Chongqing Medical University. The cells were cultured in complete Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin and added to a 25 cm2 cell culture flask with a humidified atmosphere containing 5% carbon dioxide (CO2) at 37°C. The cells in a culture flask containing 5 mL of complete medium were activated with 100 ng/mL lipopolysaccharide (LPS) for 24 h using cells in the logarithmic growth phase for the experiments. GNPs, NTNPs and TNPs (20, 40, 60, 80 and 100 μg/mL) were added to a clear-bottom 96-well plate containing RAW 264.7 cells. Next, they were incubated together for 24 h. Then, cell viability was determined by the CCK-8 method, and the absorbance was measured at 450 nm using a plate reader. In addition, GNPs, NTNPs and TNPs at a concentration of 100 μg/mL were incubated with RAW 264.7 cells for 24 h, and the apoptosis results were observed by flow cytometry.
Preparation of the Bionic System
NTNPs and TNPs (1 mg/mL, 10 μL) were incubated with LPS-activated RAW 264.7 cells (macrophages, mice) in 15-mm cell culture dishes for a certain period of time and then washed with phosphate-buffered saline (PBS) to collect the cells, thus obtaining bionic systems, ie, MNTNPs (NTNPs ingested by macrophages) and MTNPs (TNPs ingested by macrophages).
To explore the phagocytosis of the macrophages that had engulfed NPs, TNPs were stained with DiI (DiI/TNPs) and incubated with LPS-activated or nonactivated RAW 264.7 cells for 1, 2, 3 and 4 h at 37°C and 5% CO2 in an incubator. Next, the NPs were washed three times with PBS, and those that were not engulfed by the macrophages were removed. Then, the bionic systems were fixed with 4% paraformaldehyde for 30 min, and the nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) staining solution for 20 min. Finally, all the prepared samples were observed under CLSM and flow cytometry.
Then, according to the above phagocytosis results, the best time point for the incubation of NPs with macrophages was selected, and the morphology of the bionic systems (MNTNPs or MTNPs) was observed under TEM.
Effects of LIFU Irradiation on the Bionic Systems
MTNPs were cultured in 15-mm cell culture dishes, and LIFU irradiation (1, 2, 3, and 4 W/cm2) was applied at the bottom of the dish for 10 min. Then, the temperature was measured in each with a thermometer (n = 3). Next, the bionic systems were washed with PBS three times, stained with CAM/PI (Dojindo, Kumamoto Prefecture, Kyushu, Japan) and observed by CLSM.
DiI-stained NTNPs linked to CREKA-FITC peptides were engulfed by RAW 264.7 cells and then cultured in 15-mm cell culture dishes for 24 h. Next, LIFU irradiation (2 W/cm2, 10 min) was applied at the bottom of the dish. The liquid of the cell culture dishes was collected and centrifuged (1000 rpm, 5 min), and then the supernatant was taken and centrifuged again (10,000 rpm, 5 min). After the precipitates were washed three times with double-distilled water, the morphology of the precipitates in the double-distilled water suspension was observed by TEM and CLSM.
Thrombus-Targeting Performance of the Bionic System
Cell Migration Assay
Three groups of MTNPs (4 × 104) were mixed evenly in 200 μL of basal medium and then added to the upper chamber. The first group was supplemented with 500 μL of complete medium in the lower chamber; the second group was supplemented with 500 μL of complete culture medium mixed with 3 mg of platelet inflammatory factor IL-1; the last group was supplemented with 500 μL of complete culture medium mixed with 3 mg of platelet inflammatory IL-1 factor, and magnets were placed at the bottom of the lower chamber (n = 3). These groups were then placed in incubators for 24 h. Afterward, the upper chamber was washed twice with PBS and fixed with 4% paraformaldehyde for 30 min. Next, the upper chamber was stained with crystal violet staining solution for 20 min and washed twice with PBS. Finally, the migrating cells were observed under a microscope and counted.
Targeting Capability of the Bionic System in an Extracorporeal Circulation Device
Sprague–Dawley (SD) rats (200~300 g) were purchased from the Animal Center of Chongqing Medical University. The animal experiments were approved by the Animal Ethics Committee of Chongqing Medical University and conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Chongqing Medical University.
To compare the abilities of NTNPs, TNPs and MTNPs to bind with thrombi, fresh blood was obtained from the aorta abdominalis of SD rats to simulate thrombi in vitro. After 5 h of incubation in 20-mL beakers at 37°C, thrombus samples were cut into squares of 1 cm × 1 cm. As in a previous study, an internal circulation device (Figure 2) was simulated.39 This system (UIP, Kamoer, Shanghai, China) contains precision peristaltic pumps, silicone tubing, and polypropylene plastic tubing. A thrombus sample 1 cm × 1 cm in size was placed in a plastic tube, and a 3-mm-thick mesh sponge was used to secure the thrombus sample. A magnet was placed at the thrombus sample site. Next, 80 mL of DiI/NTNPs or DiI/TNPs at a concentration of 100 μg/mL or DiI/MTNPs at a concentration of 4 × 106/mL were added to the circulating model. The thrombus samples were treated with saline, NTNPs, TNPs, MTNPs, TNPs + magnet, or MTNPs + magnet + LIFU. In addition, thrombus samples were allowed to react with excess free CREKA peptides and then treated with MTNPs + magnet + LIFU. A magnet was placed at the thrombus sample site in the groups treated with TNPs + magnet and MTNPs + magnet + LIFU. In addition, the thrombus samples in the groups treated with MTNPs + magnet + LIFU (with or without excess free CREKA peptide treatment) were irradiated with LIFU (2 W/cm2, 10 min according to our LIFU irradiation experiment on the bionic system). Next, the silicone tube of the circulating device was closed, and the circulating time was set to 10 min. For venules, capillaries, transient blood flow arrest, and stenosis and embolization vessels, the flow velocity can be 0 or 20 cm/s, and a flow velocity of 40 cm/s is similar to that in a large artery.40,41 Therefore, a speed of 40 cm/s was set to approximate the flow rate of the carotid artery, and the targeting effect of the bionic system under this condition was discussed. The entire process was performed in a water tank at 37°C, and each group of experiments was repeated 3 times. At the end of each experiment, we cleaned the circulation device with double-distilled water. After the reaction, the thrombus sample was carefully washed with PBS three times, fixed with paraformaldehyde at 4°C, and then used for frozen sectioning. Finally, the results were imaged by CLSM. The fluorescence intensity of each group was analyzed by ImageJ software.
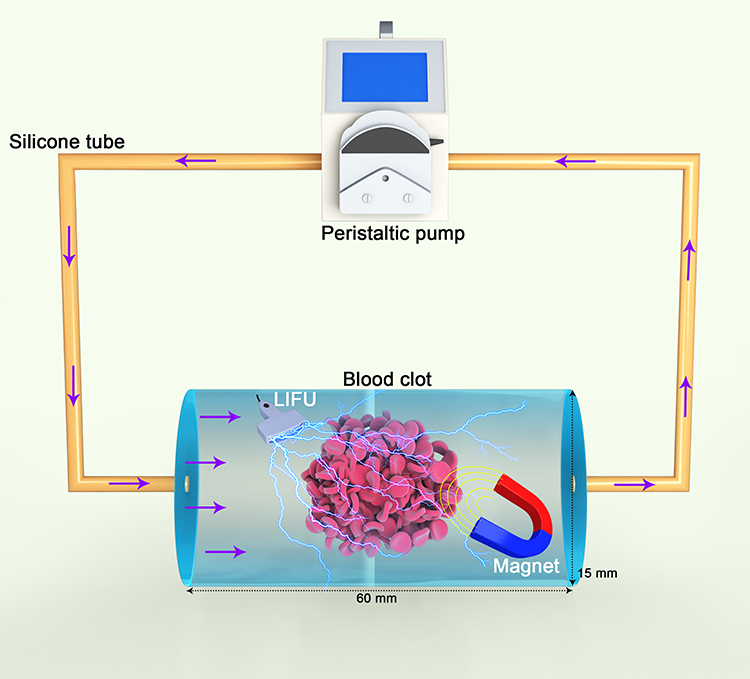 |
Figure 2 Schematic diagram of the extracorporeal circulation device. |
Targeting Capability of the Bionic System in vivo
For in vivo targeting studies, the rats were anesthetized (30 mg/kg sodium pentobarbital), and the carotid artery was exposed by removing the skin, muscle and fascia. Next, the formation of vascular thrombi was induced by placing a filter paper strip saturated with 10% FeCl3 beneath the vessels for 3 min. The filter paper was then removed, and the exposed carotid artery was rinsed with saline. Finally, thrombi were formed.
Twenty-one SD rats were randomly divided into seven groups (n = 3), followed by treatment with saline, NTNPs, TNPs, MTNPs, TNPs + magnet, MTNPs + magnet + LIFU, and MTNPs + magnet + LIFU# (#before treatment, 0.5 mL hydrated CREKA peptides (10 mg/mL) were injected into the tail vein of rats). After the thrombus model in the carotid artery was successfully established, the saline group was injected with 1 mL of saline, and the other 6 groups were injected through the tail vein with 1 mL of NTNPs or TNPs at a concentration of 100 μg/mL or MTNPs at a concentration of 4 × 106/mL. Rats in the TNPs + magnet and MTNPs + magnet + LIFU groups had magnets placed in their necks. Moreover, LIFU was used to irradiate the exposed thrombus segment of the left carotid artery in the MTNPs + magnet + LIFU group (2 W/cm2, 10 min). Afterward, the thrombus segment of the left carotid artery was extracted and rinsed twice with saline. Next, the samples were fixed with paraformaldehyde at 4°C and harvested for hematoxylin and eosin (H&E) staining. All pathological samples were examined by an experienced pathologist who was unaware of the grouping and treatment.
MR Imaging in vitro and in vivo
MR Imaging of NPs in vitro
NTNPs and TNPs (0.06, 0.07, 0.09, 0.13, 0.22, and 0.35 mM) were added to 1.5-mL EP tubes and scanned with a 3.0 T MR scanner (Magnetom Prisma, Siemens, Germany). T2WI images were collected (TR: 8340 ms; TE: 85 ms; field of view: 80 mm, matrix: 175 x 175; slice thickness: 0.9 mm). The circular region of interest (ROI) was plotted using FuncTool 2.0 software, and the T2 value of each image was obtained. Similarly, the corresponding signal strength was obtained from the ADW4.6 image workstation (ROI= 100 pixels).
MR Imaging of the Bionic System in vivo
The rats were divided into four groups (n = 3), followed by treatment with NTNPs, TNPs, TNPs + magnet, and MTNPs + magnet + LIFU. NTNPs and TNPs (100 μg/mL, 1 mL) or MTNPs (4 × 106/mL, 1 mL) were injected through the tail vein after the carotid artery thrombus model was formed. Rats in the TNPs + magnet and MTNPs + magnet + LIFU groups had magnets placed at the site of the carotid artery thrombus. In the group treated with MTNPs + magnet + LIFU, the exposed thrombus segment of the left carotid artery was irradiated with LIFU (2 W/cm2, 10 min). The necks of the rats were scanned with an MR scanner. T2WI images were collected at 0, 15, 30, 45, and 60 min (TR: 8340 ms; TE: 85 ms; field of view: 80 mm, matrix: 175x175; slice thickness: 0.9 mm). In addition, a group of SD rats (n = 3) was injected through the tail vein with 1 mL of TNPs at a concentration of 100 μg/mL or MTNPs at a concentration of 4 x 106/mL. Then, coronal MR-T2WI images of the abdomen were collected at 0 min, 10 min, 20 min, 30 min, 60 min, 2 h and 4 h. The signal-to-noise ratio (SNR) was calculated (SNR = S/N, where S = T2 signal intensity of liver, kidney, or spleen and N = T2 signal intensity of adjacent muscle tissue).
Fluorescence Imaging in vitro and in vivo
To verify the fluorescence intensities of DiR-labeled TNPs in vitro, the DiR-labeled TNPs were diluted to concentrations ranging from 60 mg/mL to 10 mg/mL for fluorescence imaging.
The rats were divided into two groups (n = 3) and treated with TNPs and MTNPs + magnet + LIFU. DiR-labeled TNPs (100 μg/mL, 1 mL) or DiR-labeled MTNPs (4 × 106/mL, 1 mL) were injected through the tail vein after the carotid artery thrombus model was formed. In the group treated with MTNPs + magnet + LIFU, a magnet was placed at the site of carotid artery thrombus in the SD rats, and the exposed thrombus segment of the left carotid artery was irradiated with LIFU (2 W/cm2, 10 min). Next, the fluorescence signal of the left carotid artery was monitored by a small-animal fluorescence imaging system at 0, 15, 30, 45, 60, and 90 min and 2 h. Then, quantitative analysis of the fluorescence intensity values of the left carotid artery regions at corresponding time points was performed. The SD rats treated as described above were sacrificed after 2 h, and the left carotid artery and major organs (heart, liver, spleen, lung, kidney) were collected for fluorescence imaging. The fluorescence intensities of these tissues and major organs were quantitatively analyzed by ImageJ.
Biocompatibility Evaluations of the Bionic System in vivo
Rat blood samples (1.0 mL) were obtained from the heart 3 h after the injection of 1 mL of saline and TNPs (concentration of 100 μg/mL) or MTNPs (concentration of 4 × 106/mL). Then, blood biochemical and hematological parameters were measured. Euthanasia was performed at 24 h, and the major organs (heart, liver, spleen, lung, kidney) were harvested and stained with H&E.
Statistical Analysis
The data were analyzed using GraphPad Prism 9. The continuous variables are presented as the mean ± the standard deviation. Two-way ANOVA was utilized for the diameter of NPs and quantitative analysis of cell migration. Quantitative analysis of the fluorescence intensity value was performed according to Student’s t test. Differences were considered significant at P<0.05.
Results and Discussion
NP Characteristics
Gelatin is a natural, highly biocompatible, biodegradable biopolymer with low immunogenicity potential and is widely used in industry, medicine, medical treatment and other fields.42,43 In the field of drug delivery systems targeting thrombi, there are no literature reports on gelatin-induced coagulation dysfunction.44,45 The structure of gelatin includes many carboxyl groups, which can be linked to the abundant amine groups of CREKA by a carbodiimide method. Therefore, we used gelatin as the carrier in this experiment. The results showed that the GNPs and TNPs were spherical, uniform in size and regular in shape (Figure 3A–D). The average particle diameter, PDI, and zeta potential are shown in Table 1. DLS analysis showed that the GNPs were small and had good dispersibility. After loading Fe3O4 and connecting CREKA peptides, the diameter of the TNPs was increased slightly compared to those of the GNPs and NTNPs (P<0.05). The ICP–MS results showed that the Fe3O4 loading rates of NTNPs and TNPs were 69.12 ± 3.41% and 57.81 ± 2.40%, respectively (Figure 3E). The NTNPs were stained by DiI and connected to CREKA-FITC peptides. Flow cytometry analysis showed that the loading rate of CREKA-FITC-conjugated NPs was 64.75% (Figure 3F). The FTIR results showed that the carboxyl termini of the NTNPs and the amino termini of CREKA peptides were dehydrated and condensed. Compared to the NTNP group, the characteristic peaks of the TNP group were at approximately 3380 and 1650 cm−1, caused by the tensile vibrations of the amide bonds (Figure 3G). CLSM images further confirmed the successful conjugation of FITC-stained CREKA peptides onto the surface of DiI-stained NTNPs, with significant overlap between green and red fluorescence in the TNP group (Figure 3H). This observation proved that the TNPs formed amide bonds and that the CREKA peptides were successfully linked to the NPs. During the observation period of 8 days, the diameters of the GNPs and TNPs fluctuated within the ranges of 0.85–7.2 nm and 2.77–8.85 nm, respectively (Figure 3I). The zeta potentials of the GNPs and TNPs fluctuated within the ranges of 0.03–1.7 mV and 0.3–1.18 mV, respectively (Figure 3J). There was no significant difference in the diameter and zeta potential changes of the GNPs and TNPs in solutions with different pH values (P>0.05), which indicates that GNPs and TNPs can maintain excellent stability in the environment for a long time and at different pH values.
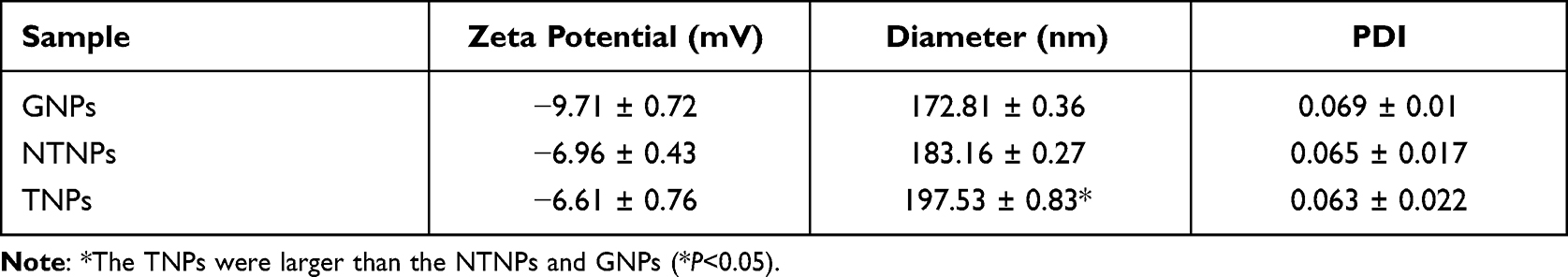 |
Table 1 Characteristics of Different NPs (n = 3) |
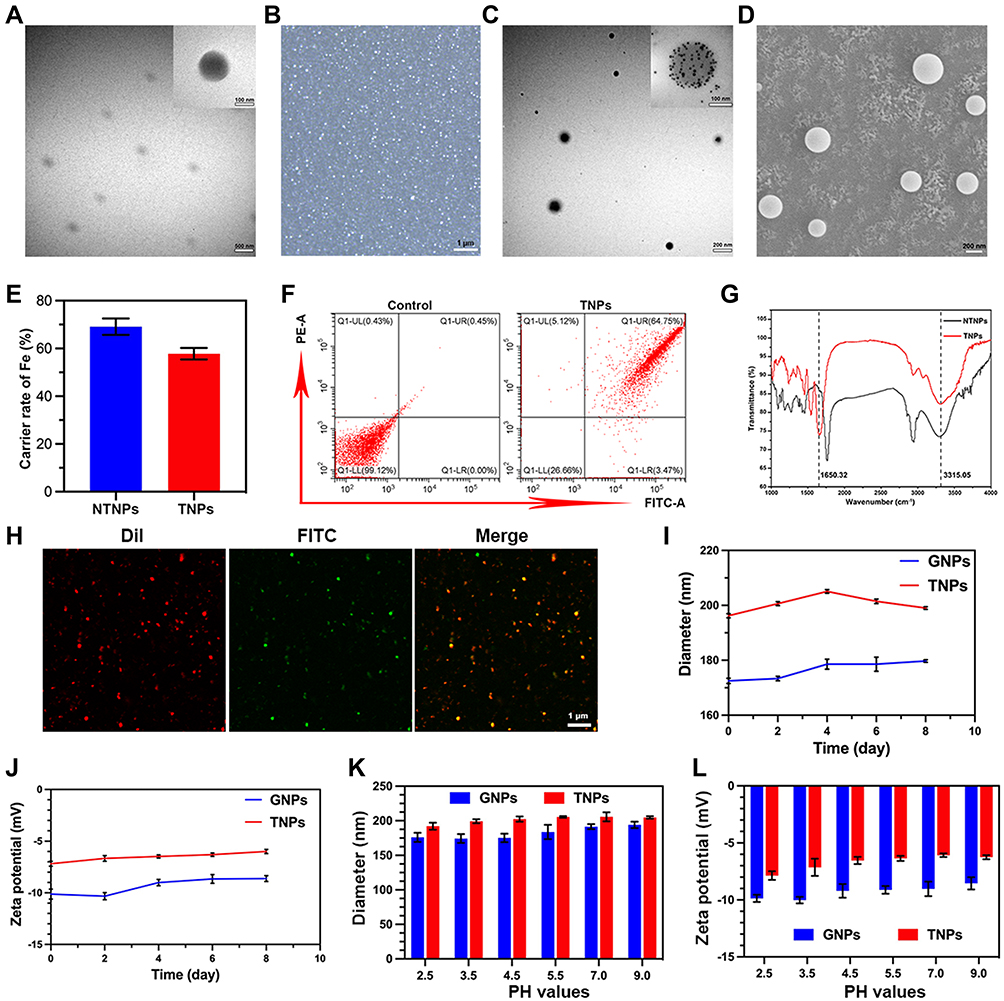 |
Figure 3 NP characterization. (A) TEM image of GNPs (scale bars: 500 nm, scale bar for enlarged TEM: 100 nm). (B) Optical microscopy image of TNPs (scale bar: 1 μm). (C) TEM image of TNPs (scale bar: 200 nm, scale bar for enlarged TEM: 100 nm). (D) SEM image of TNPs (scale bar: 200 nm). (E) Fe3O4 loading rates of NTNPs and TNPs. (F) Flow cytometry results showing the CREKA carrier rate in TNPs. (G) FTIR results of NTNPs and TNPs. (H) Results of CREKA conjugation onto TNP surfaces under CLSM (scale bar: 1 μm). Diameter (I) and zeta potential (J) changes of GNPs and TNPs in deionized water over 8 days. Diameter (K) and zeta potential (L) changes of GNPs and TNPs in solutions of different pH on day 7. |
NP Cytotoxicity
All NPs were incubated with RAW 264.7 cells for 24 h at NP concentrations ranging from 20 μg/mL to 100 μg/mL. The cell survival rate was above 80% (Figure 4A), showing that the NPs all have excellent biosafety. Flow cytometry also showed that the RAW 264.7 cells maintained excellent viability at NP concentrations up to 100 μg/mL (Figure 4B).
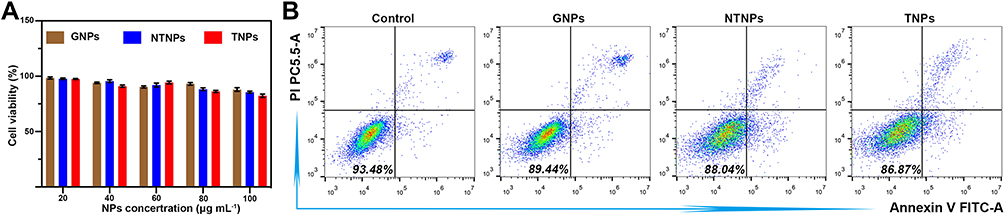 |
Figure 4 Cytotoxicity of different kinds of NPs to RAW 264.7 cells. (A) Cell viability of RAW 264.7 cells incubated with different NPs for 24 h. (B) Apoptosis results of RAW 264.7 cells incubated with different NPs for 24 h by flow cytometry. |
Construction of the Bionic System
The CLSM results showed that the red fluorescence intensity of TNPs around the DAPI-stained nucleus increased with time (Figure 5A), indicating that an increasing number of TNPs were engulfed by LPS-activated RAW 264.7 cells and subsequently accumulated around the nucleus. Moreover, the flow cytometry results displayed the same trend (Figure 5C). Nonactivated RAW 264.7 cells phagocytosed significantly fewer TNPs than LPS-activated RAW 264.7 cells during the same time period (Figure 5B). This may be because RAW 264.7 cells have a receptor on their cell surface called TLR4 that captures LPS. Once LPS binds to TLR4, the signal travels to the nucleus of the cell, affecting gene expression in the nucleus and activating the cell. As a result, LPS-activated RAW 264.7 cells were more capable of engulfing NPs.46,47 Based on the effects of the NPs on cell viability and the phagocytosis results, we selected an incubation time of 4 h and LPS-activated RAW 264.7 cells to construct the bionic systems. TEM images of the MNTNPs and MTNPs showed that the black dot-like NPs accumulated in the cytoplasm (Figure 5D). These results demonstrated the successful construction of the bionic systems.
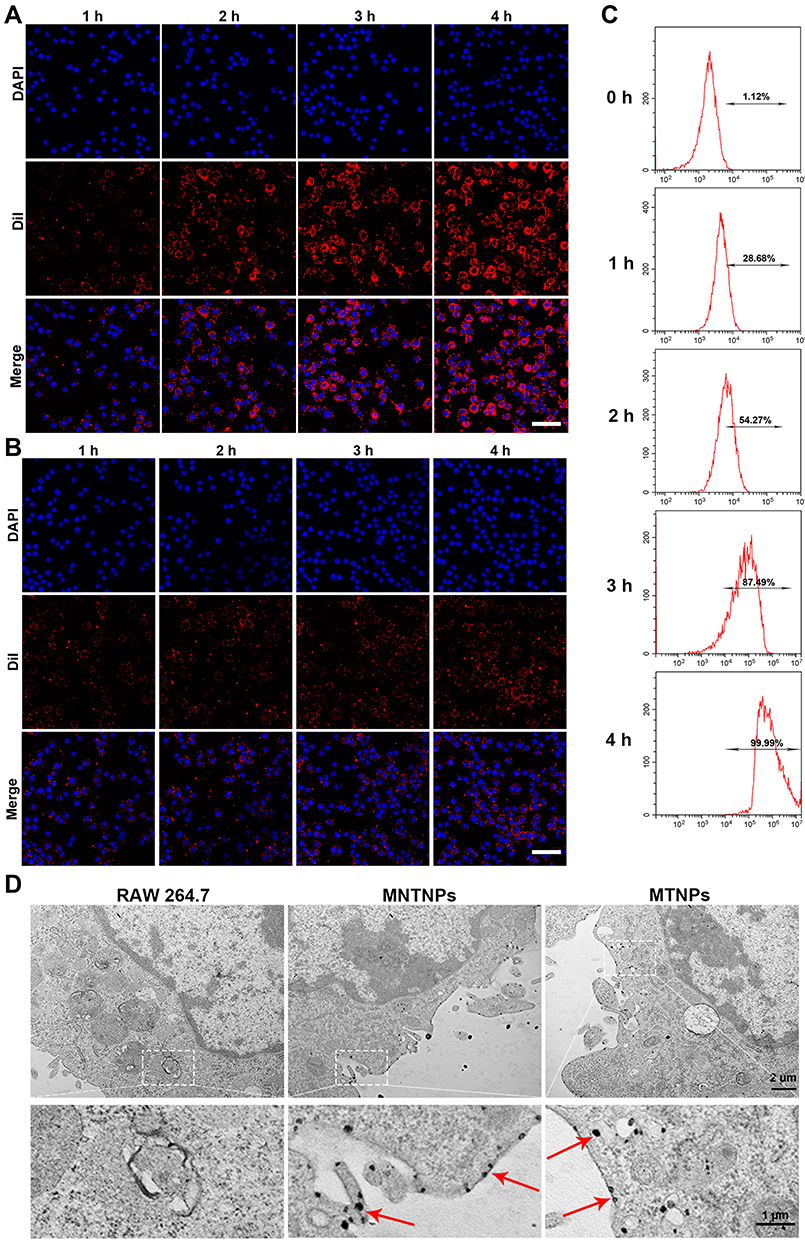 |
Figure 5 Construction of the bionic system. CLSM results of the phagocytosis of NPs by LPS-activated RAW 264.7 (A) or nonactivated RAW 264.7 (B) cells at different times (scale bar: 50 μm). (C) Flow cytometry results of phagocytosis of NPs by macrophages at different times. (D) TEM images of RAW 264.7 cells, MNTNPs and MTNPs; the red arrows represent the NPs engulfed by RAW 264.7 cells (scale bar: 2 μm, scale bar for enlarged TEM: 1 μm). |
Effects of LIFU Irradiation on the Bionic System
MTNPs were cultured in 15-mm cell culture dishes and then irradiated with LIFU at different intensities (1, 2, 3, and 4 W/cm2) for 10 min, and the effect of LIFU on the bionic system was measured. The results of live/dead staining showed that as LIFU intensity increased, the death rate of cells also increased. At 1 and 2 W/cm2, LIFU had little effect on the viability of MTNPs, and only a few MTNPs died. When the intensity of LIFU reached 3 and 4 W/cm2, all MTNPs lost viability (Figure 6A). These results suggested that the effects of LIFU on the viability of MTNPs corresponded to LIFU intensity. In addition, we explored the relationship between LIFU intensity and temperature under LIFU intensities of 0, 1, 2, 3, and 4 W/cm2 for 10 min. With increasing LIFU intensity, the temperature also increased: when the LIFU intensity was 1 W/cm2, the temperature remained at approximately human body temperature; when the LIFU intensity was 2 W/cm2, the temperature increased to 39–40°C; when the LIFU intensity was 3 W/cm2, the temperature increased to 40–41°C; and when the LIFU intensity was 4 W/cm2, the temperature reached 42.5°C (Figure 6B). Ikehara indicated that mild hyperthermia (39°C or 40°C) could cause macrophages to release 5-FU into the omentum.37 Based on our experimental results, under LIFU irradiation at 2 W/cm2 for 10 min, the temperature reached 39–40°C, which stimulated membrane and cytoplasmic enzyme activities, leading to active ion transport in macrophages and thus triggering the release of NPs.48 This temperature and the radiation intensity of LIFU will not cause harm to the human body in a short time,16,49 indicating that this method is promising for clinical application. Moreover, most of the MTNPs remained alive at 2 W/cm2 for 10 min, and the temperature did not damage the surrounding tissues, indicating the relative safety of LIFU irradiation. Therefore, we chose 2 W/cm2 to trigger the release of TNPs from macrophages for in vitro and in vivo experiments.
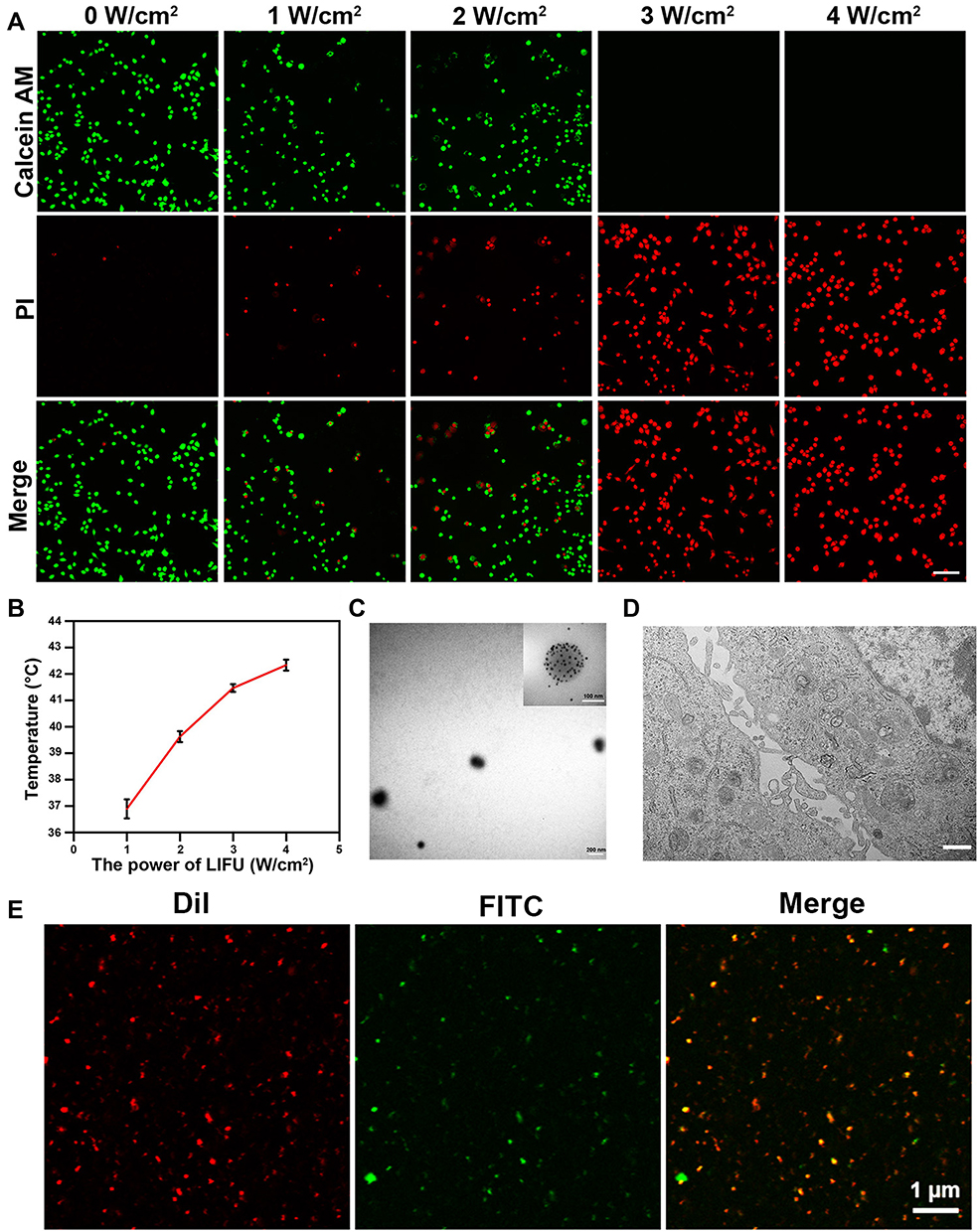 |
Figure 6 Effects of LIFU irradiation on the bionic system. (A) CLSM images of RAW 264.7 cells stained with CAM and PI after LIFU irradiation at 0, 1, 2, 3, and 4 W/cm2 for 10 min (scale bar: 50 μm). (B) Change curve of temperature with MTNPs irradiated with LIFU at different intensities (1, 2, 3, 4 W/cm2) for 10 min. (C) TEM image of NPs released by LIFU irradiation of MTNPs (scale bar: 200 nm, scale bar for enlarged TEM: 100 nm). (D) TEM image of cells treated with LIFU irradiation of MTNPs (scale bar: 2 μm). (E) CLSM images of NPs released by LIFU irradiation of MTNPs (scale bar: 1 μm). |
The TNPs released by the LIFU irradiation of MTNPs are regularly spherical in shape with a size of approximately 200 nm. Moreover, Fe3O4 still adsorbed on the surface of GNPs (Figure 6C). CLSM colocalization images showed that the CREKA peptides were still attached to the NTNP surface, while there were almost no obvious TNPs in the cytoplasm of RAW 264.7 cells (Figure 6D and E). These results suggested that TNPs can be released from MTNPs at this LIFU intensity (2 W/cm2, 10 min) and remain intact.
Thrombus-Targeting Performance of the Bionic System
Cell Migration Assay
The cell migration results showed that almost no cells in the group treated with MTNPs migrated, and only a small number of cells successfully penetrated the membrane of the upper chamber in the group treated with MTNPs + IL-1, while a large number of cells successfully penetrated the membrane in the group treated with MTNPs + IL-1 + magnet (Figure 7A and B). There were statistically significant differences among the three groups (P<0.0001). IL-1 is produced primarily by activated mononuclear macrophages and plays an important role in inflammatory responses.50 The migration of the bionic system across the upper chamber may suggest chemotaxis of the bionic system toward IL-1. The addition of magnets further reinforced the bionic system’s ability to home to the site of inflammation. Thus, the chemotaxis of macrophages and magnetic navigation can both be used to enhance the targeting capability of the drug delivery system toward thrombi. All these results may serve as a foundation for subsequent targeting experiments in vivo.
 |
Figure 7 Thrombus-targeting performance of the bionic system. (A) Images of the cell migration assay in the groups treated with MTNPs, MTNPs + IL-1 and MTNPs + IL-1 + magnet (scale bar: 50 μm). (B) Quantitative analysis of migrated cells in the groups treated with MTNPs, MTNPs + IL-1 and MTNPs +IL-1 + magnet (****P<0.0001). (C) CLSM images of frozen thrombus slices incubated with the indicated groups in an extracorporeal circulation device (# the thrombus samples were previously treated with excess free CREKA peptides; scale bar: 100 μm). (D) Average fluorescence intensity analysis of CLSM images in the groups treated with NTNPs, TNPs, MTNPs, TNPs + magnet, MTNPs + magnet + LIFU, and MTNPs + magnet + LIFU# (# the thrombus samples were previously treated with excess free CREKA peptides, *P<0.05, ****P<0.0001, ns = no significance). (E) Pathological images of carotid artery thrombus segments from the indicated groups after H&E staining; the red arrows indicate the NPs (# before treatment, 0.5 mL of hydrated free CREKA peptides (10 mg/mL) injected into the tail vein of rats; scale bar: 200 μm, scale bar for enlarged pathological images: 40 μm). |
Targeting Capability of the Bionic System in an Extracorporeal Circulation Device
After frozen sections of thrombi subjected to different treatments were prepared, the targeting effects of the bionic systems were observed under CLSM. Under the same treatment conditions without LIFU and magnetic navigation, the fluorescence signal of the TNPs was observed in the thrombus site, while the fluorescence signal of the NTNPs was not observed (Figure 7C), indicating that the NPs carrying the CREKA peptides possessed targeting ability in vitro. This finding is consistent with our previous research results.16 Some weak fluorescence signals were observed in the group treated with MTNPs (Figure 7C), possibly because a few macrophages adhered to the surface of the thrombus.
Then, the targeting properties of the bionic systems and TNPs were compared. When TNPs were not combined with magnetic navigation, the fluorescence signal was less than that of the TNPs + magnet group (Figure 7C and D). This observation proved that magnetic navigation enhances the aggregation of NPs at the thrombus site. The thrombi treated with MTNPs + magnet + LIFU and with TNPs + magnet exhibited the strongest fluorescence signals, from which it can be inferred that the greatest amounts of NPs adhered to these thrombi. This result may be due to the lack of platelet inflammatory factors in the blood clot tissues formed in vitro, which lessens the chemotaxis ability of macrophages in response to platelet inflammatory factors. Thus, there was no significant difference in the abilities of the two groups to target blood clots in vitro (Figure 7D, P>0.05). The thrombus samples were allowed to react with excess free CREKA peptides, and only a small amount of NPs released by macrophages bound to the thrombus sample. This may be because excessive free CREKA peptides preoccupy fibrin targets, leaving little remaining fibrin sites to bind to the CREKA peptides of TNPs. Compared with the group treated with MTNPs + magnet + LIFU, the difference in average fluorescence intensity was statistically significant (P<0.0001). It is showed that the CREKA peptides have a good and specific targeting to fibrin on the surface of thrombus.
Thrombus-Targeting Performance of the Bionic System in vivo
The blood vessels under different treatments were stained with H&E. The red arrows indicate the NPs. Since the surfaces of the thrombi are not smooth, NPs are more likely to accumulate in thrombus crevices, and NPs might also accumulate in the process of fixation, staining and sectioning, producing large black dots in the figure The results showed that more NPs accumulated around the thrombus site in the group treated with TNPs than in the groups treated with NTNPs or MTNPs without LIFU irradiation, and the effect was further enhanced by magnetic navigation (Figure 7E). The reason that only a small number of NPs aggregated at the thrombus site in the MTNP group may be that the NPs were not released from the MTNPs without LIFU irradiation. The results observed for this group in vivo were similar to those in vitro (Figure 7C). However, in the MTNPs + magnet + LIFU group, more NPs were deposited at the thrombus site than in the TNPs + magnet group, which was different from the in vitro results. This suggested that the natural ability of macrophages to target platelet inflammatory factors plays a role in vivo. On the other hand, macrophages may resist complicated blood flow situations in vivo, evade RES capture, and facilitate the transport of MTNPs to the thrombus site, creating conditions that maximize the subsequent release of NPs. Therefore, the group treated with MTNPs + magnet + LIFU showed more NPs accumulated at the thrombus site after LIFU irradiation. Compared with the group treated with MTNPs + magnet + LIFU, the group treated with MTNPs + Magnet + LIFU# did not accumulate many NPs at the thrombus site. This is probably because the free CREKA peptides preoccupied a part of the target binding sites on fibrin, leading to the failure of CREKA peptide binding in TNPs. However, the natural chemotaxis of macrophages to platelet factors still transports part of the NPs near the thrombi. This finding indicates that the CREKA peptide can bind specifically to fibrin and that MTNPs can specifically target thrombi.
MR Imaging in vitro and in vivo
MR Imaging of NPs
By recording the T2 data of NPs at different concentrations, we found that the MR signal decreased gradually with increasing NP concentration (Figure 8A), indicating that NTNPs and TNPs produced a high magnetic field gradient with a slight signal difference. This finding was consistent with our previous results showing that Fe3O4 has a strong negative enhancement effect.16 In addition, the slight difference between the results of the TNPs and NTNPs indicated that the conjugation of NTNPs to CREKA peptides did not affect the MR properties (Figure 8B).
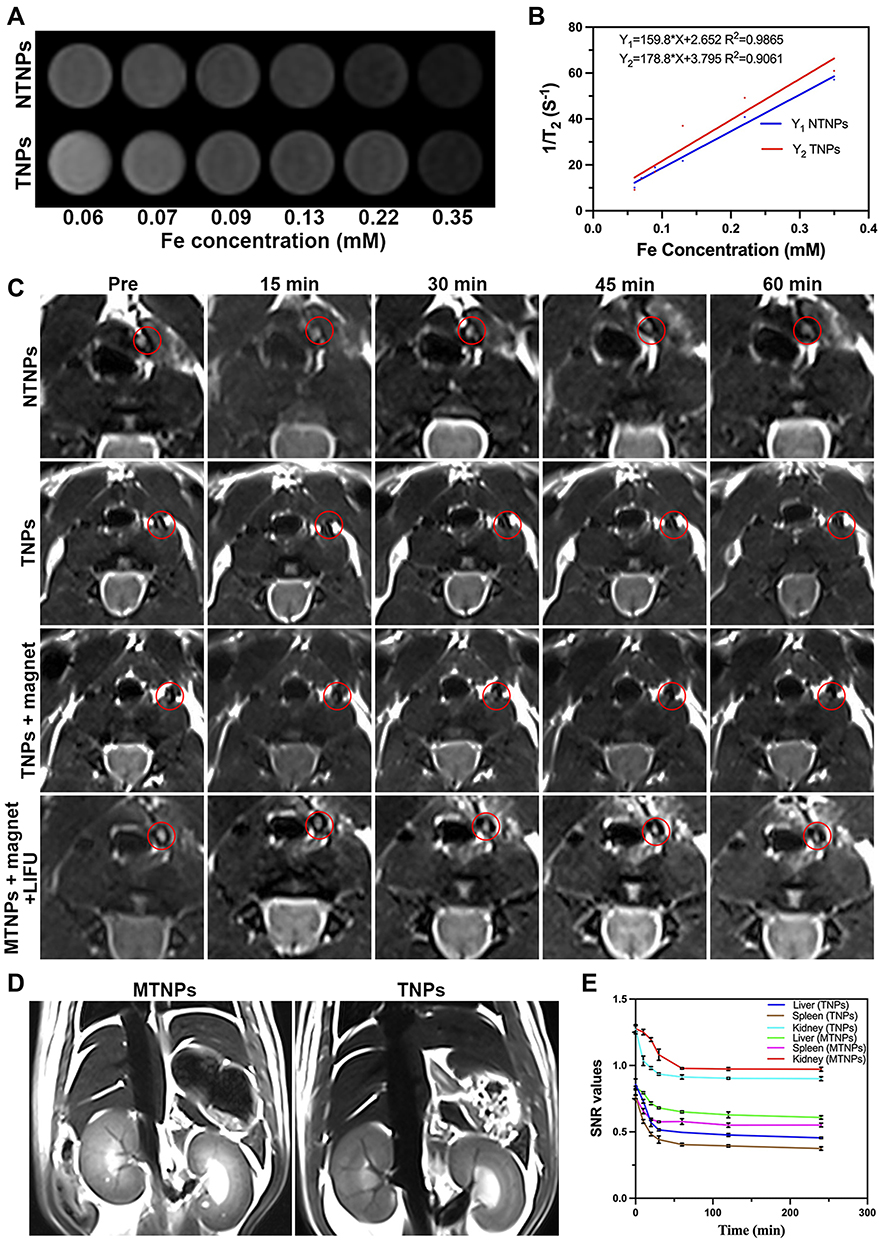 |
Figure 8 MR images in vitro and in vivo. MR-T2WI images (A) and 1/T2 (B) of NTNPs and TNPs with different Fe concentrations (0.06, 0.07, 0.09, 0.13, 0.22, and 0.35 mM, from left to right). (C) MR-T2WI images of thrombi at 0, 15, 30, 45, and 60 min after injection of NTNPs, TNPs (with or without magnet), MTNPs (with or without magnet + LIFU). The thrombus is shown in the red circle. (D) MR-T2WI images of the abdomen after injection of MTNPs and TNPs. (E) SNR curves of the liver, spleen, and kidney before and after injection of TNPs and MTNPs at 0 min, 10 min, 20 min, 30 min, 60 min, 2 h and 4 h. |
MR Imaging of the Bionic System in vivo
A thrombus model in the carotid artery was constructed and showed high signals on MR-T2WI images (Figure 8C). There was no significant signal change around the thrombus before and after NTNP injection. In the group treated with TNPs, the signal of the carotid artery decreased slightly after injection. Additionally, the signal reduction around the thrombus was more obvious in the group treated with TNPs + magnet than in the group treated with TNPs. For the group treated with MTNPs + magnet + LIFU, the signal around the thrombus decreased significantly, and the degree of reduction was significantly higher than those of all the previous groups. The reason for this may be that MTNPs are attracted to the thrombus site by the natural platelet-targeting ability of macrophages combined with magnetic navigation. The targeting performance was further enhanced by the release of TNPs from MTNPs after LIFU irradiation, which exposed the CREKA peptides, causing the NPs to continuously target fibrin. These results suggest that under the camouflage of macrophages and with magnetic guidance, MTNPs may escape capture by the RES and transport more TNPs to the thrombus site, increasing NP accumulation at the thrombus under LIFU irradiation, which improves thrombus-targeting ability in vivo.
Coronary MR-T2WI images of the abdomen demonstrated that the signals in the liver, kidney and spleen decreased significantly after TNP injection (Figure 8D). However, the SNR reductions in the liver, kidney and spleen of the MTNP group were significantly smaller than those of the TNP group (Figure 8E). The results showed that in the same period of time, the TNPs were more easily cleared by the liver, kidney and spleen, while MTNPs could escape capture by the RES because of the protection offered by the macrophages, improving the accumulation of the bionic system at the site of the thrombus and ensuring the safety of this application in vivo. This finding is consistent with the results of other studies.21,24,25
Fluorescence Imaging in vitro and in vivo
The fluorescence signal of TNPs was continuously enhanced and approached equilibrium as the concentration increased in vitro, and fluorescence quantitative analysis revealed the same results (Figure 9A).
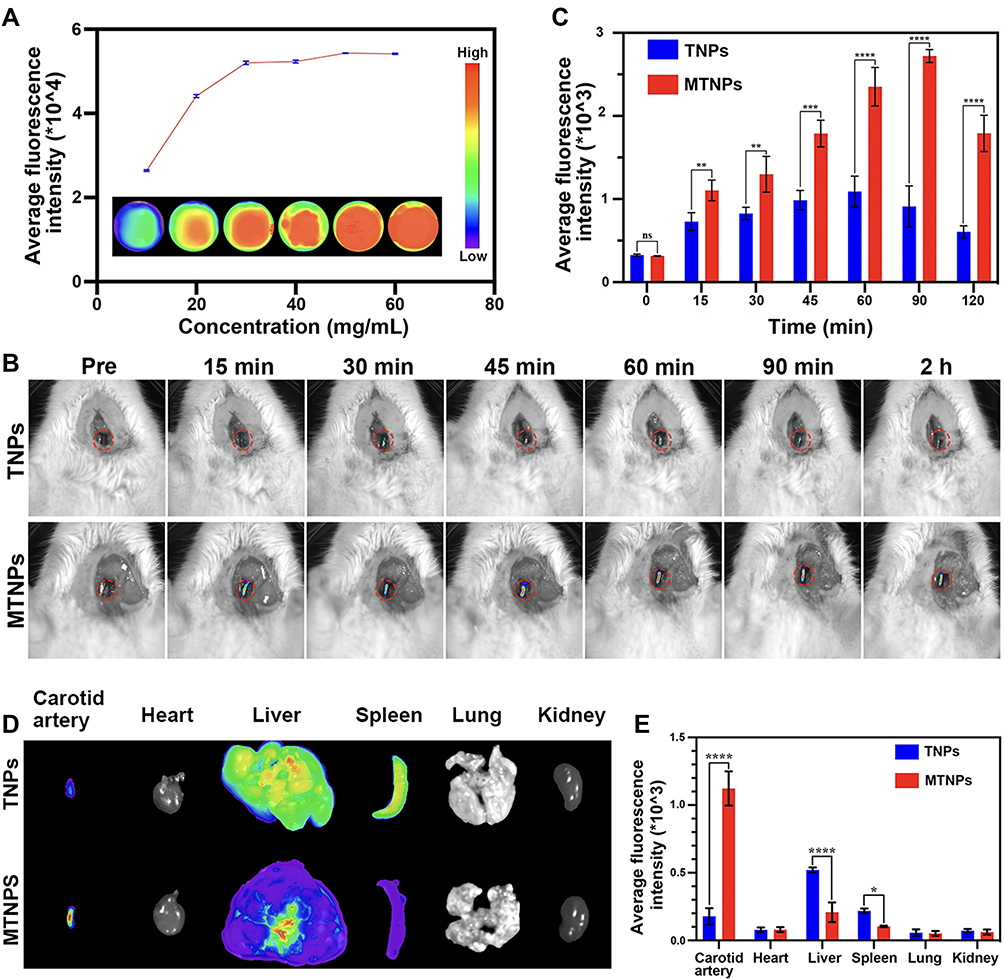 |
Figure 9 Fluorescence images in vitro and in vivo and biodistribution. (A) Fluorescence images and average fluorescence intensity values of TNPs at different concentrations in vitro. (B) Fluorescence images of the left carotid artery before and after injections of TNPs and MTNPs (with magnetic navigation and LIFU irradiation; the thrombus segment of the left carotid artery is shown in the red circle in each image) at different time points. (C) Quantitative analysis of the fluorescence intensity values of the left carotid artery regions at different time points (**P <0.01, ***P <0.001, ****P <0.0001). (D) Ex vivo fluorescence intensity images of the left carotid artery and major organs (heart, liver, spleen, lung, kidney) of the groups treated with TNPs and MTNPs (with magnetic navigation and LIFU irradiation). (E) Quantitative analysis of the fluorescence intensity values of ex vivo tissue and major organs for the groups treated with TNPs and MTNPs (with magnetic navigation and LIFU irradiation) (*P<0.05, ****P <0.0001). |
The image showed that the fluorescence intensity of carotid thrombosis increased gradually with increasing time from 0 to 90 min (Figure 9B). This result suggested that after the MTNPs target the thrombus site, due to LIFU irradiation and magnet attraction, an increasing number of NPs accumulate at the thrombus site over time. The fluorescence signal in the carotid artery was the strongest at 90 min. At 2 h, the fluorescence signal was slightly weaker than before, possibly caused by the removal of some NPs by the circulatory system. Quantitative analysis showed that at 60 min, 90 min and 120 min, the fluorescence intensity of the group treated with MTNPs + magnet + LIFU was significantly higher than that of the group treated with TNPs (P<0.0001, Figure 9C). This observation indicates that after LIFU irradiation and magnetic navigation attraction, the group treated with MTNPs + magnet + LIFU accumulated more NPs at the site of the thrombus than the group treated with TNPs. This finding was consistent with the MR results in vivo, which suggested that the magnet-guided bionic system with LIFU responsiveness had better targeting performance and higher specificity.
After two hours, euthanasia was performed, the left carotid artery and major organs of the rats were removed, and the fluorescence intensity signal of the left carotid artery for the group treated with TNPs was found to be significantly weaker than that for the group treated with MTNPs + magnet + LIFU (Figure 9D and E). However, the fluorescence signals of the liver and spleen for the TNPs group were significantly stronger than those for the group treated with MTNPs + magnet + LIFU. This indicates that more NPs were removed from the liver and spleen for the group treated with TNPs. However, there was no obvious fluorescence signal of the kidney in the group treated with TNPs, which was slightly different from the results of MR imaging. This may be because the imaging medium of MR is superparamagnetic Fe3O4, which can yield sensitive imaging results even at a low concentration (Figure 8A). However, DiR-labeled NPs are less sensitive to fluorescence imaging, and relatively high concentrations of NPs can be used for fluorescence imaging (Figure 9A). This may indicate that MTNPs have suitably prolonged systemic circulation time, creating more opportunities for the bionic system to target the thrombus site. In addition, these results indicated that the NPs are excreted mainly through the liver and spleen in vivo. This is consistent with other literature results.51,52
In conclusion, the bionic system has a remarkable thrombus-targeting effect in SD rats, but it may face many difficulties in clinical application. For larger superficial vessels, magnetic field and ultrasound conditions may be relatively easy to achieve; for smaller deeper vessels where ultrasound cannot easily penetrate, such as coronary arteries, it presents a real challenge. However, it has been reported that the implantation of diluted microferromagnetic wires into blood vessels can provide simultaneous delivery of ferromagnetic NPs under the action of an external magnetic field.53 Clinical intravascular ultrasound is a method used in coronary arteries. It is believed that with the development of molecular imaging technology, it will be possible to implant ultrasonic probes into blood vessels with microferromagnetic wires in vitro, which can effectively contain the attenuation of LIFU. In addition, the external magnetic field can effectively enhance the aggregation of Fe3O4 nanoparticles at the thrombus site.
Biocompatibility Evaluation of the Bionic System in vivo
No differences were found with regard to complete blood count (WBC, RBC, HGB, and PLT) or biochemical detection (ALT, AST, CREA, and UA), and all the results were within the normal range (Figure 10A). These results indicate that TNPs and MTNPs have good biocompatibility and nontoxicity and can be used safely as drug delivery systems. Among the tissue sections, there was no cell integrity damage or necrosis, nor were there inflammatory reactions in the major organs (heart, liver, spleen, lung, and kidney) (Figure 10B), indicating the superior biosafety of TNPs and MTNPs within 24 h of circulation in vivo.
 |
Figure 10 In vivo biocompatibility evaluation. (A) In vivo complete blood count and biochemical detection 3 h after injection of the indicated treatment groups. (B) H&E staining of major organs (heart, liver, spleen, lung and kidney) 24 h after injection of the indicated treatments (scale bar: 100 μm). |
Conclusion
In this study, we successfully constructed a bionic system and demonstrated the targeting effect of this system in vitro and in vivo. The bionic system guided by magnetic navigation with LIFU responsiveness showed better targeting ability than TNPs alone. The combination of active (macrophage chemotaxis to platelets) and passive (magnetic navigation) targeting enhanced bionic system aggregation around the thrombus, and the macrophages could evade capture by RES in vivo. NPs were released from the macrophages by LIFU irradiation and continually deposited at the thrombus site under the targeting of the CREKA peptide to fibrin; thus, the overall thrombus-targeting capability of the system was significantly enhanced. Superior biocompatibility and low toxicity were demonstrated and showed that this bionic system can be used as a drug carrier for targeting thrombi or other diseases and provides a new paradigm for thrombolytic therapy. Although our experiment lacks clinical proof of efficacy, given the development of molecular imaging and the reliability of our experimental data, many problems will be solved, and this system might be used clinically in the near future. Currently, the bionic system has not yet been used to deliver drugs, and it is not clear whether it will affect drug activity. Next, we will load thrombolytic drugs into the system to explore the thrombolytic effect.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Grant Nos. 81971608, 81901807, 81701650, and 82102063), the Kuanren Talents Program of the Second Affiliated Hospital of Chongqing Medical University (CQYC2020030389, 2020-7), the Chongqing Natural Science Foundation (cstc2021jcyj-msxmX0040) and the Chongqing Talents Program (cstc2021ycjh-bgzxm0168). The authors are grateful to American Journal Experts (AJE) for their assistance with language editing.
Disclosure
We declare that we have no conflicts of interest.
References
1. Anderson JL, Halperin JL, Albert NM, et al. Management of patients with peripheral artery disease (compilation of 2005 and 2011 ACCF/AHA guideline recommendations): a report of the American college of cardiology foundation/American heart association task force on practice guidelines. Circulation. 2013;127(13):1425–1443. doi:10.1161/CIR.0b013e31828b82aa
2. Refaat A, Del Rosal B, Palasubramaniam J, et al. Near-infrared light-responsive liposomes for protein delivery: towards bleeding-free photothermally-assisted thrombolysis. J Control Release. 2021;337:212–223. doi:10.1016/j.jconrel.2021.07.024
3. Sun M, Miyazawa K, Pendekanti T, et al. Combination targeting of ‘platelets + fibrin’ enhances clot Anchorage efficiency of nanoparticles for vascular drug delivery. Nanoscale. 2020;12(41):21255–21270. doi:10.1039/d0nr03633a
4. Chung TW, Wang SS, Tsai WJ. Accelerating thrombolysis with chitosan-coated plasminogen activators encapsulated in poly-(lactide-co-glycolide) (PLGA) nanoparticles. Biomaterials. 2008;29(2):228–237. doi:10.1016/j.biomaterials.2007.09.027
5. Lu TY, Chiang CY, Fan YJ, et al. Dual-targeting glycol chitosan/heparin-decorated polypyrrole nanoparticle for augmented photothermal thrombolytic therapy. ACS Appl Mater Interfaces. 2021;13(8):10287–10300. doi:10.1021/acsami.0c20940
6. Wang Y, Xu X, Zhao X, Yin Z. Functionalized polymeric hybrid micelles as an efficient nanotheranostic agent for thrombus imaging and thrombolysis. Acta Biomater. 2021;122:278–290. doi:10.1016/j.actbio.2020.10.015
7. de Saint Victor M, Crake C, Coussios CC, Stride E. Properties, characteristics and applications of microbubbles for sonothrombolysis. Expert Opin Drug Deliv. 2014;11(2):187–209. doi:10.1517/17425247.2014.868434
8. Wang S, Guo X, Xiu W, et al. Accelerating thrombolysis using a precision and clot-penetrating drug delivery strategy by nanoparticle-shelled microbubbles. Sci Adv. 2020;6(31):eaaz8204. doi:10.1126/sciadv.aaz8204
9. Doelare SAN, Jean Pierre DM, Nederhoed JH, et al. Microbubbles and ultrasound accelerated thrombolysis for peripheral arterial occlusions: the outcomes of a single arm phase II trial. Eur J Vasc Endovasc Surg. 2021;62(3):463–468. doi:10.1016/j.ejvs.2021.05.030
10. Chen JP, Yang PC, Ma YH, et al. Characterization of chitosan magnetic nanoparticles for in situ delivery of tissue plasminogen activator. Carbohydr Polym. 2011;84(1):364–372. doi:10.1016/j.carbpol.2010.11.052
11. Moghanizadeh A, Ashrafizadeh F, Varshosaz J, Ferreira A. Study the effect of static magnetic field intensity on drug delivery by magnetic nanoparticles. Sci Rep. 2021;11(1):18056. doi:10.1038/s41598-021-97499-7
12. Ouyang H, Zheng Z, Chen Y, et al. A magnetically modified black phosphorus nanosheet-based heparin delivery platform for preventing DVT accurately. J Mater Chem B. 2019;7(40):6099–6108. doi:10.1039/c9tb01459d
13. Zhang Y, Zhou J, Guo D, Ao M, Zheng Y, Wang Z. Preparation and characterization of gadolinium-loaded PLGA particles surface modified with RGDS for the detection of thrombus. Int J Nanomedicine. 2013;8:3745–3756. doi:10.2147/IJN.S49835
14. Zhou J, Guo D, Zhang Y, Wu W, Ran H, Wang Z. Construction and evaluation of Fe3O4-based PLGA nanoparticles carrying rtPA used in the detection of thrombosis and in targeted thrombolysis. ACS Appl Mater Interfaces. 2014;6(8):5566–5576. doi:10.1021/am406008k
15. Xu J, Zhou J, Zhong Y, et al. Phase transition nanoparticles as multimodality contrast agents for the detection of thrombi and for targeting thrombolysis: in vitro and in vivo experiments. ACS Appl Mater Interfaces. 2017;9(49):42525–42535. doi:10.1021/acsami.7b12689
16. Zhong Y, Zhang Y, Xu J, et al. Low-intensity focused ultrasound-responsive phase-transitional nanoparticles for thrombolysis without vascular damage: a synergistic nonpharmaceutical strategy. ACS Nano. 2019;13(3):3387–3403. doi:10.1021/acsnano.8b09277
17. Ta HT, Li Z, Hagemeyer CE, et al. Molecular imaging of activated platelets via antibody-targeted ultra-small iron oxide nanoparticles displaying unique dual MRI contrast. Biomaterials. 2017;134:31–42. doi:10.1016/j.biomaterials.2017.04.037
18. Ta HT, Prabhu S, Leitner E, et al. Enzymatic single-chain antibody tagging: a universal approach to targeted molecular imaging and cell homing in cardiovascular disease. Circ Res. 2011;109(4):365–373. doi:10.1161/CIRCRESAHA.111.249375
19. Zhang Y, Zhong Y, Ye M, et al. Polydopamine-modified dual-ligand nanoparticles as highly effective and targeted magnetic resonance/photoacoustic dual-modality thrombus imaging agents. Int J Nanomedicine. 2019;14:7155–7171. doi:10.2147/IJN.S216603
20. Korin N, Kanapathipillai M, Matthews BD, et al. Shear-activated nanotherapeutics for drug targeting to obstructed blood vessels. Science. 2012;337(6095):738–742. doi:10.1126/science.1217815
21. Zhang W, Wang M, Tang W, et al. Nanoparticle-laden macrophages for tumor-tropic drug delivery. Adv Mater. 2018;30(50):e1805557. doi:10.1002/adma.201805557
22. Wang Q, Cheng H, Peng H, Zhou H, Li PY, Langer R. Non-genetic engineering of cells for drug delivery and cell-based therapy. Adv Drug Deliv Rev. 2015;91:125–140. doi:10.1016/j.addr.2014.12.003
23. Gao WJ, Liu JX, Liu MN, et al. Macrophage 3D migration: a potential therapeutic target for inflammation and deleterious progression in diseases. Pharmacol Res. 2021;167:105563. doi:10.1016/j.phrs.2021.105563
24. Yao Q, Yang G, Wang H, et al. Aging erythrocyte membranes as biomimetic nanometer carriers of liver-targeting chromium poisoning treatment. Drug Deliv. 2021;28(1):1455–1465. doi:10.1080/10717544.2021.1949075
25. Ding N, Dou C, Wang Y, et al. Antishear stress bionic carbon nanotube mesh coating with intracellular controlled drug delivery constructing small-diameter tissue-engineered vascular grafts. Adv Healthc Mater. 2018;7(11):e1800026. doi:10.1002/adhm.201800026
26. Albalawi AE, Khalaf AK, Alyousif MS, et al. Fe3O4@piroctone olamine magnetic nanoparticles: synthesize and therapeutic potential in cutaneous leishmaniasis. Biomed Pharmacother. 2021;139:111566. doi:10.1016/j.biopha.2021.111566
27. Li Q, Liu X, Chang M, Lu Z. Thrombolysis enhancing by magnetic manipulation of fe3o4 nanoparticles. Materials. 2018;11(11):2313. doi:10.3390/ma11112313
28. Heid S, Unterweger H, Tietze R, et al. Synthesis and characterization of tissue plasminogen activator-functionalized superparamagnetic iron oxide nanoparticles for targeted fibrin clot dissolution. Int J Mol Sci. 2017;18(9):1837. doi:10.3390/ijms18091837
29. Chen JP, Yang PC, Ma YH, Tu SJ, Lu YJ. Targeted delivery of tissue plasminogen activator by binding to silica-coated magnetic nanoparticle. Int J Nanomedicine. 2012;7:5137–5149. doi:10.2147/IJN.S36197
30. Ma YH, Wu SY, Wu T, Chang YJ, Hua MY, Chen JP. Magnetically targeted thrombolysis with recombinant tissue plasminogen activator bound to polyacrylic acid-coated nanoparticles. Biomaterials. 2009;30(19):3343–3351. doi:10.1016/j.biomaterials.2009.02.034
31. Zan P, Yang C, Sun H, Zhao L, Lv Z, He Y. One-pot fabricating Fe3O4/graphene nanocomposite with excellent biocompatibility and non-toxicity as a negative MR contrast agent. Colloids Surf B Biointerfaces. 2016;145:208–216. doi:10.1016/j.colsurfb.2016.04.049
32. Burtea C, Laurent S, Vander Elst L, Muller RN. Contrast agents: magnetic resonance. Handb Exp Pharmacol. 2008;(185 Pt 1):135–165. doi:10.1007/978-3-540-72718-7_7
33. Bowary P, Greenberg BD. Noninvasive focused ultrasound for neuromodulation: a review. Psychiatr Clin North Am. 2018;41(3):505–514. doi:10.1016/j.psc.2018.04.010
34. Wang S, Li B, Li X, et al. Low-intensity ultrasound modulation may prevent myocardial infarction-induced sympathetic neural activation and ventricular arrhythmia. J Cardiovasc Pharmacol. 2020;75(5):432–438. doi:10.1097/FJC.0000000000000810
35. Bai S, Liao J, Zhang B, et al. Multimodal and multifunctional nanoparticles with platelet targeting ability and phase transition efficiency for the molecular imaging and thrombolysis of coronary microthrombi. Biomater Sci. 2020;8(18):5047–5060. doi:10.1039/d0bm00818d
36. Song F, Gao H, Li D, et al. Low intensity focused ultrasound responsive microcapsules for non-ablative ultrafast intracellular release of small molecules. J Mater Chem B. 2021;9(10):2384–2393. doi:10.1039/d0tb02788j
37. Ikehara Y, Niwa T, Biao L, et al. A carbohydrate recognition-based drug delivery and controlled release system using intraperitoneal macrophages as a cellular vehicle. Cancer Res. 2006;66(17):8740–8748. doi:10.1158/0008-5472.CAN-06-0470
38. Li X, Bottini M, Zhang L, et al. Core-satellite nanomedicines for in vivo real-time monitoring of enzyme-activatable drug release by fluorescence and photoacoustic dual-modal imaging. ACS Nano. 2019;13(1):176–186. doi:10.1021/acsnano.8b05136
39. Xu J, Zhou J, Zhong Y, et al. EWVDV-mediated platelet-targeting nanoparticles for the multimodal imaging of thrombi at different blood flow velocities. Int J Nanomedicine. 2020;15:1759–1770. doi:10.2147/IJN.S233968
40. Hathcock JJ. Flow effects on coagulation and thrombosis. Arterioscler Thromb Vasc Biol. 2006;26(8):1729–1737. doi:10.1161/01.ATV.0000229658.76797.30
41. Clifford PS. Local control of blood flow. Adv Physiol Educ. 2011;35(1):5–15. doi:10.1152/advan.00074.2010
42. Sahoo N, Sahoo RK, Biswas N, Guha A, Kuotsu K. Recent advancement of gelatin nanoparticles in drug and vaccine delivery. Int J Biol Macromol. 2015;81:317–331. doi:10.1016/j.ijbiomac.2015.08.006
43. Echave MC, Saenz Del Burgo L, Pedraz JL, Orive G. Gelatin as biomaterial for tissue engineering. Curr Pharm Des. 2017;23(24):3567–3584. doi:10.2174/0929867324666170511123101
44. Uesugi Y, Kawata H, Saito Y, Tabata Y. Ultrasound-responsive thrombus treatment with zinc-stabilized gelatin nano-complexes of tissue-type plasminogen activator. J Drug Target. 2012;20(3):224–234. doi:10.3109/1061186X.2011.633259
45. Kawata H, Uesugi Y, Soeda T, et al. A new drug delivery system for intravenous coronary thrombolysis with thrombus targeting and stealth activity recoverable by ultrasound. J Am Coll Cardiol. 2012;60(24):2550–2557. doi:10.1016/j.jacc.2012.08.1008
46. Ye M, Zhou J, Zhong Y, et al. SR-A-targeted phase-transition nanoparticles for the detection and treatment of atherosclerotic vulnerable plaques. ACS Appl Mater Interfaces. 2019;11(10):9702–9715. doi:10.1021/acsami.8b18190
47. Yi BG, Park OK, Jeong MS, et al. In vitro photodynamic effects of scavenger receptor targeted-photoactivatable nanoagents on activated macrophages. Int J Biol Macromol. 2017;97:181–189. doi:10.1016/j.ijbiomac.2017.01.037
48. Roszkowski W, Szmigielski S, Janiak M, Wrembel JK, Roszkowski K, Hryniewicz W. Effect of hyperthermia on rabbit macrophages. Immunobiology. 1980;157(2):122–131. doi:10.1016/S0171-2985(80)80094-5
49. Jiang N, Hu B, Cao S, et al. Stable low-dose oxygen release using h2o2/perfluoropentane phase-change nanoparticles with low-intensity focused ultrasound for coronary thrombolysis. Ultrasound Med Biol. 2020;46(10):2765–2774. doi:10.1016/j.ultrasmedbio.2020.06.004
50. Pyrillou K, Burzynski LC, Clarke MCH. Alternative pathways of IL-1 activation, and its role in health and disease. Front Immunol. 2020;11:613170. doi:10.3389/fimmu.2020.613170
51. Sah H, Thoma LA, Desu HR, Sah E, Wood GC. Concepts and practices used to develop functional PLGA-based nanoparticulate systems. Int J Nanomedicine. 2013;8:747–765. doi:10.2147/IJN.S40579
52. Rezvantalab S, Drude NI, Moraveji MK, et al. PLGA-based nanoparticles in cancer treatment. Front Pharmacol. 2018;9:1260. doi:10.3389/fphar.2018.01260
53. Hournkumnuard K, Natenapit M. Magnetic drug targeting by ferromagnetic microwires implanted within blood vessels. Med Phys. 2013;40(6):062302. doi:10.1118/1.4805097
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.