")
Back to Journals » OncoTargets and Therapy » Volume 12
M2 macrophages confer resistance to 5-fluorouracil in colorectal cancer through the activation of CCL22/PI3K/AKT signaling
Authors Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, Xiong B
Received 13 December 2018
Accepted for publication 12 March 2019
Published 18 April 2019 Volume 2019:12 Pages 3051—3063
DOI https://doi.org/10.2147/OTT.S198126
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Chen Wei,1–4* Chaogang Yang,1–4* Shuyi Wang,1–4 Dongdong Shi,1–4 Chunxiao Zhang,1–4 Xiaobin Lin,1–4 Bin Xiong1–4
1Department of Gastrointestinal Surgery, Zhongnan Hospital of Wuhan University, Wuhan 430071, People’s Republic of China; 2Department of Gastric and Colorectal Surgical Oncology, Zhongnan Hospital of Wuhan University, Wuhan 430071, People’s Republic of China; 3Hubei Key Laboratory of Tumor Biological Behaviors, Wuhan 430071, People’s Republic of China; 4Hubei Cancer Clinical Study Center, Wuhan 430071, People’s Republic of China
*These authors contributed equally to this work
Background: M2 macrophages are crucial components of tumor microenvironment that frequently associated with the resistance of therapeutic treatments in human cancers, but their role in the chemosensitivity of colorectal cancer (CRC) to 5-fluorouracil (5-FU) is still obscure.
Methods: In our study, we clarified the biological functions of M2 macrophages and their mechanism on the chemosensitivity of CRC cells to 5-FU. Then, we analyzed the correlation between CCL22 and CD68+, and CD163+, tumor-associated macrophages (TAMs), and further elucidated the prognostic value of CCL22 and CD163+, M2 macrophages in clinical CRC samples.
Results: M2 macrophages decreased the inhibitory effect of 5-FU on CRC cells migration and invasion by secreting CCL22, and declined the apoptosis induced by 5-FU. Treated with a neutralizing anti-CCL22 antibody destroyed these effects. We further illuminated that M2 macrophages regulated 5-FU resistance of CRC cells through epithelial-mesenchymal transition (EMT) program, PI3K/AKT pathway, and caspase-mediated apoptosis. Clinically, CCL22 was found to have elevated expression in CRC tissue samples, and was positively associated with CD163+, TAMs. Furthermore, the patients with higher CD163+, M2 macrophages and higher expression of CCL22 in CRC tissues had a lower overall survival (OS) rate compared with lower ones.
Conclusion: Our findings indicate that M2 macrophage regulated 5-FU-mediated CRC chemoresistance via the EMT program, PI3K/AKT pathway, and caspase-mediated apoptosis by releasing CCL22.
Keywords: M2 macrophages, colorectal cancer, 5-fluorouracil, chemotherapy resistance, CCL22
Introduction
Colorectal cancer (CRC) is one of the most prevalent malignancies and the second leading cause of cancer-related deaths worldwide.1 This highly invasive disease is characterized by hepatic or pulmonary metastasis, and poor prognosis.2 The standard treatment for CRC is based on 5-fluorouracil (5-FU) regimen (oxaliplatin, irinotecan, and cetuximab).3 Although adjuvant chemotherapy before and after operation has been proved to increase patients’ survival, however, 5-FU resistance is still the most important problem to affect the efficiency of chemotherapy in CRC. So far, the specific mechanism of 5-FU resistance has not yet been elucidated. Therefore, getting a better understanding of the molecular mechanisms to 5-FU resistance is rather critical for improving the prognosis of CRC patients.
Tumor microenvironment consists of a variety of tumor cells and stromal cells like endothelial cells, mesenchymal stem cells, as well as tumor-associated macrophages (TAMs), which provides support for tumor progression.4 As the most important components of the tumor microenvironment, TAMs has been shown to play a pivotal role in tumor progression and chemoresistance.5–7 Macrophages are plastic cells that undergo different functional reprogramming depending on various environmental cues.8 Generally, TAMs can be divided into two distinct polarized types: the classically activated (M1 macrophages) and the alternative activated (M2 macrophages) phenotypes.9 Rather than acting anti-tumor effect, M2 macrophages express high level of hemoglobin scavenger receptor (CD163) and anti-inflammatory cytokines (IL-10) to favor tumor cell progression.10 The M2 macrophages promote multiple tumors progression by enhancing proliferation, invasion, metastasis, angiogenesis, and immunosuppression.11,12 There is increasing evidence that M2 macrophages can mediate tumor chemoresistance, and immunotherapy against M2 macrophages might be a novel combination for cancer treatment.13 However, the detailed interaction and molecular mechanisms between chemoresistance and M2 macrophages remain unclear.
Based on the above research status, we speculated that cytokines or chemokines released from M2 macrophages might affect the efficiency of 5-FU treatment on CRC cells. In the present study, we investigated the mechanisms of M2 macrophages in the development of resistance to 5-FU chemotherapy, and the results revealed a significant role of CCL22 derived from M2 macrophages in these processes.
Materials and methods
Patients and tissue samples
Primary CRC tissue samples were obtained from 68 patients who underwent curative resection at Zhongnan Hospital of Wuhan University (Wuhan, China). All included patients were identified as adenocarcinoma of colorectal by histopathology and had available survival data. Moreover, all patients were devoid of neoadjuvant chemotherapy or radiotherapy before surgical resection and did not be diagnosed with autoimmune diseases. Formalin-fixed, paraffin-embedded (FFPE) cancer tissue specimens were obtained from these patients after surgery. This study was conducted in accordance with the Declaration of Helsinki, and all related procedures were approved by the ethics committee of Zhongnan Hospital of Wuhan University. All included patients provided written informed consent.
Immunohistochemistry
To examine the level of heterogeneous macrophages and CCL22 in CRC tissue, paraffin-embedded cancer samples were serially sectioned at 4 μm thickness. Antigen retrieval was performed by a pressure cooker for 30 mins in 0.01 M citrate buffer (pH 6.0), followed by treatment with 3% hydrogen peroxide for 5 mins. Specimens were incubated with monoclonal antibodies against human CD68 (1:500; Abcam, USA), CD163 (1:50; Abcam, USA), and CCL22 (1:200; CST, USA) overnight at 4 degree, followed by an incubation with secondary antibody (HRP-labeled anti-mouse antibody, DAKO). For negative control, isotype-matched antibodies were applied. After washing with PBS, the sections were visualized using diaminobenzidine (DAB) system and hematoxylin re-dying, observed and analyzed with microscope. Three high-magnification fields were chosen randomly under optical microscope and more than 3×100 cells were analyzed. Sections were scored and grouped with positive staining rate and intensity. The positive staining rate was defined according to the proportion of positively stained cancer cells: “negative” was 0, “1–25%” for 1, “26–50%” for 2, “51–75%” for 3, “76–100%” for 4. The score of staining intensity: “negative” was 0, “1+” for 1, “2+” for 2, “3+” for 3. Patients were divided into two groups according to the scores “positive staining rate score” × “staining intensity score”. Equal or less than 3 were classified into low expression, more than 3 were classified into high expression group.
Cell culture
The human monocyte cell line THP-1 and CRC cell lines, DLD1 and HT29 cells were purchased from the Chinese of Sciences in Shanghai. Cells were cultured in RPMI 1640 medium (Gibco) with 10% fetal bovine serum (FBS) (Gibco) at 37°C in a humidified atmosphere with 5% CO2. Macrophages and CRC cell lines co-cultivation were conducted using the non-contact co-culture transwell system (Corning). For cell growth, 3×105 THP-1 cells were seeded in 0.4 μm sized pore inserts and polarized into M2 macrophages. Inserts containing M2 polarized THP-1 macrophages were transferred to 6-well plate seeded with DLD1 or HT29 cells (1×105 cells per well) in advance and co-cultured. After 48 hrs of co-culture, CRC cells were harvested for further analyses.
Macrophage generation and differentiation
The protocols of M2 macrophages polarization from THP-1 were performed according to previously established method. Briefly, to obtain M2 phenotype, THP-1 cells were first treated with 100 nM PMA (Sigma-Aldrich) for 24 hrs, followed by cultured by the addition of IL-4 (Invitrogen) and IL-13 (R&D) (20 ng/ml) for another 24 hrs.
RNA isolation and quantitative real-time PCR (RT-PCR)
The total RNA was isolated using the Trizol Reagent (Invitrogen) according to the manufacturer’s instructions. After detection of RNA concentration, 1 μg of total RNA was reversely transcribed with random primers, using the PrimeScript™ RT reagent kit (Toyobo, Osaka). RT-PCR was performed using the SYBR-Green PCR Master Mix (Takara). Relative expression was calculated using the 2−ΔΔCt method.
Quantification of chemokine by enzyme-linked immunosorbent assay (ELISA)
The concentrations of chemokine were estimated for each experimental condition by ELISA, using commercial kits purchased from R&D Systems (Minneapolis, USA), according to the manufacturer’s instructions.
Cell counting kit-8 (CCK-8) assay
CCK-8 assays were performed to determine the viability of cells. Briefly, CRC cells, after corresponding treatment in the study, were seeded into 96 well plates, cultured overnight. Subsequently, 10 μL CCK-8 solution was added into 100 μL culture medium in each well and the mixture was incubated according to the manufacturer’s instructions.
Colony formation
For colony formation detection, 500 cells were planted in 6-well plates and cultured for 2 weeks. Cells were then fixed with 4% paraformaldehyde and stained with 0.5% crystal violet. The assay was performed three times for each treatment.
Wound healing assay
A wound-healing assay was applied to assess the migration ability of CRC cells following co-cultured with M2 macrophages. Cells were grown to 90–100% confluence in 6-well plates, and a wound was scratched by a plastic pipette tip. In order to remove cellular debris, the remaining cells of the 6-well plates were washed away in PBS and incubated with serum-free medium at 37°C. After 24 hrs, migrating cells at the wound front were photographed. The area of the wound was measured with Image J software.
Invasion assay
Cell invasion assays were performed using 24-well Transwells (8 µm pore size; Corning) pre-coated with Matrigel (Falcon 354480; BD Biosciences). In total, 1×105 cells were suspended in 500 µl RPMI 1640 containing 1% FBS and added to the upper chamber, while 750 µl RPMI 1640 containing 10% FBS was placed in the lower chamber. After 48 hrs of incubation, Matrigel and the cells remaining in the upper chamber were removed using cotton swabs. Cells on the lower surface of the membrane were fixed in 4% paraformaldehyde and stained with 0.5% crystal violet. Cells in 5 microscopic fields (at ×200 magnification) were counted and photographed. All experiments were performed in triplicate.
Western blot
Cells were lysed using a RIPA buffer, including a protease inhibitor cocktail (Thermo Scientific, USA). The proteins were separated by SDS-PAGE gels and transferred to PVDF membranes (Millipore, USA). After blocking with 5% non-fat milk, the membranes were incubated with primary antibodies at 4°C overnight. The membranes were washed three times and incubated for 2 hrs at room temperature with HRP-conjugated secondary antibodies. The antibodies used were the anti-E-cadherin (1:1,000, Cell Signaling), the anti-Vimentin (1:1,000, Cell Signaling), the anti-snail (1:1,000, Cell Signaling), the anti-PI3K p85 (1:1,000, Abacm), the anti-phospho-PI3K p85 (1:1,000, Cell Signaling), the anti-cleaved caspase-3 (1:1,000, Cell Signaling), the anti-cleaved caspase-8 (1:1,000, Cell Signaling), the anti-PARP (1:1,000, Abacm), the anti-total-AKT (1:1,000, Abacm), the anti-phospho-AKT (1:1,000, Cell Signaling), and anti-β-actin antibody (1:5,000, Santa cruz). Proteins were detected using a Bio-Rad ChemiDoc XRS+ System. Bio-Rad Image Lab software was used for densitometric analysis.
Cell apoptosis assay
Apoptosis cells were stained using Annexin V-FITC apoptosis detection kit (Caltag laboratories, USA). The cells were incubated with 5 mL of Annexin V and 5 mL of PI for 15 min at room temperature as recommended by the manufacturer, and the cells were analyzed with the FACS flow cytometer.
Statistical analysis
Patient characteristics were shown as count and percentages for categorical data, and median with ranges (minimum, maximum) or mean ± standard deviation (mean ± SD) for continuous data. Comparisons among groups were analyzed using Kruskal–Wallis tests with pair-wise comparisons or Mann–Whitney U test if the data were not normally distributed. The Pearson χ2 test or Fisher’s exact test was used to compare qualitative variables. Correlations were analyzed by Spearman correlation. Kaplan–Meier method was used for survival analysis and drawing the survival curves, and difference among patients’ subgroups was calculated by log-rank test. All statistical analyses were performed with SPSS statistical software (version 21.0, IBM SPSS) and GraphPad Prism software (version 6.0, GraphPad Software) for Windows. Two-sided P<0.05 was considered as statistically significant.
Results
M2 macrophages induce the resistance of CRC cells to 5-FU
Previous research revealed that M2 macrophages are mainly TAMs subtypes infiltrated in CRC microenvironment. We first generated M2 macrophages from human monocyte cell line THP-1 treated with IL-4 and IL-13. The M2 macrophages were validated on the basis of M2 macrophage surface antigens, including CD206, CD163, arginase-1 (Figure 1A). In addition, M2 macrophages related-gene expression was detected using RT-PCR (Figure 1B). The expression of classic M2 markers (CD163, CD206, IL-10, TGF-β, CD204, Arg1) was increased compared with unactivated macrophages (PMA-treated THP-1), indicating that we successfully generated M2 macrophages from human monocyte cell line THP-1.
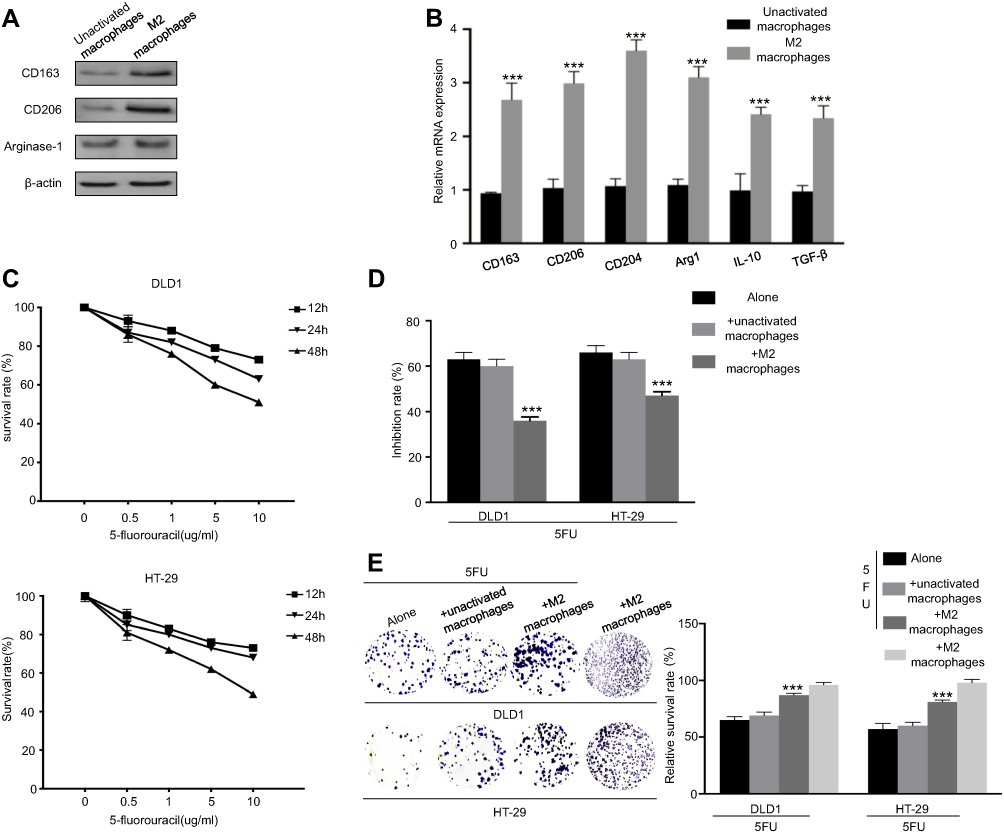 | Figure 1 M2 macrophages induce the resistance of CRC cells to 5-FU. (A) Western blot analysis was used to detect M2 marker CD163, CD206, and arginase-1 in M2 macrophages or unactivated macrophages. (B) Macrophages treated with IL13 and IL4 were generated to M2 macrophages. After 48 hrs, RT-PCR was applied using primers for M2 markers (CD206, CD163, CD204, IL10, arginase-1, and TGF-β). (C) Colorectal cancer cells were treated with different concentrations of 5-FU (0 µg/ml, 0.5 µg/ml, 1 µg/ml, 5 µg/ml, 10 µg/ml) for different time points (12 hrs, 24 hrs, 48 hrs) for the CCK8 assay. (D) Colorectal cancer cells were cocultured with M2 macrophages or unactivated macrophages. Then, cells were treated with 10 µg/ml 5-FU for 48 hrs. CCK-8 was used to analyze inhibition rate. (E) Colorectal cancer cells were cultured alone, cocultured with M2 macrophages or unactivated macrophages. Then, cells were treated with or without 10 µg/ml 5-FU for 48 hrs. Clone formation was used to analyze the survival rate. ***P<0.001. n=3 independent experiments. Error bars, SEM. |
In order to clarify the optimal dose for 5-FU applications, the CRC cell lines DLD1 and HT-29 were treated with various concentrations of 5-FU for different time points (12, 24, and 48 hrs). CCK8 assay showed the cell viability decreased with increasing concentration and continued time (Figure 1C). In the subsequent drug resistance experiments, we applied the following dosage of 5-FU: DLD1 (10 µg/ml, 48 hrs), HT-29 (10 µg/ml, 48 hrs). Then, we co-cultured M2 macrophages with CRC cells in a non-contact transwell system, which allowed the exchange of soluble factors, but prevented direct cell-cell contact. After co-culture, these CRC cells were treated with 5-FU for 48 hrs. Inhibition rate assays demonstrated that M2 macrophages enhanced DLD1 and HT-29 cells resistance to 5-FU (Figure 1D), together with an enhanced colony forming ability (Figure 1E). These results demonstrate that M2 macrophages mediated the resistance of CRC cells to 5-FU.
CCL22 secreted from M2 macrophages induces the resistance of CRC cells to 5-FU
Previous studies have demonstrated that chemokine secretion represents the major functional response of macrophages; it was speculated that a signaling mechanism between M2 macrophages and CRC cells exists that accounts for the previously described chemo-resistance.14,15 We then applied RT-PCR-based chemokine array analysis to globally identify inflammatory mediators, and found that the most abundant chemokine was CCL22 in M2 macrophages compared with control (Figure 2A). ELISA assay further showed that CCL22 levels were significantly increased in the media from M2 macrophages compared to those from PMA-treated THP-1 macrophages (Figure 2B). As previous study has identified CCL22 as an independent marker affecting the prognosis of gastric cancer patients with 5-FU-based adjuvant chemotherapy,12 therefore, we focused on CCL22 in the further studies. To demonstrate the role of CCL22 in the resistance of CRC cells to 5-FU, CRC cells were treated with an exogenous recombinant CCL22 for 48 hrs, and then treated with 5-FU for another 48 hrs. CCK8 and colony formation assay demonstrated that CCL22 significantly decreased the 5-FU sensitivity of CRC cells, and increased relative survival rate (Figure 2C and D). Furthermore, a CCL22 neutralizing antibody was used to confirm M2 macrophages induced the resistance to 5-FU in CRC cells through CCL22. After applying CCL22 neutralizing antibody in M2 macrophages co-culture medium, the CRC cells were treated with 5-FU for 48 hrs. The cell viability assays demonstrated that CCL22 neutralizing antibody decreased M2 macrophages-meditated CRC cells resistance to 5-FU (Figure 2C), together with a reduced colony formation (Figure 2D). These results demonstrate that M2 macrophages-derived CCL22 is one of the major chemokines that may mediate the interplay between M2 macrophages and CRC cells.
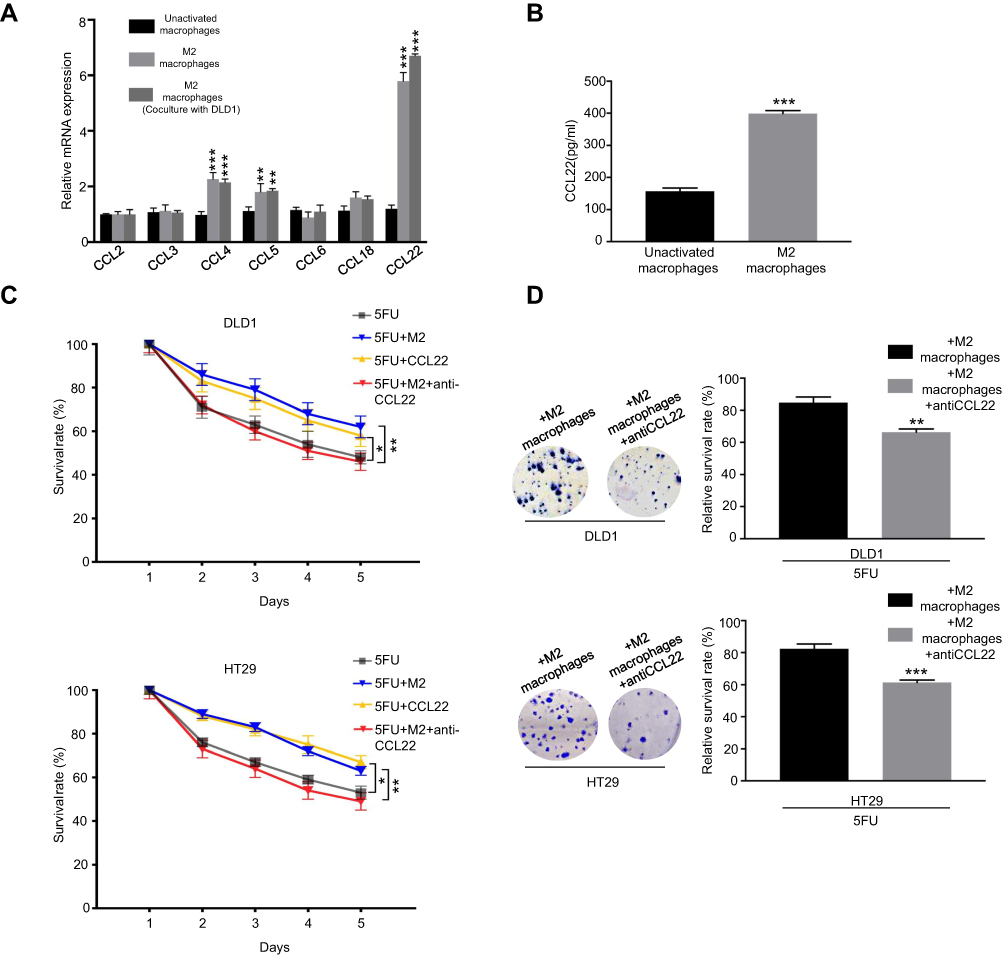 | Figure 2 CCL22 derived from M2 macrophages induce the resistance of CRC cells to 5-FU. (A) RT-PCR for analyzing the expression of chemokines in unactivated macrophages and M2 macrophages co-cultured with or without DLD1. (B) ELISA assay of CCL22 protein secretion from M2 macrophages or unactivated macrophages. (C) Colorectal cancer cells were cocultured with M2 macrophages with or without anti-CCL22 antibody. Then, cells were treated with 10 µg/ml 5-FU for 48 hrs. CCK-8 was used to analyze survival rate. (D) Colorectal cancer cells were cocultured with M2 macrophages with or without anti-CCL22 antibody. Then, cells were treated with 10 µg/ml 5-FU for 48 hrs. CCK-8 was used to analyze survival rate. *P<0.05; **P<0.01; ***P<0.001. n=3 independent experiments. Error bars, SEM. |
M2 macrophages inhibit apoptosis in CRC cells by affecting expression of apoptosis-associated proteins
Various genetic, functional, and biochemical researches revealed that CRC cells are resistant to 5-FU by utilizing intrinsic and acquired resistance to apoptosis that is a critical process of reprogrammed cell proliferation and death. 5-FU exerts anti-tumor function mainly to mediate apoptosis of tumor cells. Therefore, we supposed that M2 macrophages reduced the effect of 5-FU through the regulation of apoptosis signaling pathway. Annexin V/PI double staining showed that M2 macrophages reduced 5-FU-induced apoptosis compared to 5-FU treatment group (Figure 3A). CCL22 neutralizing antibody increased the number of apoptotic cells induced by 5-FU compared to M2 macrophages coculture to the contrary (Figure 3A). Considering the biochemical features of apoptosis including the activation of caspase, western blot was used to detect key pro-apoptotic proteins such as cleaved caspase-3, cleaved caspase-8, and cleaved PARP, which were the downstream of caspase-dependent apoptosis signaling pathway. Coculture of CRC cells with M2 macrophages protected CRC cells from 5-FU-induced apoptosis by inhibiting the activation of cleavage of caspase-3, cleaved caspase-8, and cleaved PARP, and a contrary effect was exhibited in applied CCL22 neutralizing antibody cells (Figure 3B).
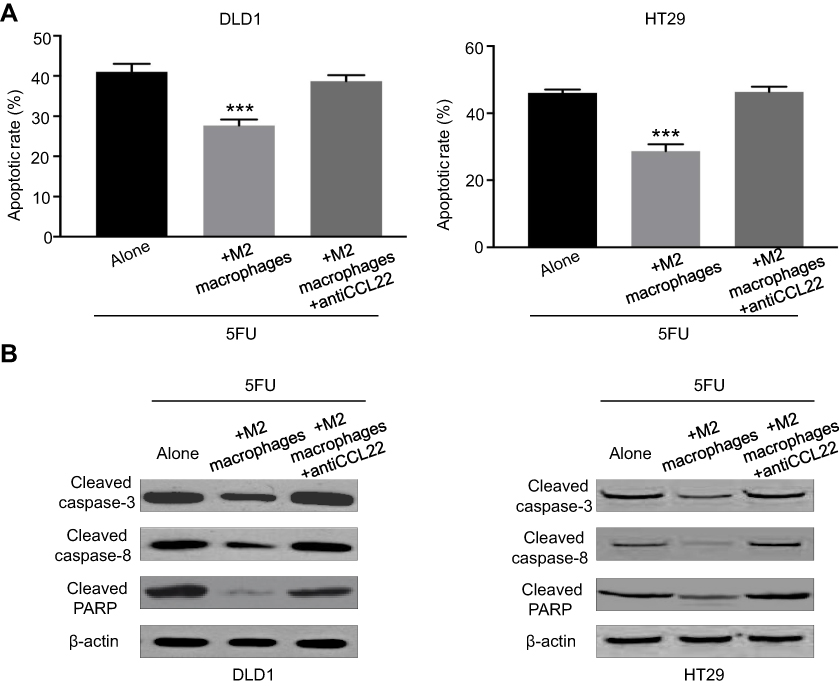 | Figure 3 M2 macrophages inhibit apoptosis in CRC cells by altering expression of apoptosis-associated proteins. (A) Colorectal cancer cells were cocultured with M2 macrophages with or without anti-CCL22 antibody. Then, cells were treated with 10 µg/ml 5-FU for 48 hrs. Flow cytometry was used to analyze cell apoptosis. (B) Colorectal cancer cells were cocultured with M2 macrophages with or without anti-CCL22 antibody. Then, cells were treated with 10 µg/ml 5-FU for 48 hrs. Western blots of apoptosis-associated proteins were analyzed. ***P<0.001. n=3 independent experiments. Error bars, SEM. |
M2 macrophages regulate the CCL22/PI3K/AKT signaling pathway to enhance the anti-apoptotic ability of CRC cells
The ATP-binding cassette transporters (ABC transporters) proteins play a key role in chemotherapy resistance for tumor therapy. Eight ABC transporter genes, including ABCG1, ABCC1, ABCG2, and so on, were applied for RT-PCR detection to determine whether M2 macrophages affected chemoresistance by regulation of ABC transporter genes expression. However, none of the genes was differently expressed in DLD1 cells co-cultured with M2 macrophages compared with DLD1 cells alone (Figure 4A), suggesting that M2 macrophages have no regulation on the expression of ABC transporter genes in CRC cells.
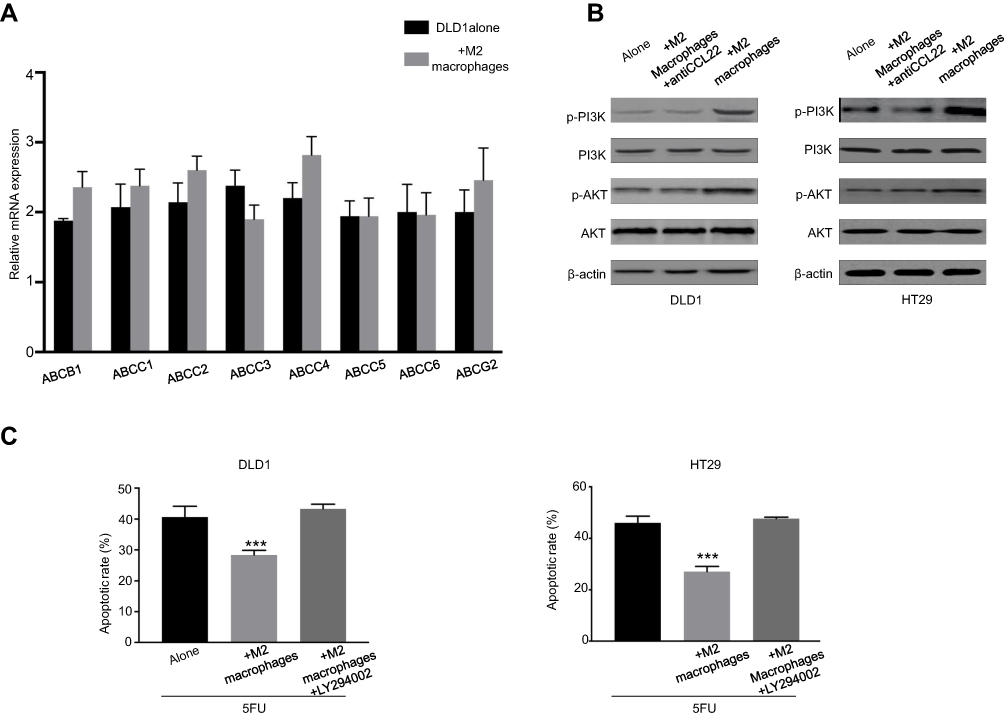 | Figure 4 CCL22 regulates the PI3K/AKT signaling pathway to enhance the anti-apoptotic ability of CRC cells. (A) RT-PCR for analyzing the expression of ABC transporter genes in DLD1 cells cocultured with or without M2 macrophages. (B) Colorectal cancer cells were cocultured with M2 macrophages with or without anti-CCL22 antibody. Western blots of PI3K/AKT pathway proteins were analyzed. (C) Colorectal cancer cells were cocultured with M2 macrophages with or without LY294002. Then, cells were treated with 10 µg/ml 5-FU for 48 hrs. Flow cytometry was used to analyze cell apoptosis. ***P<0.001. n=3 independent experiments. Error bars, SEM. |
Previous studies revealed that the PI3K/AKT pathway plays a critical role in regulation of cell proliferation, progression, apoptosis, angiogenesis, and chemoresistance.16,17 Therefore, we further investigated the potential role of PI3K/AKT pathway in M2 macrophages-derived CCL22-mediated 5-FU resistance in CRC cells. The results found that CRC cells co-cultured with M2 macrophages increased the expressions of p-PI3K and p-AKT, whereas treatment of CCL22 neutralizing antibody inhibited co-cultured-induced expressions of p-PI3K and p-AKT ( Figure 4B). Furthermore, treatment of LY294002, an AKT inhibitor, markedly blocked M2 macrophages-regulated sensitivity of CRC cells toward 5-FU and strikingly increased apoptosis (Figure 4C).
M2 macrophages regulate 5-FU-mediated inhibition of cell migration and invasion through epithelial-mesenchymal transition (EMT) in CRC cells
Tumor metastasis is commonly responsible for CRC-related mortality, and potential functions of anticancer drugs such as 5-FU include inhibition of tumor invasion and metastasis. To investigate whether M2 macrophages coculture contributes to 5-FU resistance by mediating EMT to enhance cell migration and invasion, wound healing assay and transwell invasion assay were performed. As shown in Figure 5A, M2 macrophages coculture reversed the inhibition of migration induced by 5-FU, which was also confirmed by transwell assay (Figure 5B). As EMT is the basis of tumor metastasis, we then detected the expression level of EMT-related proteins (vimentin, snail, and E-cadherin) in CRC cells treated with different conditions. The results showed that 5-FU significantly increased the expression of E-cadherin and reduced the expression of snail and vimentin after applying 5-FU in CRC cells culture medium, and these effects could be reversed by M2 macrophages (Figure 5C). Collectively, these results indicate that M2 macrophages induce EMT to offset 5-FU-mediated inhibition of cell migration and invasion in CRC cells.
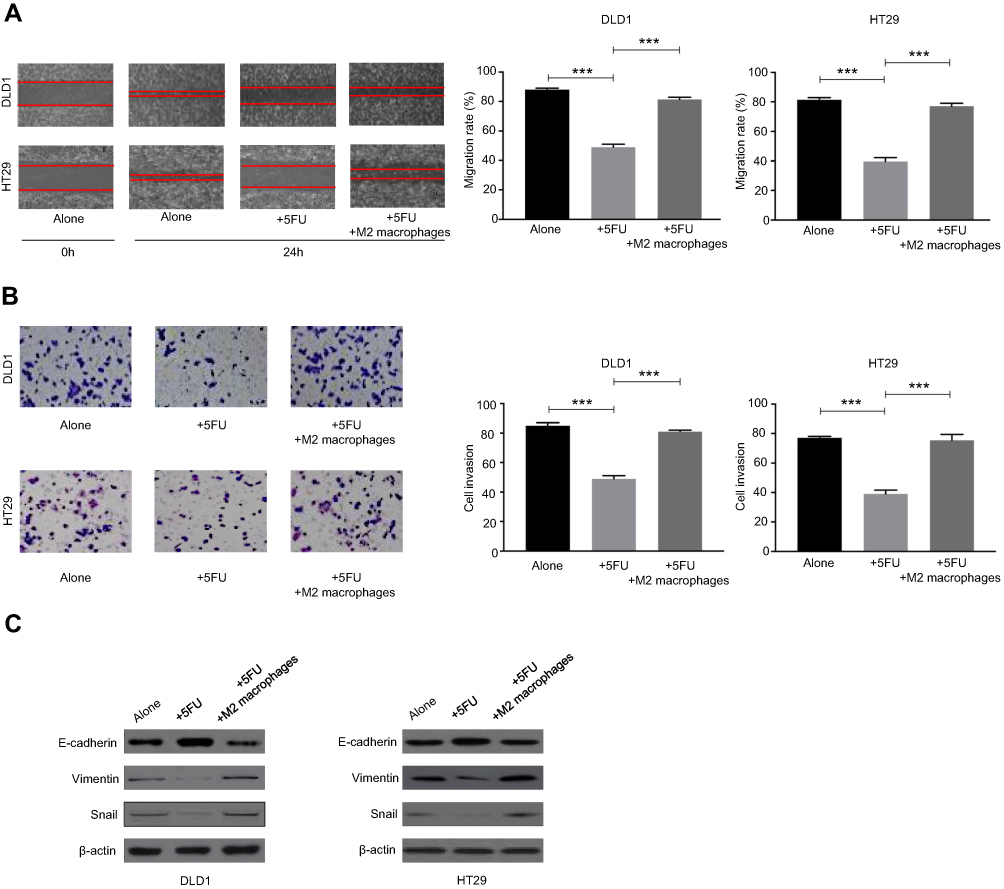 | Figure 5 M2 macrophages regulated 5-FU-mediated inhibition of cell migration and invasion through epithelial-mesenchymal transition (EMT) in CRC cells. (A) Colorectal cancer cells were cocultured with or without M2 macrophages and then treated with or without 5-FU. Cell migration was analyzed by a wound-healing assay. (B) Colorectal cancer cells were cocultured with or without M2 macrophages and then treated with or without 5-FU. Cell invasion was analyzed by transwell assay. (C) Colorectal cancer cells were cocultured with or without M2 macrophages and then treated with or without 5-FU. Western blots of EMT-associated proteins were analyzed. ***P<0.001. n=3 independent experiments. Error bars, SEM. |
Relationship between CCL22 expression and M2 macrophages in CRC tissues
Firstly, we analyzed the expression of CCL22 in CRC tissues and adjacent non-tumor tissues by RT-PCR; the result found that the expression of CCL22 was significantly increased in CRC tissues compared to that in para-cancer tissues (P<0.001, Figure 6A). Then, the expression of CCL22, CD68+ TAMs, and CD163+ TAMs in CRC tissues was also examined by IHC (Figure 6B) and further analyzed the association of CCL22 with TAMs in CRC tissues. Interestingly, we found that CCL22 expression was more abundant in areas with a high CD163+ TAMs count than in areas with a low CD163+ TAMs count (r=0.536, P<0.001, Figure 6D). There was no correlation between the expression of CCL22 and CD68+ TAMs in CRC tissues (r=0.231, P=0.058, Figure 6C). Furthermore, we found that patients with higher CD163+ M2 macrophages and higher expression of CCL22 in CRC tissues had a lower overall survival (OS) rate compared with lower expression ones (P=0.006, Figure 6E; P<0.001, Figure 6F). These data indicate CCL22 could be used as an effective biomarker for predicting 5-FU resistance and prognosis in CRC.
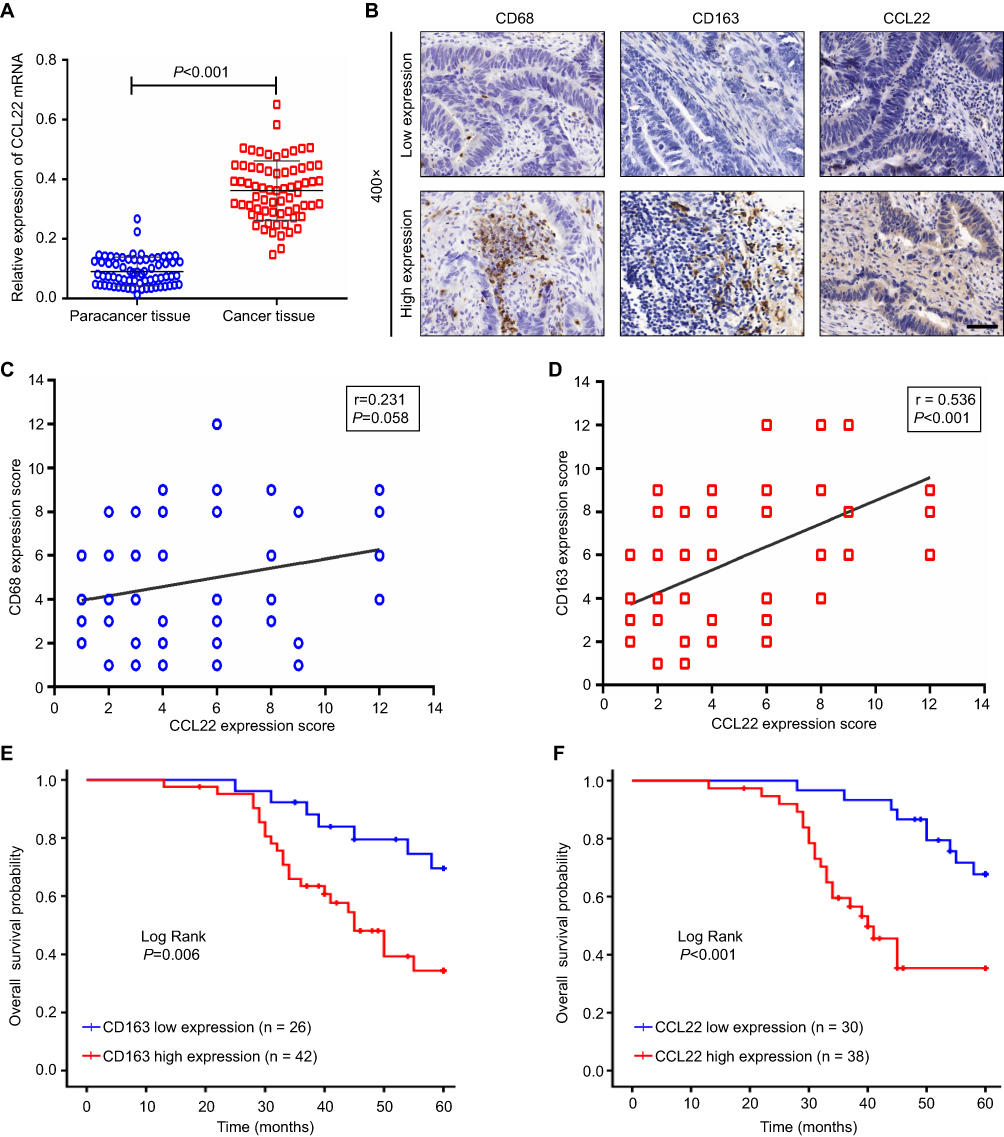 | Figure 6 Relationship between CCL22 expression and M2 macrophages in CRC tissue (n=68). (A) RT-PCR detected CCL22 expression in CRC and adjacent non-tumor tissues. (B) Representative IHC staining for CCL22, CD68, and CD163 in human CRC tissues. (C) Association of CD68 and CCL22 expression in CRC tissues. (D) Association of CD163 and CCL22 expression in CRC tissues. (E) Association of CD163+ M2 macrophages expression in CRC tissues with the patients’ overall survival. (F) Association of CCL22 expression in CRC tissues with the patients’ overall survival.Error bars, SEM. Scale bar, 400×. |
Discussion
In the past decade, the incidence of CRC has increased year by year.1 Although some progress with the clinical application of molecule-targeted drugs has been made in the treatment of CRC, more efforts are needed to reduce the chemoresistance and improve the treatment effect.18 5-FU is widely used for the clinical treatment of various malignant tumors, such as colorectal,19 breast,20 and gastric cancers.21 However, during chemotherapy in CRC patients, 5-FU resistance often occurs.22 Therefore, it is necessary to thoroughly investigate the specific molecular mechanisms of CRC 5-FU resistance, which is important for reversing the treatment of chemoresistance.
Recently, accumulating evidence have suggested that the tumor microenvironment plays an important role in the development of tumor chemoresistance.23,24 M2 macrophages are prominent component in the CRC microenvironment associated with tumor progression and poor prognosis.25 However, the molecular mechanism for intercellular communication between M2 macrophages and CRC cells response to 5-FU treatment is poorly understood. In our study, we found that there was a positive correlation between CCL22 expression and CD163+ M2 macrophages distribution in CRC tissue. Therefore, we speculated this chemokine CCL22, generating by M2 macrophages, could play a critical role in tumor progression and chemoresistance. Furthermore, in vitro experiments demonstrated our clinical hypothesis that M2 macrophages could mediate CRC cells resistance to 5-FU by secreting CCL22. In contrast, Malesci et al. demonstrated that CD68+ macrophages could increase the combinatorial treatment response of 5-FU in stage III CRC.26 CD68 was a pan-macrophage marker which did not represent the different TAMs subpopulations. The sub-classification of TAMs is variably represented in distinct tumor microenvironments, and expression of M1 and M2 polarization markers are considerably heterogeneous in CRC.27 We therefore considered that these conflicting conclusions could be the result of macrophages heterogeneity, meant that the way to evaluate the effect of CD68+ TAMs on chemoresistance was lack of variety. Currently, the phenotypic changes of macrophages after phagocytosis of tumor cells, especially antibody-dependent cellular phagocytosis (ADCP), and the effects of such changes on immune effector cells are still poorly understood. Recently, Song et al. reported that ADCP could induce immunosuppression of macrophages, and immune checkpoint inhibitors (ICIs) against this immunosuppression could promote the therapeutic effect of anti-tumor antibodies.28 Especially, trastuzumab acts as a neoadjuvant therapy to up-regulate PD-L1 and IDO expression in TAMs in HER2-expressing breast cancer patients and is associated with poor response to trastuzumab.28 These findings provide supporting evidence for the synergistic therapeutic effect of ICIs and anti-tumor targeted therapy in preclinical level, which might provide new ideas for exploring the effects of targeted therapeutic drugs (such as cetuximab, panitumumab) for tumor immune microenvironment and immunotherapy in CRC.
Previously, various studies have shown that CCL22 plays an important role in the regulation of tumor microenvironment. Wu et al. observed that CCL22 expression in gastric cancer tissues was closely associated with high infiltration of Foxp3+ Tregs, which formed the immunosuppression of tumor microenvironment.29 Takumi et al. reported similar results that tumor-secreted CCL22 plays a critical role in immunosuppression by binding to Foxp3+ Tregs, the surface of which was highly expressed CC chemokine receptor 4 (CCR4).30 Collectively, most of these researches on CCL22 in tumor microenvironment had focused on the regulation of Foxp3+ Treg recruitment. Herein, we demonstrated that M2 macrophages transmitted CCL22 to cancer cells that prompted the development of 5-FU resistance and EMT program in CRC cells. Similar to our findings, recent studies showed that CCL22 and its receptor CCR4 could mediate tumor migration and invasion in breast and gastric cancer cells.31,32 A consistent finding was also reported in a study where M2 macrophage-derived CCL22 was proven to enhance tumor migration capacities and correlated with venous infiltration in hepatocellular carcinoma patients.33 Our study also defined that CCL22, as an adverse prognostic biomarker, could be applied to stratify CRC patients. Of special interests, CCL22 could be used as a new molecular biological factor to evaluate tumor chemotherapy and progression. Thus, we advocated the application of CCL22 expression to better regulate the adjuvant chemotherapy management in pre-operational and post-operational evaluation.
The PI3K/AKT pathway, downstream of cytokine receptor, EGFR, and receptor tyrosine kinase, is the most commonly activated signaling pathway in various solid tumors, which is involved in tumor progression and chemoresistance.16,17 The PI3K/Akt pathway contributes to chemoresistance in different types of cancers by regulating proliferation, apoptosis, autophagy, angiogenesis, and EMT.34 Cell survival pathways such as EGFR or PI3K/AKT have been reported to be activated by chemotherapy drugs like docetaxel, paclitaxel, and 5-FU.35,36 Similar to the results from esophageal squamous cell carcinoma cells (SCCs),35 our results showed that 5-FU inhibited CRC cell growth with decreased phosphorylation of PI3K and AKT. Because the activated PI3K/AKT pathway could partly counteract the inhibition of cell growth, combinatorial cocultured with M2 macrophages and treatment with 5-FU that activates the PI3K/AKT pathway will impair the inhibition effect of 5-FU. Our results also demonstrated that CCL22 secreted from M2 macrophages offset with the anti-tumor effect of 5-FU by activating the PI3K/AKT pathway. Thus, studying the interaction between tumor cells and TAMs in tumor microenvironment might offer novel clues to immunological therapy. With the research continuing to deepen, we believe that the use of anti-CCR4 monoclonal antibodies or blocking of the CCL22/PI3K/AKT pathway may be a potential treatment for CRC therapy in the future.
Conclusion
Taken together, our findings indicate that M2 macrophages regulate 5-FU-mediated chemoresistance via the EMT program, PI3K/AKT pathway, and caspase-mediated apoptosis with the release of CCL22 in CRC. Our study introduces M2 macrophages as important components to the field of chemoresistance and indicates that the CCL22/PI3K/AKT pathway may be a potential therapeutic target for combating 5-FU-induced chemoresistance in CRC.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81572874), National Natural Science Fund Youth Fund of China (81702411), Zhongnan Hospital of Wuhan University, Technology and Innovation Seed Found (znpy 2016058).
Author contributions
Concept and study design: B Xiong. Experimental implementation: C Wei, CG Yang and XB Lin. Data collection: SY Wang, DD Shi and CX Zhang. Statistical analyses: C Wei and CG Yang. Picture drawing: C Wei and CG Yang. Manuscript writing: C Wei and CG Yang. Manuscript revision: B Xiong. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67(3):104–117.
2. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66(4):683–691. doi:10.1136/gutjnl-2015-310912
3. Gamelin EC, Danquechin-Dorval EM, Dumesnil YF, et al. Relationship between 5-fluorouracil (5-FU) dose intensity and therapeutic response in patients with advanced colorectal cancer receiving infusional therapy containing 5-FU. Cancer. 2015;77(3):441–451. doi:10.1002/(SICI)1097-0142(19960201)77:3<441::AID-CNCR4>3.0.CO;2-N
4. Hanahan D, Coussens LM. Accessories to the crime: functions of Cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. doi:10.1016/j.ccr.2012.02.022
5. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;41(34):1065–1073. doi:10.1189/jlb.0609385
6. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. doi:10.1016/j.immuni.2014.06.010
7. Steidl C, Lee T, Shah S, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. New Engl J Med. 2010;362(10):875–885. doi:10.1056/NEJMoa0905680
8. Chen JJW, Lin YC, Yao PL, et al. Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol. 2005;23(5):953. doi:10.1200/JCO.2005.12.172
9. Murray PJ. Macrophage polarization. Annu Rev Physiol. 2017;79(1):541. doi:10.1146/annurev-physiol-022516-034339
10. Edin S, Wikberg ML, Dahlin AM, et al. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLoS One. 2012;7(10):e47045. doi:10.1371/journal.pone.0047045
11. Ghosh S, Mukherjee S, Choudhury S, et al. Reactive oxygen species in the tumor niche triggers altered activation of macrophages and immunosuppression: role of fluoxetine. Cell Signal. 2015;27(7):1398–1412. doi:10.1016/j.cellsig.2015.03.013
12. Li X, Yao W, Yuan Y, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66(1):157–167. doi:10.1136/gutjnl-2015-310514
13. Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-associated macrophages: potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev. 2016;99(Pt B):180–185. doi:10.1016/j.addr.2015.11.009
14. Lee GT, Kwon SJ, Kim J, et al. WNT5A induces castration-resistant prostate cancer via CCL2 and tumour-infiltrating macrophages. Br J Cancer. 2018;118(5):670–678. doi:10.1038/bjc.2017.451
15. Usman MW, Gao J, Zheng T, et al. Macrophages confer resistance to PI3K inhibitor GDC-0941 in breast cancer through the activation of NF-kappaB signaling. Cell Death Dis. 2018;9(8):809. doi:10.1038/s41419-018-0849-6
16. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170(4):605–635. doi:10.1016/j.cell.2017.07.029
17. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13(2):140–156. doi:10.1038/nrd4204
18. Chung KY, Saltz LB. Adjuvant therapy of colon cancer: current status and future directions. Cancer J. 2007;13(3):192–197. doi:10.1097/PPO.0b013e318074d26e
19. De GA, Tournigand C, André T, Larsen AK, Louvet C. Targeted agents for adjuvant therapy of colon cancer. Semin Oncol. 2007;6(1):46–51.
20. Walde D. Adjuvant docetaxel for node-positive breast cancer. New Engl J Med. 2005;353(9):954. doi:10.1056/NEJMc051802
21. MacDonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. Cancer/Radiothérapie. 2002;6(4):266–267. doi:10.1016/S1278-3218(02)00178-6
22. Shi L, Wu L, Chen Z, et al. MiR-141 activates Nrf2-dependent antioxidant pathway via down-regulating the expression of Keap1 conferring the resistance of hepatocellular carcinoma cells to 5-fluorouracil. Cell Physiol Biochem. 2015;35(6):2333–2348. doi:10.1159/000374036
23. Binenbaum Y, Na’Ara S, Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist Updat. 2015;23(3):55–68. doi:10.1016/j.drup.2015.10.002
24. Mcmillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov. 2013;12(3):217–228. doi:10.1038/nrd3870
25. Herrera M, Herrera A, Domínguez G, et al. Cancer‐associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. 2013;104(4):437–444. doi:10.1111/cas.12096
26. Malesci A, Bianchi P, Celesti G, et al. Tumor-associated macrophages and response to 5-flourouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunology. 2017;6(12). doi:10.1080/2162402X.2017.1342918
27. Algars A, Irjala H, Vaittinen S, et al. Type and location of tumor-infiltrating macrophages and lymphatic vessels predict survival of colorectal cancer patients. Int J Cancer. 2012;131(4):864–873. doi:10.1002/ijc.26457
28. Su S, Zhao J, Xing Y, et al. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell. 2018;175(2):442–457 e423. doi:10.1016/j.cell.2018.09.007
29. Wu S, He H, Liu H, et al. C-C motif chemokine 22 predicts postoperative prognosis and adjuvant chemotherapeutic benefits in patients with stage II/III gastric cancer. Oncoimmunology. 2018;7(6):e1433517. doi:10.1080/2162402X.2018.1490854
30. Kumai T, Nagato T, Kobayashi H, et al. CCL17 and CCL22/CCR4 signaling is a strong candidate for novel targeted therapy against nasal natural killer/T-cell lymphoma. Cancer Immunol Immunother. 2015;64(6):697. doi:10.1007/s00262-015-1675-7
31. Cao L, Hu X, Zhang J, Huang G, Zhang Y. The role of the CCL22-CCR4 axis in the metastasis of gastric cancer cells into omental milky spots. J Transl Med. 2014;12(1):267. doi:10.1186/s12967-014-0267-1
32. B O P, Dolgor B, Monica B, et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009;69(14):5996. doi:10.1158/0008-5472.CAN-08-3660
33. W H Y O, Chung-Mau L, Chang-Chun L, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol. 2015;62(3):607–616. doi:10.1016/j.jhep.2014.10.029
34. Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol. 2017;45:62–71. doi:10.1016/j.ceb.2017.02.007
35. Lu YX, Chen DL, Wang DS, et al. Melatonin enhances sensitivity to fluorouracil in oesophageal squamous cell carcinoma through inhibition of Erk and Akt pathway. Cell Death Dis. 2016;7(10):e2432. doi:10.1038/cddis.2016.330
36. Brunelle JK, Zhang B. Apoptosis assays for quantifying the bioactivity of anticancer drug products ☆. Drug Resist Updat. 2010;13(6):172–179. doi:10.1016/j.drup.2010.09.001
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.