")
Back to Journals » Clinical Ophthalmology » Volume 10
Lysyl oxidase-like 1 polymorphisms in a southwestern Greek cataract population with pseudoexfoliation syndrome
Authors Panoutsopoulos A, Gartaganis V, Giannakopoulos M, Goumas P, Anastassiou E, Gartaganis S
Received 19 June 2015
Accepted for publication 13 August 2015
Published 21 January 2016 Volume 2016:10 Pages 161—166
DOI https://doi.org/10.2147/OPTH.S90789
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Alexios A Panoutsopoulos,1 Vassiliki S Gartaganis,2 Marios P Giannakopoulos,1 Panos D Goumas,3 Evangelos D Anastassiou,4 Sotirios P Gartaganis1
1Department of Ophthalmology, School of Medicine, University of Patras, Achaia, Greece; 2Protein Chemistry Group, Institute of Molecular Oncology, BSRC “Al Fleming”, Vari, Greece; 3Department of Otolaryngology, Head and Neck Surgery, 4Department of Microbiology, School of Medicine, University of Patras, Achaia, Greece
Purpose: The aim of this study was to determine the possible association of rs1048661 and rs3825942 single nucleotide polymorphisms (SNPs) in the lysyl oxidase-like 1 (LOXL1) gene of cataract patients from southwestern Greece with pseudoexfoliation (PEX) syndrome.
Patients and methods: Ninety-three patients with PEX syndrome and 74 without PEX syndrome were recruited with the principal diagnosis being cataract. LOXL1 SNPs, rs1048661 and rs3825942, were genotyped by using polymerase chain reaction.
Results: The G allele of rs1048661 was found in 96.7% in the PEX group as compared to 80.5% of non-PEX alleles (P=19×10-4; Odds ratio [OR] =5.37; 95% confidence interval [CI] =1.68–17.12). Similarly, the G allele of rs3825942 was found in 72.1% of the PEX group as compared to 41.8% of non-PEX alleles (P=4×10-5; OR =3.78; 95% CI =1.98–7.23). The T and A allele frequencies of rs1048661 and rs3825942, respectively, were underrepresented in the PEX group patients as compared to non-PEX group.
Conclusion: Our data confirm previously reported association between LOXL1 polymorphisms and PEX syndrome in a southwestern Greek population. A significant association was found for the G allele of rs1048661 and rs3825942 demonstrating that the GG haplotype is a high-risk factor for the development of PEX syndrome.
Keywords: exfoliative syndrome, PEX syndrome, single nucleotide polymorphisms
Introduction
Pseudoexfoliation (PEX) syndrome is an age-related systemic disease of the extracellular matrix characterized by the widespread deposition of an abnormal extracellular fibrillary material on many intra- and extraocular tissues. It is widely recognized that this syndrome is associated with severe chronic secondary open-angle glaucoma in up to 50% of the affected eyes and an increased risk of intraoperative and postoperative complications during cataract surgery.1 Moreover, it is also associated with various other complications such as angle-closure glaucoma, blood–aqueous barrier dysfunction, or corneal endothelial decompensation.2 Despite extensive research, the pathogenesis of PEX syndrome remains obscure. Similar pathological changes to those in the eye have been identified in many visceral organs such as lung, heart, gallbladder, brain, as well as in the skin.3–6 Although the exact composition of the abnormal extracellular material and the mechanisms responsible for its excessive production and accumulation still remain unknown, the concept of an elastotic process has recently been established. Molecular, biological, and biochemical data support the pathogenetic concept of PEX syndrome as a type of stress-induced elastic fibrillopathy.7 The etiopathogenesis involves genetic and nongenetic factors. Several genetic factors have been associated with development and heredity of PEX syndrome. The most important evidence for genetic causes is the identification of single nucleotide polymorphisms (SNPs) in the lysyl oxidase-like 1 (LOXL1) gene on chromosome 15q24.1, the noncoding intronic rs2165241 SNP and two nonsynonymous coding SNPs (rs1048661 and rs3825942). They have been strongly correlated with PEX syndrome and PEX glaucoma in Icelandic and Swedish populations.8 Moreover, the presence of certain apolipoprotein E gene variants,9 fibrillin-1, MMP-2, TIMP-1 variants, and several region variations such as the 30 cM 18q12.1–21.33 locus on chromosome 1810 showed an increased risk for developing PEX syndrome in certain populations. However, nongenetic factors including ultraviolet light exposure, dietary factors, infectious agents, trauma, as well as oxidative stress, hypoxia, and inflammation have also been suggested to act as comodulating external factors.11
Additionally, high oxidative stress and products of lipid peroxidation may promote abnormal extracellular matrix metabolism and cause disease progression.12,13
LOXL1 is a member of the lysyl oxidase family of enzymes that catalyze the covalent cross-linking of collagen and elastin in connective tissues through oxidative deamination of lysine or hydroxylysine side chains and is required for elastic fiber formation.7,14 It is formed as a propeptide which accumulates in sites of elastogenesis, participating in the formation of a microfibril scaffold. There, it cleaves off to its active form and deaminates lysine residues of tropoelastin monomers. These are cross-linked to each other and lead to formation of elastin polymers in the extracellular matrix of several structures of the eye, such as iris, cornea, or zonules.14–16 The two risk coding SNPs (rs1048661 and rs3825942) are located in the N-terminal part of pro-LOXL1 which was suggested to be critical in enzyme activation and substrate identification.15,17,18 Our study was designed to investigate the incidence of the two nonsynonymous coding SNPs (rs1048661 and rs3825942) in LOXL1 gene in a southwestern Greek cataract population with or without PEX syndrome.
Material and methods
Patient population and clinical diagnosis
A prospective study was designed. Patients with or without evidence of ocular PEX syndrome were selected from individuals admitted to the Ophthalmology Clinic of the University Hospital of Patras, Greece, with the principal diagnosis of cataract to be operated in a 24-month period. The hospital mainly admits patients from southwestern Greece, consisting of about 50% urban and 50% rural areas, with a population of 780,000 inhabitants. Only patients with senile cataract and those belonging to the abovementioned geographic region were included. The study was approved by the ethics committee of the University Hospital of Patras, Greece, and was conducted in accordance with the ethical principles of the Declaration of Helsinki.
All patients gave informed consent and full medical history before entering the study. They underwent detailed ophthalmologic examination that included slit-lamp biomicroscopic examination with dilated pupil, applanation tonometry, gonioscopy, and fundus examination. Moreover, they were examined independently by two medical doctors (ophthalmologists), with the same instruments, in a dark examination room. Patients with PEX syndrome were defined as those with clinical evidence of PEX material and intraocular pressure of less than 21 mmHg with no clinical evidence of glaucomatous optic neuropathy in either eye. Following examination, they were classified either in the PEX group or in the non-PEX group. Patients with aphakia or pseudophakia in the fellow eye were excluded because of difficulties in making a definite diagnosis of PEX syndrome. Patients and controls with diabetes mellitus or other malignant diseases were also excluded from the study.
Sample collection and genotyping
Peripheral blood samples (2–5 mL) were collected in EDTA-containing tubes and immediately stored at −80°C until further processing. Genomic DNA was extracted from peripheral blood nucleated cells using the QIAamp DNA blood mini kit (Cat. No 51104, Qiagen, Hilden, Germany), in accordance with the manufacturer’s instructions, and stored in 50 μL of elution buffer at -20°C (240 ng/μL). It was genotyped for two nonsynonymous SNPs located in exon 1 of LOXL1: rs1048661 (R141L) (c.758G>T; p.Arg141Leu) and rs3825942 (G153D) (c.794G>A; p.Gly153Asp) by polymerase chain reaction (PCR) using specific primers (forward: 5′-CAACGGGCAGGTGTACAGCTT-3′, reverse: 5′-GCGGGGTCGTAGTTCTCGTAC-3′). The primers amplify a 441 bp product which spans both the polymorphic sites in exon 1 of LOXL1. The PCR mixture contained 20 μL of genomic DNA, 2 μL of each primer (10 μM), 0.5 μL proofreading (Pfu) DNA polymerase (product M7741, Promega, Madison, WI, USA), 1 μL PCR nucleotide mix (product C1141, Promega), and 3 μL of ultrapure DMSO (Sigma-Aldrich, St Louis, MO, USA, compendia quality level product D2650) in a total volume of 50 μL per reaction. PCR amplification was performed with an initial denaturation at 94°C for 5 minutes, followed by 10 cycles of denaturation at 94°C for 30 seconds, primer annealing from 65°C to 60°C (touchdown PCR) for 1 minute with 0.5°C decrement, extension at 72°C for 45 seconds, followed by 30 cycles of denaturation at 94°C for 30 seconds, annealing at 60°C for 1 minute, extension at 72°C for 45 seconds, followed by a final extension at 72°C for 1 minute. PCR products were separated on a 2% low-melt agarose gel containing ethidium bromide and purified using the Wizard SV Gel and PCR Clean-Up System (product A9281, Promega), according to the manufacturer’s protocol. Purified PCR products were analyzed by Sanger sequencing using an automated DNA sequencer (Big Dye Terminator chemistry, version 3 on a 3730XL capillary sequencer; Applied Biosystems, Inc., Foster City, CA, USA). Sequences were analyzed by using a sequencing analysis software BioEdit (BioEdit graphical sequence alignment editor, version 7.0.9.0).
Statistical analysis
Data were analyzed using analytical statistical software (SPSS version 16.0; SPSS, Chicago, IL, USA). Hardy–Weinberg equilibrium and the significance between the allele and genotype frequencies were assessed using a standard chi-square (χ2) test. Fisher’s exact test was used to compare the PEX group with the non-PEX group. Odds ratios (ORs) for the allele and genotype frequencies of PEX group compared to non-PEX group were calculated using the χ2 test, taking a 95% confidence interval (95% CI) into account. A P-value <0.05 was considered as statistically significant. All results were adjusted using the Yates correction.
Results
A total of 93 patients with PEX syndrome (PEX group) and 74 patients without PEX syndrome (non-PEX group) were recruited for this study from among patients with the principal diagnosis of cataract to be operated in a 24-month period. All PEX group and non-PEX group patients originated from the southwestern province of Greece. The PEX group patients had a mean age of 76.5±7.52 years (range, 47–94 years) and included 46 males and 47 females. The non-PEX group patients had a mean age of 73.1±8.2 years (range, 46–92 years) and included 36 males and 38 females. There were no significant differences in mean age and sex between the PEX and non-PEX groups (P>0.05). Unilateral involvement of PEX was found in 13 (14%) and bilateral involvement in 80 (86%) patients among those with PEX syndrome. The mean age 64.46±6.51 years (range, 47–69 years) of the unilateral PEX syndrome involvement was significantly different from that of 78.45±5.43 years (range, 70–94 years) of the bilateral involvement (P=0.000). The demographic characteristics of the study population are presented in Table 1. The distribution of the LOXL1 allele and genotype frequencies of SNPs rs1048661 and rs3825942 was analyzed in patients with and without PEX syndrome. The genotype distribution of both SNPs in PEX and non-PEX group was consistent with the Hardy–Weinberg equilibrium. The detailed count of the allele and genotype frequencies of rs1048661 and rs3825942 polymorphisms of LOXL1 are presented in Table 2. For both SNPs a significant association with PEX syndrome was identified for the allele and the genotype frequencies. The G allele of rs1048661 was found in 96.7% in PEX group alleles as compared to 80.5% of non-PEX group alleles (P=1.9×10−3; OR =5.37; 95% CI =1.68–17.12). In addition, the G allele of rs3825942 was significantly more frequent in PEX group, as it was found in 72.1% of PEX group alleles as compared to 41.8% of non-PEX group alleles (P=4×10−5; OR =3.78; 95% CI =1.98–7.23). Instead, the T allele of rs1048661 appeared at a significantly lower frequency, and the A allele of rs3825942 was underrepresented in the PEX group as compared to non-PEX group. The genotype GG of rs1048661 was found in 25.2% in PEX group as compared to 13.9% in non-PEX group (P=5×10−2; OR =2.23; 95% CI =0.99–5.02). The genotype GG of rs3825942 was found in 20.2% in the PEX group as compared to 10.8% in non-PEX group (P=3×10−2; OR =2.56; 95% CI =1.06–6.14). For rs1048661, the GT genotype was underrepresented, whereas for rs3825942, the GA genotype was even more underrepresented in the PEX group as compared to non-PEX group. Haplotype association analysis identified four common haplotypes (GG, GA, TG, and TA) defined by the two SNPs (rs1048661–rs3825942). Accounted haplotype frequencies are presented in Table 3. The haplotypes were significantly associated with the PEX group either as a risk or as a protective haplotype. The high-risk haplotype GG was more frequent (69.9%) in the PEX group as compared to the non-PEX group (29.7%) (P=2.5×10−7), while the haplotype GA was underrepresented in the PEX group (8.5%) than in non-PEX group (37.9%) (P=5×10−6). Haplotypes TG and TA were present at lower levels in both groups without any statistical significance.
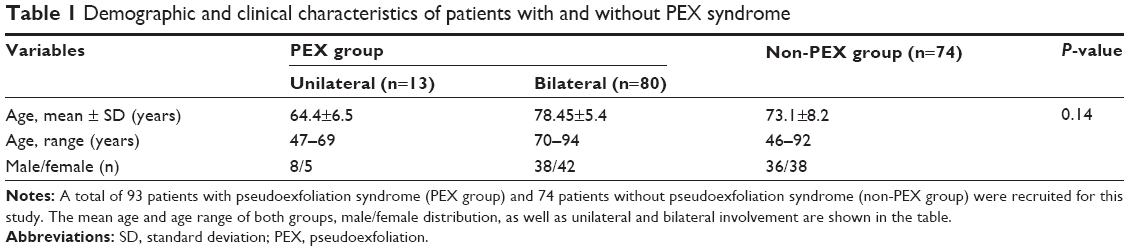 | Table 1 Demographic and clinical characteristics of patients with and without PEX syndrome |
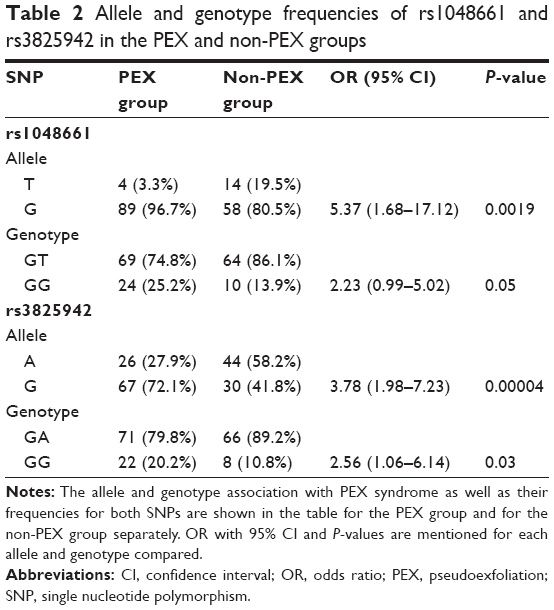 | Table 2 Allele and genotype frequencies of rs1048661 and rs3825942 in the PEX and non-PEX groups |
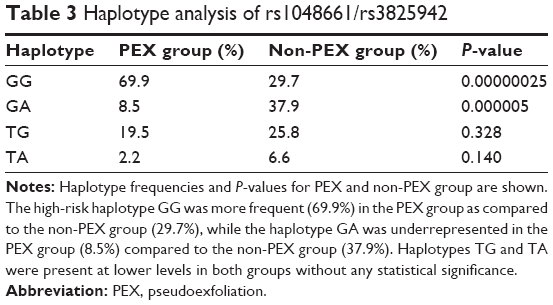 | Table 3 Haplotype analysis of rs1048661/rs3825942 |
Discussion
The potential roles of SNPs of LOXL1 gene in PEX syndrome were suggested by Thorleifsson et al.8 Their study established a strong association between three SNPs in the LOXL1 gene on chromosome 15q24.1 with both PEX syndrome and PEX glaucoma in Swedish and Icelandic populations. LOXL1 is a key enzyme involved in elastic fiber synthesis and homeostasis supporting a role of elastogenesis and elastosis in the pathophysiology of PEX syndrome.17 Various studies in populations from the United States,19–23 Europe,18,24 and Australia25 highlighted genetic susceptibility of LOXL1 polymorphisms to PEX syndrome and showed that the LOXL1 gene is the principal genetic risk factor for PEX syndrome. The risk for PEX disease in Caucasians was associated in a large number of cases with the G allele and GG genotype of rs1048661 and the G allele and GG genotype of rs3825942. A common haplotype (GG) formed by two nonsynonymous SNPs, rs1048661 and rs3825942 located in exon 1 of LOXL1, has been reported as a risk factor for the vast majority of PEX patients.
We studied 167 cataract patients, 93 with PEX syndrome (PEX group) and 74 without PEX syndrome (non-PEX group) from a southwestern Greek population. We confirmed a strong association with LOXL1 variants in our PEX group patients. The frequency of allele G of SNPs rs1048661 and rs3825942 was higher in the PEX group than in the non-PEX group. Also, the frequencies of genotype GG at SNP rs1048661 and the same genotype at SNP rs3825942 were higher in the PEX group than in the non-PEX group. These findings indicate that the G allele and GG genotype of rs1048661 had an increased risk for PEX group, while the G allele and GG genotype of rs3825942 had an even stronger risk for PEX group. In accordance with our results, these frequencies are comparable to the rates observed in most Caucasians including Swedish, Icelandic, American, German, Italian, and Australian populations. Our results indicate that the prevalence of the G allele of SNP rs1048661 in PEX group (0.967) was higher than in the non-PEX group (0.79) and that this difference was statistically significant (P=19×10−4). In addition, the prevalence of the G allele of SNP rs3825942 in PEX group (0.721) was higher than in the non-PEX group (0.418) and that this difference was statistically significant (P=4×10−5). The association of the high-risk G alleles with PEX syndrome, for the two SNPs examined in the current study, seems to be largely consistent across most of the tested populations, with similar results. However, there are significant exceptions where G allele of rs1048661 is not statistically significant for the occurrence of PEX syndrome in Indian26 or Chinese27 populations. Moreover, the G allele of rs1048661 was found to be actually protective in one Chinese population27 as in several Japanese populations.28–32 Finally, the A allele and not the G allele of rs3825942 seems to be the high-risk allele for PEX syndrome in South African populations.33,34 These inconsistencies show that the connection between LOXL1 and PEX syndrome may possibly involve another mechanism independent of the alleles of the abovementioned SNPs or could be associated with other SNPs in the LOXL1 or other genes.35–37
Focusing on the results of the genotype frequencies, GG genotype of rs1048661 (P=5×10−2) and GG genotype of rs3825942 (P=3×10−2) show less significance than the equivalent statistics of G allele of rs1048661 (P=1.9×10−3) and rs3825942 (P=4×10−5). The high-risk haplotype GG formed by the two nonsynonymous coding SNPs, rs1048661 and rs3825942, was found in 69.9% in PEX group and 29.7% of the non-PEX group (P=2.5×10−7; OR =5.49; 95% CI =2.82–10.69), whereas the haplotype GA defined by rs1048661 and rs3825942 was found in 8.5% in the PEX group as compared to 37.9% in the non-PEX group (P=5×10−6; OR =6.47, 95% CI =2.73–15.34). In our study, the GA haplotype, defined by rs1048661 and rs3825942, was found to have a potential protective effect on developing PEX syndrome. However, many cataract patients carrying LOXL1 risk alleles (and genotypes) do not have the PEX syndrome phenotype. Consequently, it must be assumed that additional genetic factors may influence the manifestation of the PEX syndrome. Additionally, a number of nongenetic factors have also been evaluated but not yet identified for the possible implication in the development of PEX syndrome. These include ultraviolet light, autoimmunity, slow virus infection, and trauma.38
Conclusion
In summary, we showed that the G allele of LOXL1 SNPs rs1048661 and rs3825942 is associated with the PEX syndrome population of this study in a similar manner to the Icelandic, Swedish, American, Australian, German, Italian, and Finnish populations. Finally, our results demonstrate that the SNP rs3825942 in the LOXL1 gene constitutes a significant risk factor to development of PEX syndrome in the southwestern Greek population.
Disclosure
The authors report no conflicts of interest in this work.
References
Ritch R, Schlötzer-Schrehardt U. Exfoliation syndrome. Survey Ophthalmol. 2001;45:265–315. | ||
Naumann GO, Schlötzer-Schrehardt U, Küchle M. Pseudoexfoliation syndrome for the comprehensive ophthalmologist. Intraocular and systemic manifestations. Ophthalmology. 1998;105:951–968. | ||
Streeten BW, Dark AJ, Wallace RN, Li ZY, Hoepner JA. Pseudoexfoliative fibrillopathy in the skin of patients with ocular pseudoexfoliation. Am J Ophthalmol. 1990;110:490–499. | ||
Streeten BW, Li ZY, Wallace RN, Eagle RC Jr, Keshgegian AA. Pseudoexfoliative fibrillopathy in visceral organs of a patient with pseudoexfoliation syndrome. Arch Ophthalmol. 1992;110:1757–1762. | ||
Schlötzer-Schrehardt UM, Koca MR, Naumann GO, Volkholz H. Pseudoexfoliation syndrome. Ocular manifestation of a systemic disorder? Arch Ophthalmol. 1992;110:1752–1756. | ||
Schumacher S, Schlötzer-Schrehardt U, Martus P, Lang W, Naumann GO. Pseudoexfoliation syndrome and aneurysms of the abdominal aorta. Lancet. 2001;357:359–360. | ||
Schlötzer-Schrehardt U, Pasutto F, Sommer P, et al. Genotype-correlated expression of lysyl oxidase-like 1 in ocular tissues of patients with pseudoexfoliation syndrome/glaucoma and normal patients. Am J Pathol. 2008;173:1724–1735. | ||
Thorleifsson G, Magnusson KP, Sulem P, et al. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science. 2007;317:1397–1400. | ||
Yilmaz A, Tamer L, Ateş NA, Camdeviren H, Değirmenci U. Effects of apolipoprotein E genotypes on the development of exfoliation syndrome. Exp Eye Res. 2005;80:871–875. | ||
Lemmelä S, Forsman E, Sistonen P, Eriksson A, Forsius H, Järvelä I. Genome-wide scan of exfoliation syndrome. Invest Ophthalmol Vis Sci. 2007;48:4136–4142. | ||
Schlötzer-Schrehardt U. Pseudoexfoliation syndrome: the puzzle continues. J Ophthalmic Vis Res. 2012;7:187–189. | ||
Gartaganis SP, Georgakopoulos CD, Patsoukis NE, Gotsis SS, Gartaganis VS, Georgiou CD. Glutathione and lipid peroxide changes in pseudoexfoliation syndrome. Curr Eye Res. 2005;30:647–651. | ||
Gartaganis SP, Patsoukis NE, Nikolopoulos DK, Georgiou CD. Evidence for oxidative stress in lens epithelial cells in pseudoexfoliation syndrome. Eye. 2007;21:1406–1411. | ||
Csiszar K. Lysyl oxidases: a novel multifunctional amine oxidase family. Prog Nucleic Acid Res Mol Biol. 2001;70:1–32. | ||
Liu X, Zhao Y, Gao J, et al. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet. 2004;36:178–182. | ||
Kielty CM, Wess TJ, Haston L, Ashworth JL, Sherratt MJ, Shuttleworth CA. Fibrillin-rich microfibrils: elastic biopolymers of the extracellular matrix. J Muscle Res Cell Motil. 2002;23:581–596. | ||
Thomassin L, Werneck CC, Broekelmann TJ, et al. The pro-regions of lysyl oxidase and lysyl oxidase-like 1 are required for deposition onto elastic fibers. J Biol Chem. 2005;280:4284–4286. | ||
Pasutto F, Krumbiegel M, Mardin CY, et al. Association of LOXL1 common sequence variants in German and Italian patients with pseudoexfoliation syndrome and pseudoexfoliation glaucoma. Invest Ophthalmol Vis Sci. 2008;49:1459–1463. | ||
Fingert JH, Alward WL, Kwon YH, et al. LOXL1 mutations are associated with exfoliation syndrome in patients from the midwestern United States. Am J Ophthalmol. 2007;144:974–975. | ||
Aragon-Martin JA, Ritch R, Liebmann J, et al. Evaluation of LOXL1 gene polymorphisms in exfoliation syndrome and exfoliation glaucoma. Mol Vis. 2008;14:533–541. | ||
Challa P, Schmidt S, Liu Y, et al. Analysis of LOXL1 polymorphisms in a United States population with pseudoexfoliation glaucoma. Mol Vis. 2008;14:146–149. | ||
Fan BJ, Pasquale L, Grosskreutz CL, et al. DNA sequence variants in the LOXL1 gene are associated with pseudoexfoliation glaucoma in a U.S. clinic-based population with broad ethnic diversity. BMC Med Genet. 2008;9:5. | ||
Yang X, Zabriskie NA, Hau VS, et al. Genetic association of LOXL1 gene variants and exfoliation glaucoma in a Utah cohort. Cell Cycle. 2008;7:521–524. | ||
Mossböck G, Renner W, Faschinger C, Schmut O, Wedrich A, Weger M. Lysyl oxidase-like protein 1 (LOXL1) gene polymorphisms and exfoliation glaucoma in a Central European population. Mol Vis. 2008;14:857–861. | ||
Hewitt AW, Sharma S, Burdon KP, et al. Ancestral LOXL1 variants are associated with pseudoexfoliation in Caucasian Australians but with markedly lower penetrance than in Nordic people. Hum Mol Genet. 2008;17:710–716. | ||
Ramprasad VL, George R, Soumittra N, Sharmila F, Vijaya L, Kumaramanickavel G. Association of non-synonymous single nucleotide polymorphisms in the LOXL1 gene with pseudoexfoliation syndrome in India. Mol Vis. 2008;14:318–322. | ||
Lee KY, Ho SL, Thalamuthu A, et al. Association of LOXL1 polymorphisms with pseudoexfoliation in the Chinese. Mol Vis. 2009;15:1120–1126. | ||
Fuse N, Miyazawa A, Nakazawa T, Mengkegale M, Otomo T, Nishida K. Evaluation of LOXL1 polymorphisms in eyes with exfoliation glaucoma in Japanese. Mol Vis. 2008;14:1338–1343. | ||
Hayashi H, Gotoh N, Ueda Y, Nakanishi H, Yoshimura N. Lysyl oxidase-like 1 polymorphisms and exfoliation syndrome in the Japanese population. Am J Ophthalmol. 2008;145(3):582–585. | ||
Mabuchi F, Sakurada Y, Kashiwagi K, Yamagata Z, Iijima H, Tsukahara S. Lysyl oxidase-like 1 gene polymorphisms in Japanese patients with primary open angle glaucoma and exfoliation syndrome. Mol Vis. 2008;14:1303–1308. | ||
Mori K, Imai K, Matsuda A, et al. LOXL1 genetic polymorphisms are associated with exfoliation glaucoma in the Japanese population. Mol Vis. 2008;14:1037–1040. | ||
Ozaki M, Lee KY, Vithana EN, et al. Association of LOXL1 gene polymorphisms with pseudoexfoliation in the Japanese. Invest Ophthalmol Vis Sci. 2008;49(9):3976–3981. | ||
Rautenbach RM, Bardien S, Harvey J, Ziskind A. An investigation into LOXL1 variants in black South African individuals with exfoliation syndrome. Arch Ophthalmol. 2011;129(2):206–210. | ||
Williams SE, Whigham BT, Liu Y, et al. Major LOXL1 risk allele is reversed in exfoliation glaucoma in a black South African population. Mol Vis. 2010;16:705–712. | ||
Ferrell G, Lu M, Stoddard P, et al. A single nucleotide polymorphism in the promoter of the LOXL1 gene and its relationship to pelvic organ prolapse and preterm premature rupture of membranes. Reprod Sci. 2009;16(5):438–446. | ||
Krumbiegel M, Pasutto F, Mardin CY, et al. Exploring functional candidate genes for genetic association in German patients with pseudoexfoliation syndrome and pseudoexfoliation glaucoma. Invest Ophthalmol Vis Sci. 2009;50(6):2796–2802. | ||
Krumbiegel M, Pasutto F, Schlotzer-Schrehardt U, et al. Genome-wide association study with DNA pooling identifies variants at CNTNAP2 associated with pseudoexfoliation syndrome. Eur J Hum Genet. 2011;19(2):186–193. | ||
Damji KF, Bains HS, Stefansson E, et al. Is pseudoexfoliation syndrome inherited? A review of genetic and nongenetic factors and a new observation. Ophthalmic Genet. 1998;19:175–185. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.