")
Back to Journals » OncoTargets and Therapy » Volume 11
Lymphangioleiomyomatosis: a case report and review of diagnosis and treatment
Authors Liu Y, Guo Z , Zhao C, Li X, Liu H, Chen J
Received 3 January 2018
Accepted for publication 7 May 2018
Published 31 August 2018 Volume 2018:11 Pages 5339—5347
DOI https://doi.org/10.2147/OTT.S161360
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Yi Liu,* Zhibin Guo,* Chenlong Zhao, Xin Li, Hongyu Liu, Jun Chen
Department of Lung Cancer Surgery, Tianjin Key Laboratory of Lung Cancer Metastasis and Tumor Microenvironment, Tianjin Lung Cancer Institute, Tianjin Medical University General Hospital, Tianjin 300052, China
*These authors contributed equally to this work
Abstract: Lymphangioleiomyomatosis (LAM) is a rare disease that generally affects young women and involves the abnormal proliferation of smooth muscle-like cells (LAM cells) in the lungs (pulmonary LAM) and extrapulmonary sites (extrapulmonary LAM). This disease is rare in males. It is hard to distinguish between lung cancer and pulmonary LAM, especially during early stages. Herein, we present a case of a 66-year-old man with a small nodule in the right upper lobe that was first diagnosed as a lung malignancy using a chest CT scan. After a wedge dissection, a pathologist performed a histologic and immunohistochemical examination, and a diagnosis of pulmonary LAM was made. We further performed a 518-gene panel analysis using next-generation sequencing, and only three genes, BARD1, BLM, and BRCA2, were found to have mutations. We also provide a summary of the diagnosis and treatment of this disease.
Keywords: lymphangioleiomyomatosis, lung cancer, diagnosis and treatment
Introduction
Lymphangioleiomyomatosis (LAM) is a rare disease affecting almost exclusively women and involves many organs. It is characterized by cystic lung lesions and extrapulmonary features consisting of renal angiomyolipomas and lymphatic involvement such as lymphangioleiomyomas and chylous effusions.1 There are two forms of LAM: one is a sporadic form that occurs in 3.3–7.7 million women and the other is an inherited form that occurs in 30%–80% of women with tuberous sclerosis complex (TSC).2,3 Although it has been reported in men, it is extremely rare. The causes of LAM remain unclear; therefore, currently, most patients with LAM around the world could not obtain an effective treatment. Herein, we report the case of a male patient with LAM and an analysis of a 518-gene panel using next-generation sequencing (NGS).
Case presentation
A 66-year-old man was admitted with the chief complaint of cough with bloodstained phlegm for approximately 3 days. No other complaint or remarkable past medical history was revealed. He had an approximate 15-year history of smoking. A family history of malignancy comprised only his mother who died of lung cancer. As shown in Figure 1, a chest CT scan revealed an isolated solid nodule with a vacuole and pleural traction signs in the right upper lobe, indicating a malignant nodule. The system review was noncontributory and normal, including peripheral blood count, baseline serum chemistry screening, and tumor biomarker tests (carcinoembryonic antigen, antigen 19-9, carbohydrate antigen 24-2, CYFRA21-1, and squamous cell carcinoma antigen). Upper abdominal CT scan and brain MRI were normal. Therefore, a clinical diagnosis of an upper right lung nodule, most possibly a malignant tumor, was given.
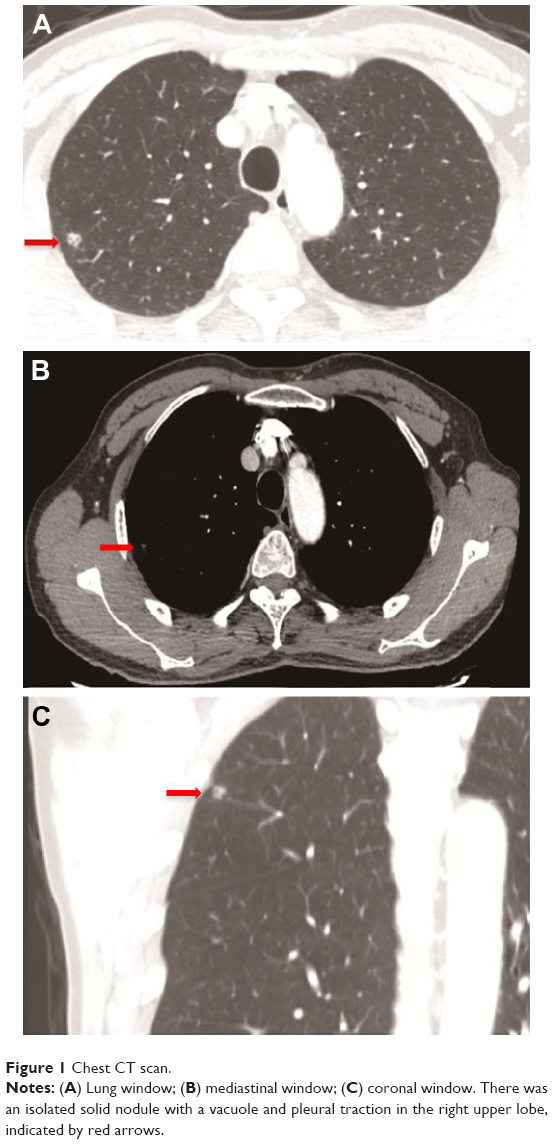 | Figure 1 Chest CT scan. |
Then, a wedge resection was performed using video-assisted thoracoscopy. As shown in Figure 2, a histopathologic examination showed a piece of dense fibrous connective tissue with some differently shaped aqueducts. Immunohistochemical staining was positive for CD34, SMA, and desmin, and negative for HMB-45. Therefore, a diagnosis of primary lung LAM was made by the pathologists. The patient recovered after the operation and was doing well during a 6-month follow-up.
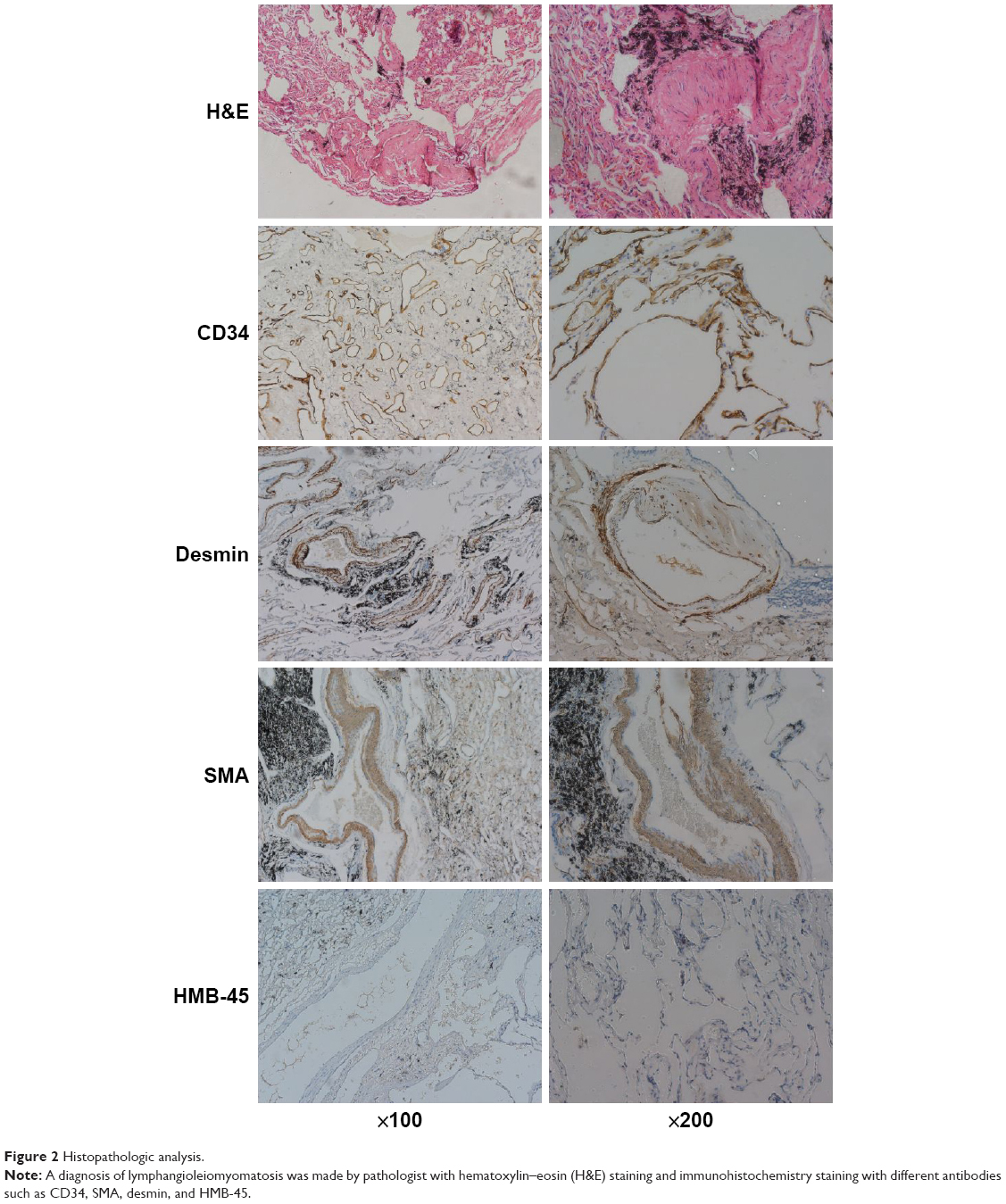 | Figure 2 Histopathologic analysis. |
As this was a rare case involving a male patient with primary lung LAM, we performed an analysis of the lesion tissues, using a 518 tumor-related gene panel, to explore possible molecular abnormalities using NGS (Zhongyuan Xiehe Gene Technology Co., Ltd., Tianjin, China; www.vcanbio.com); gene names are shown in Table S1. DNA in the patient’s blood was considered baseline to reduce the influence of leukocyte, but for the germline mutation-related genes, we reviewed the sequencing results from both sides of blood and tissues to avoid missing the important germline mutations. These genes are broadly divided into four categories: targeted drug-related genes (such as EGFR, ALK, KRAS, BRAF, MET, ROS1, RET, HER2, NRAS, KIT, and PDGFRA), chemotherapy efficacy and side effect-related genes, genetic susceptibility genes (including 78 related genes), and other genes, which were called Catalogue of Somatic Mutations in Cancer (COSMIC). In these detected genes, only three uncommon mutations were found that were from three genetic susceptibility genes named BARD1, BLM, and BRCA2. All these three mutations were germline mutations, since the mutations were found from both sides of blood and tissues sequencing analysis, as shown in Figure 3. The mutation in the BARD1 gene was in exon 6 with a mutation that has an unknown meaning at encoding sequence c.1518_1 519CA; BLM and BRCA2 had missense mutations at encoding sequence c.2371C>T (p.R791C) and c.4136A>G (p.Q1379R) in exon 11, respectively. We also further checked the mutation of TSC1 and TSC2 genes from both sides of blood and tissues sequencing results and did not find the mutation for these two genes. These results indicated that the tumor mutation burden in this case of primary lung LAM was low and that no remarkable gene mutation was found.
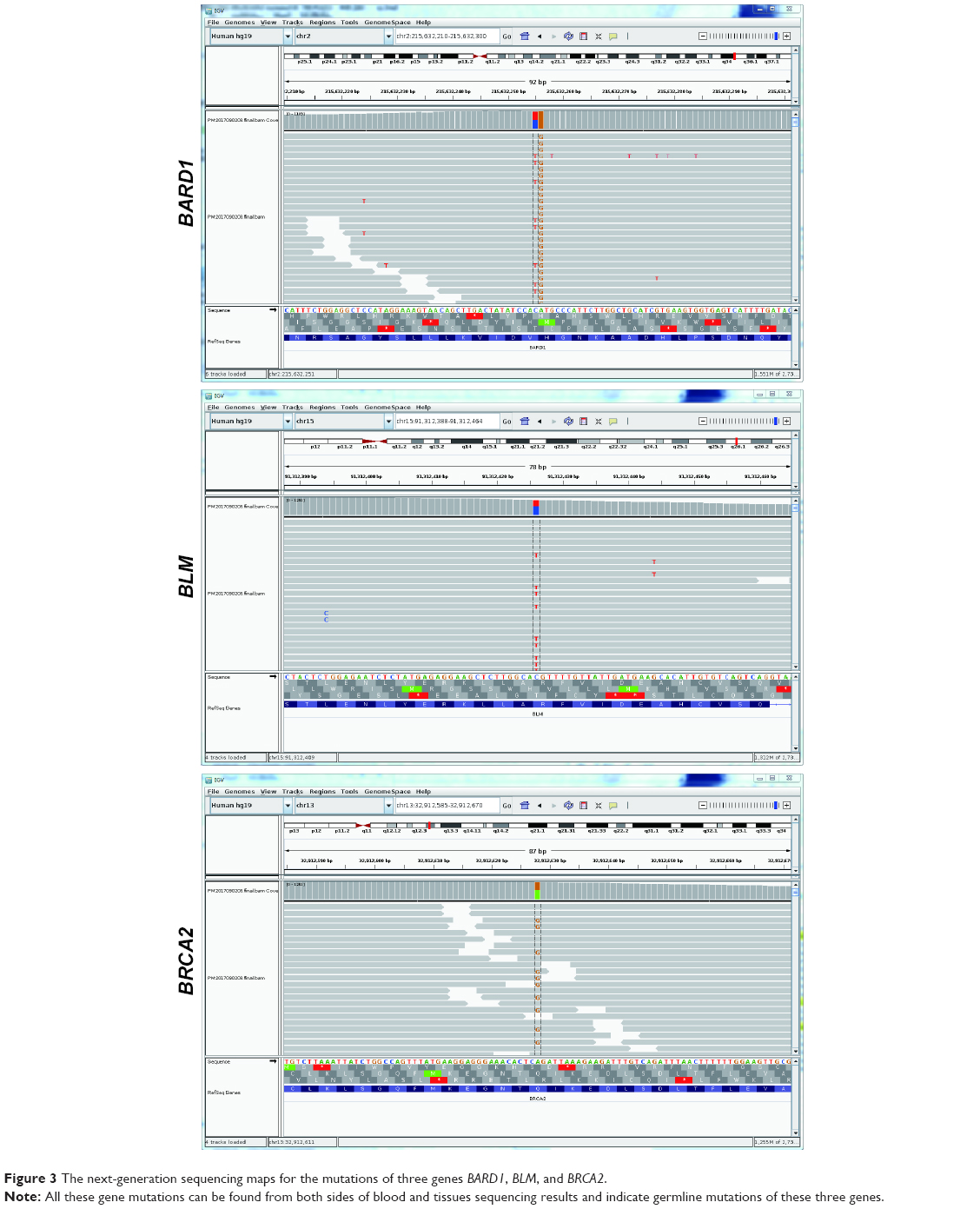 | Figure 3 The next-generation sequencing maps for the mutations of three genes BARD1, BLM, and BRCA2. |
Written informed consent was obtained from the patient for the publication of this case report and the accompanying images.
Discussion
LAM is a devastating female-predominant pulmonary disease characterized by the accumulation of abnormal smooth muscle-like cells in the lung parenchyma and severe emphysema-like lung destruction that can lead to respiratory failure and death.4–7 LAM may be related to changes of female hormones in the body. LAM occurs both sporadically (sLAM) and in association with TSC (TSC-LAM). sLAM has a rare prevalence of 1:1,000,000, whereas TSC-LAM is more common, as approximately 30% of women with TSC have coexisting LAM.8,9 To a certain extent, the biological behavior of LAM is similar to that of low-grade neoplasms, and LAM can also metastasize.10
To diagnose LAM is difficult. In general, there are no specific symptoms at an early stage in patients with LAM, and many symptoms are similar to those of other lung conditions (such as asthma, chronic obstructive pulmonary disease, and bronchitis). Symptoms vary, but the two most frequent clinical presentations of LAM are recurrent pneumothoraces and dyspnea. Less frequently, the first manifestation of LAM is a chylous effusion, abdominal or pelvic tumor, hemoptysis, abdominal hemorrhage caused by a renal angiomyolipoma, or the incidental discovery of lung cysts and/or abdominal tumors.2,3,11–14 To our knowledge, Ye et al reported in a study of 120 patients with LAM that there was dyspnea in 97.5%, cough in 48.3%, pneumothoraces in 42.5%, bloodstained phlegm in 40.8%, chylothorax in 28.3%, chest pain in 19.2%, and chyloperitoneum in 5.8%. Patients with LAM mostly experience an airflow obstruction and/or reduced lung-diffusing capacity.15 According to an article reported by Zhang et al, among 86 patients, the most common form of pulmonary dysfunction was an obstructive ventilation dysfunction (65.1%, 56/86) and a diffusion dysfunction (38.4%, 33/86).16 In our case, the patient experienced a cough with bloodstained phlegm for 3 days. His pulmonary function test result was normal since the patient was at an early stage.
It is important to make a proper diagnosis. The European Respiratory Society guidelines state that we need to have a CT scan showing lung cysts, as well as another piece of evidence, for example, tuberous sclerosis, a kidney tumor, or a chylous effusion. It is helpful to obtain a blood test showing a high level of a VEGF-D protein. In the presence of consistent clinical data and characteristic radiologic findings, serum levels of VEGF-D equal to or greater than 800 pg/mL are now accepted as being diagnostic of LAM because they are extremely rare in other cystic lung diseases.17–19 Unfortunately, we do not have an efficient method to test for VEGF-D. In rare cases in which a diagnosis cannot be made from a combination of the abovementioned symptoms, it is necessary to obtain a biopsy. Tissue samples are collected from the lungs and tested, wherein the tissue may show the histological features of LAM that include immunoreactivity of LAM cells with monoclonal antibody HMB-45.20 In addition, LAM cells exhibit positive immunoreactivity for ER and PR, β-catenin, E-cadherin, and EGFR.20–25 As indicated in Zhang et al’s report on 81 patients, the most common form of immunoreactivity was observed for HMB-45 (69.1%, 56/81) and SMA (46.9%, 38/81), followed by PR (43.2%, 35/81), ER (42.0%, 34/81), actin (34.6%, 28/81), desmin (29.6%, 34/81), CD34 (12.3%, 10/81), F-VIII (9.9%, 8/81), gp100 (6.2%, 5/81), and vimentin (4.9%, 4/81).16 Immunohistochemical staining of our patient was positive for CD34, D2-40, SMA, and desmin. In addition, the antipeptide antibody αPEP13h reacting with a C-peptide terminal of the Pmel17 protein identifies over 82% of the LAM cells in lung nodules, but HMB-45 identifies only 25%.26 As LAM is such a rare condition, it is important to obtain a proper opinion from a specialist as early as possible and to discuss your individual case with an expert on the condition. Once LAM is diagnosed, effective treatment and management should be offered. Supportive treatment could be administered first for patients with an airflow obstruction and/or reduced lung-diffusing capacity that includes the use of inhalers to make breathing easier and oxygen to help the patients with breathlessness.27 For early pulmonary LAM with a nodule, a wedge/segmental dissection or lobectomy is the first choice. As the disease progresses, most patients will experience more than one pneumothorax (lung collapse) during their lives. Pleurodesis may be recommended to prevent this from reoccurring which can improve the quality of life for women with LAM.27 A total pleural covering with sheets composed of an oxidized regenerated cellulose mesh to wrap the entire visceral pleura can successfully prevent the recurrence of a pneumothorax in LAM; it is minimally invasive and rarely causes a restrictive ventilatory impairment.28 Lung transplant may be an option for some women with advanced LAM, for which no other treatment option exists. Many women with LAM have undergone a successful lung transplant and, as a result, experienced an improved quality of life.27
During treatment, LAM arises from mutations in the TSC genes (TSC1 and TSC2) that normally do not get activated, and the TSC genes encode the proteins, hamartin and tuberin, which combine to form a complex (TSC1–TSC2 complex). Through the mTORC1 pathway, the complex can regulate the mTOR protein that is a necessary factor to regulate cell proliferation and lymphangiogenesis. In LAM, the absence of the TSC1–TSC2 complex leads to the uncontrolled activation of mTOR.29 As pharmacologic inhibitors of mTOR, such as everolimus and sirolimus, can directly inhibit T-lymphocyte proliferation, there is ample evidence that mTOR inhibitors are effective for the treatment of pulmonary and renal LAM.30–34 Li et al reported that IGFBP2 could bind insulin, IGF1, and IGF2 in the circulation to modulate cell survival, migration, and invasion of neoplasms. Targeting IGFBP2 may serve as a potential therapeutic strategy for women with LAM and other female gender-specific neoplasms.35 Another study demonstrated that Syk inhibition is a potential therapy for patients with LAM.36 Recent research involving cells derived from patients with LAM showed that estrogen increased cell resistance to anoikis in vitro that was accompanied by a decreased accumulation of the proapoptotic protein, Bim, an activator of anoikis. Therefore, targeting Bim may be a plausible future treatment strategy for patients with LAM.37
There were few reports so far on the role of the germline mutations of BARD1, BLM, and BRCA2 in LAM. BARD1 is a kind of tumor suppressor gene. BARD protein can combine with BRCA1 protein to form a heterodimer, which can repair the DNA damage and maintain stable cytogenetics.38,39 Mutations in any part of the heterodimer result in decreasing tumor suppressor activity, leading to tumorigenesis. As reported, mutations of the heterodimer can regulate the expression of aromatase to participate in the development of breast cancer.40 BRCA2 is also a kind of tumor suppressor gene. Predominantly presenting in mammary gland cells, BRCA2 gene mutation can lead to the failure of gene repair and the tumorigenesis of breast cancer. Aromatase is an important enzyme in the synthesis of estrogen in vivo. As reported above, estrogen is related to LAM. In the case of our patient, the gene mutations may have been caused by the abnormal level of aromatase, leading to the synthesis of estrogen, and finally the development of LAM. BLM gene is one of the members of the RecQ DNA helicase family. The RecQ DNA helicase family plays an important role in inhibiting human tumorigenesis.41,42 Bloom syndrome is related to the mutation of BLM gene, but the interaction between BLM and various proteins in the DNA metabolism pathway is not yet clear.
Moreover, LAM is a rare female-predominant pulmonary disease with nonspecific symptoms. The diagnosis of LAM must combine a CT scan showing lung cysts, evidence of tuberous sclerosis or a kidney tumor, a chylous effusion, and a serum examination, such as serum levels of VEGF-D. Treatment depends on the stage of the disease, and new targeted drugs are anticipated.
Acknowledgments
This work was financially supported by grants from the National Natural Science Foundation of China (81773207), the Science and Technology Support Key Program of Tianjin (17YFZCSY00840), and Tianjin Key Project of Natural Science Foundation (16JCZDJC34200, 16PTSYJC00160). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
Yi Liu, Zhibin Guo, and Jun Chen wrote this manuscript and analyzed all data. Yi Liu, Zhibin Guo, Chenlong Zhao, and Xin Li provided medical care for the patients and collected the data. Hongyu Liu and Jun Chen revised the article. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Taveira-DaSilva AM, Moss J. Epidemiology, pathogenesis and diagnosis of lymphangioleiomyomatosis. Expert Opin Orphan Drugs. 2016;4(4):369–378. | ||
Cudzilo CJ, Szczesniak RD, Brody AS, et al. Lymphangioleiomyomatosis screening in women with tuberous sclerosis. Chest. 2013;144(2):578–585. | ||
Harknett EC, Chang WY, Byrnes S, et al. Use of variability in national and regional data to estimate the prevalence of lymphangioleiomyomatosis. QJM. 2011;104(11):971–979. | ||
Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer. 2014;14(5):329–341. | ||
Schiavina M, Contini P, Fabiani A, et al. Efficacy of hormonal manipulation in lymphangioleiomyomatosis. A 20-year-experience in 36 patients. Sarcoidosis Vasc Diffuse Lung Dis. 2007;24(1):39–50. | ||
Hammes SR, Krymskaya VP. Targeted approaches toward understanding and treating pulmonary lymphangioleiomyomatosis (LAM). Horm Cancer. 2013;4(2):70–77. | ||
Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817–824. | ||
Freitas CS, Baldi BG, Araújo MS, Heiden GI, Kairalla RA, Carvalho CR. Use of sirolimus in the treatment of lymphangioleiomyomatosis: favorable responses in patients with different extrapulmonary manifestations. J Bras Pneumol. 2015;41(3):275–280. | ||
Johnson SR. Lymphangioleiomyomatosis. Eur Respir J. 2006;27(5):1056–1065. | ||
Wahid S, Chiang PC, Luo HL, Huang SC, Tsai EM, Chiang PH. Pelvic lymphangioleiomyomatosis treated successfully with everolimus: two case reports with literature review. Medicine (Baltimore). 2017;96(10):e4562. | ||
Ryu JH, Moss J, Beck GJ, et al. The NHLBI lymphangioleiomyomatosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med. 2006;173(1):105–111. | ||
McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest. 2008;133(2):507–516. | ||
Ferrans VJ, Yu ZX, Nelson WK, et al. Lymphangioleiomyomatosis (LAM): a review of clinical and morphological features. J Nippon Med Sch. 2000;67(5):311–329. | ||
Matsui K, Tatsuguchi A, Valencia J, et al. Extrapulmonary lymphangioleiomyomatosis (LAM): clinicopathologic features in 22 cases. Hum Pathol. 2000;31(10):1242–1248. | ||
Ye L, Jin M, Bai C. Clinical analysis of patients with pulmonary lymphangioleiomyomatosis (PLAM) in mainland China. Respir Med. 2010;104(10):1521–1526. | ||
Zhang LJ, Liang Y, Zhong XN, et al. Literature review of clinical and pathological features of pulmonary lymphangioleiomyomatosis for 130 cases in China in the last thirty years. Chin Gen Pract. 2015;18(3):329–334. | ||
Glasgow CG, Avila NA, Lin JP, Stylianou MP, Moss J. Serum vascular endothelial growth factor-D levels in patients with lymphangioleiomyomatosis reflect lymphatic involvement. Chest. 2009;135(5):1293–1300. | ||
Young LR, Vandyke R, Gulleman PM, et al. Serum vascular endothelial growth factor-D prospectively distinguishes lymphangioleiomyomatosis from other diseases. Chest. 2010;138(3):674–681. | ||
Young L, Lee HS, Inoue Y, et al. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med. 2013;1(6):445–452. | ||
Matsumoto Y, Horiba K, Usuki J, Chu SC, Ferrans VJ, Moss J. Markers of cell proliferation and expression of melanosomal antigen in lymphangioleiomyomatosis. Am J Respir Cell Mol Biol. 1999;21(3):327–336. | ||
Grzegorek I, Lenze D, Chabowski M, et al. Immunohistochemical evaluation of pulmonary lymphangioleiomyomatosis. Anticancer Res. 2015;35(6):3353–3360. | ||
Flavin RJ, Cook J, Fiorentino M, Bailey D, Brown M, Loda MF. β-Catenin is a useful adjunct immunohistochemical marker for the diagnosis of pulmonary lymphangioleiomyomatosis. Am J Clin Pathol. 2011;135(5):776–782. | ||
Barnes EA, Kenerson HL, Mak BC, Yeung RS. The loss of tuberin promotes cell invasion through the β-catenin pathway. Am J Respir Cell Mol Biol. 2010;43(5):617–627. | ||
Barnes EA, Kenerson HL, Jiang X, Yeung RS. Tuberin regulates E-cadherin localization: implications in epithelial-mesenchymal transition. Am J Pathol. 2010;177(4):1765–1778. | ||
Lesma E, Grande V, Ancona S, Carelli S, Di GAM, Gorio A. Anti-EGFR antibody efficiently and specifically inhibits human TSC2−/− smooth muscle cell proliferation. Possible treatment options for TSC and LAM. PLoS One. 2008;3(10):e3558. | ||
Valencia JC, Steagall WK, Zhang Y, et al. Antibody αPEP13h reacts with lymphangioleiomyomatosis cells in lung nodules. Chest. 2015;147(3):771–777. | ||
Lymphangioleiomyomatosis (LAM). Breathe (Sheff). 2017;13(1):64–71. | ||
Kurihara M, Mizobuchi T, Kataoka H, et al. A total pleural covering for lymphangioleiomyomatosis prevents pneumothorax recurrence. PLoS One. 2016;11(9):e0163637. | ||
Huang J, Manning BD. The TSC1–TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412(2):179–190. | ||
McCormack FX, Inoue Y, Moss J, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364(17):1595–1606. | ||
Neurohr C, Hoffmann AL, Huppmann P, et al. Is sirolimus a therapeutic option for patients with progressive pulmonary lymphangioleiomyomatosis. Respir Res. 2011;12:66. | ||
Mavroudi M, Zarogoulidis P, Katsikogiannis N, et al. Lymphangioleiomyomatosis: current and future. J Thorac Dis. 2013;5(1):74–79. | ||
Courtwright AM, Goldberg HJ, Henske EP, El-Chemaly S. The effect of mTOR inhibitors on respiratory infections in lymphangioleiomyomatosis. Eur Respir Rev. 2017;26(143). | ||
Kennedy BK, Pennypacker JK. Mammalian target of rapamycin: a target for (lung) diseases and aging. Ann Am Thorac Soc. 2016;13 Suppl 5:S398–S401. | ||
Li X, Liu X, Zhang L, et al. Insulin growth factor binding protein 2 mediates the progression of lymphangioleiomyomatosis. Oncotarget. 2017;8(22):36628–36638. | ||
Cui Y, Steagall WK, Lamattina AM, et al. Aberrant SYK kinase signaling is essential for tumorigenesis induced by TSC2 inactivation. Cancer Res. 2017;77(6):1492–1502. | ||
Li C, Li N, Liu X, et al. Proapoptotic protein Bim attenuates estrogen-enhanced survival in lymphangioleiomyomatosis. JCI Insight. 2016;1(19):e86629. | ||
Xia Y, Pao GM, Chen HW, Verma IM, Hunter T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J Biol Chem. 2003;278(7):5255–5263. | ||
Atipairin A, Canyuk B, Ratanaphan A. Substitution of aspartic acid with glutamic acid at position 67 of the BRCA1 RING domain retains ubiquitin ligase activity and zinc(II) binding with a reduced transition temperature. J Biol Inorg Chem. 2011;16(2):217–226. | ||
Lu Y, Kang T, Hu Y. BRCA1/BARD1 complex interacts with steroidogenic factor 1 – a potential mechanism for regulation of aromatase expression by BRCA1. J Steroid Biochem Mol Biol. 2011;123(1–2):71–78. | ||
Doerfler L, Harris L, Viebranz E, Schmidt KH. Differential genetic interactions between Sgs1, DNA-damage checkpoint components and DNA repair factors in the maintenance of chromosome stability. Genome Integr. 2011;2:8. | ||
Singh DK, Ahn B, Bohr VA. Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology. 2009;10(3):235–252. |
Supplementary material
 | Table S1 List of the detected 518 genes |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.