")
Back to Journals » International Journal of Nanomedicine » Volume 12
Lymph cancer chemotherapy: delivery of doxorubicin-gemcitabine prodrug and vincristine by nanostructured lipid carriers
Authors Ni S, Qiu L, Zhang G, Zhou H, Han Y
Received 25 August 2016
Accepted for publication 21 November 2016
Published 27 February 2017 Volume 2017:12 Pages 1565—1576
DOI https://doi.org/10.2147/IJN.S120685
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Shuqin Ni, Lei Qiu, Guodong Zhang, Haiyan Zhou, Yong Han
Department of Internal Medicine Oncology, Shandong Cancer Hospital Affiliated to Shandong University, Shandong Academy of Medical Science, Ji’nan, Shandong, People’s Republic of China
Purpose: Radiation and chemotherapy are the most common course of treatment for B-cell lymphoma. Doxorubicin (DOX), gemcitabine (GEM), and vincristine (VCR) are the commonly used antilymphoma chemotherapeutic drugs. The aim of this study is to construct a novel drug delivery system for the combination delivery of the three drugs on lymphoma.
Materials and methods: DOX–GEM prodrug was synthesized. Novel nanostructured lipid carriers (NLCs) containing DOX–GEM prodrug and VCR were prepared and used to treat B-cell lymphoma through in vivo treatment to a lymph cancer animal model. The systemic toxicity of the nanomedicine was also evaluated during the treatment.
Results: DOX–GEM prodrug and VCR-loaded NLCs (DOX–GEM VCR NLCs) exhibited the highest antitumor effect in B-cell lymphoma cells and lymphoma animal xenografts when compared with the single drug-loaded NLCs and the drug solutions.
Conclusion: It could be concluded that the highest antitumor effect can be achieved by the system due to the stable drug-loading capacity, attractive anticancer therapeutic effects, and reduced toxicities in human Burkitt’s lymphoma cell line and mice-bearing cancer model. The resulting DOX–GEM VCR NLCs could be an efficient antilymph cancer agent and could be developed further for the treatment of other tumors.
Keywords: combination chemotherapy, B-cell lymphoma, prodrug, nanostructured lipid carriers, drug delivery
Introduction
Lymphoma is the fifth most common cancer that originates within the lymphocytes of secondary lymphoid organs and extranodal tissue.1 Lymphoma cases are nominally classified into the following two types: Hodgkin lymphoma and non-Hodgkin lymphoma (NHL). NHL (90% from B cells and 10% from T cells) is about seven times more prevalent than Hodgkin lymphoma.2 Until recently, radiation and chemotherapy are the most common courses of treatment for B-cell lymphoma. Based on the NCCN (National Comprehensive Cancer Network) NHL clinical practice guidelines, the chemotherapy regimen (CHOP) (cyclophosphamide, doxorubicin [DOX], vincristine [VCR] sulfate, and prednisone) is the first-line therapy for B-cell lymphomas, accepted worldwide. Although the overall survival outcomes of the traditional chemotherapy regimen are up to 70%, relapse and drug resistance remain a clinical challenge; even more, there are still 50%–60% of aggressive NHL patients existing with recurrence and lack of novel regimens to treat.3 Therefore, novel combination therapies are urgently needed to enhance therapeutic efficacies and reduce adverse effects.
Chemotherapeutics for lymphoma are typically administered systemically and act through diverse intracellular mechanisms, including DNA cross-linking, mitosis inhibition, and DNA strand breakage.1,4–7 The single drug regimen, such as liposomal DOX,1,8 gemcitabine (GEM),9–14 and liposomal VCR,1,15 has been proven to treat B-cell lymphoma in clinical trials. Based on their (DOX, GEM and VCR [DGV]) different mechanisms of action, the combination chemotherapies of DGV are effective and well tolerated in relapse and refractory NHL.16–18 However, free drugs without nano-based drug delivery system have severe toxicity or drug resistance: free DOX has side effects of irreversible cardiomyopathy and heart failure, which limit its clinical use;19–21 free GEM has a short half-life (ranged from 42 to 92 min); and free VCR has severe side effects, such as dose-dependent neurotoxicity and MDR.22,23 Therefore, prodrug-based nanodrug delivery systems (DOX–GEM prodrug and VCR nanostructured lipid carriers [NLCs]) are designed in our group and anticipated to solve these problems.
Compared to combinational therapy with the physical technology, combinatorial drug conjugation strategy, covalently conjugating multiple therapeutic agents through hydrolyzable linkers to form drug conjugates, has the following advantages: unifying the pharmacokinetics and cellular uptake of various drug molecules and maximizing the combinatorial effects.24 In this study, DOX–GEM was synthesized as the prodrug and delivered by lipid nanoparticles.
Solid lipid nanoparticles (SLNs) have come up as the latest development in the arena of lipid-based colloidal delivery systems after nanoemulsion and liposomes ever since their introduction in the early 1990s.25 SLNs combine the advantages of the traditional systems and avoid some of their major disadvantages, including controllable drug release, enhanced bioavailability of poorly water-soluble drugs, and alleviated drug efflux to overcome multidrug resistance.26,27 NLCs, a new generation of SLNs, consist of a solid lipid matrix with a certain amount of liquid lipid. NLCs have some advantages over SLNs: increasing the payload of drugs, preventing the drug expulsion, and improving the drug stability.28–30
Accordingly, DOX–GEM prodrug was synthesized, and DOX–GEM- and VCR-loaded NLCs (DOX–GEM VCR NLCs) were designed and examined in mice-bearing human Burkitt’s lymphoma cell line (Raji) model. This system was expected to achieve stable drug-loading (DL) capacity, attractive anticancer therapeutic effects, and reduced toxicities.
Materials and methods
Materials, cells, and animals
DOX, GEM, VCR, glutaric anhydride (GA), pyridine, and dimethylaminopyridine (DMAP) were purchased from Sigma-Aldrich (St Louis, MO, USA). Distearoyl phosphatidyl-ethanolamine (DSPE) was purchased from Avanti Polar Lipids (Alabaster, AL, USA). Precirol ATO 5 was generously provided by Gattefossé (Paramus, NJ, USA). Injectable soya lecithin was obtained from Shanghai Taiwei Pharmaceutical Co, Ltd (Shanghai, People’s Republic of China). All other chemicals used were at least reagent grade and obtained from Sigma-Aldrich or Merck (Darmstadt, Germany) and were used without further purification.
A human Burkitt’s lymphoma cell line, Raji, was obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). Raji cells were cultured in RPMI-1640 supplemented with 10% heat-inactivated fetal bovine serum (Gibco, Waltham, MA, USA). Cells were grown as suspension cultures and maintained in a humidified atmosphere at 37°C and 5% CO2. The cells were washed twice with phosphate-buffered saline (PBS) before use. The VCR-resistant lymphoma cell line, Raji/VCR, was generated from Raji cells following the way of exposing the cells to VCR at a concentration of 2 μg/mL.31
BALB/c nude mice (4–6 weeks old, 18–22 g weight) were purchased from the Medical Animal Test Center of Shandong University (Ji’nan, People’s Republic of China) and were maintained under specific pathogen-free conditions. All animal-handling procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and followed the guidelines of the Animal Welfare Act. All animal experiments were approved by the Experimental Animal Ethical Committee of Shandong University. A suspension of 5×107 Raji cells in 100 μL PBS was injected subcutaneously on the right flanks of a nude mouse. Tumor volumes were measured in two dimensions by using a vernier caliper. Tumor volumes were calculated using the formula: (length × width2)/2.
Synthesis of DOX–GEM prodrug
GEM (0.1 mmol) was dissolved in 10 mL N, N-dimethylformamide (DMF). GEM solution was added into GA–pyridine solution (0.1 mmol/mL, 3 mL) (Figure 1).24 Then, DMAP (0.01 mmol) dissolved in pyridine (0.2 mL) was added into the GEM–GA solution and stirred under 600 rpm at room temperature for 4 h. Then, the reaction was quenched by diluting the solution with dichloromethane (DCM). DMAP and pyridine were removed by extracting with deionized water. Finally, crude GEM–GA was obtained under drying in vacuum. The crude product was purified by high performance liquid chromatography (HPLC).
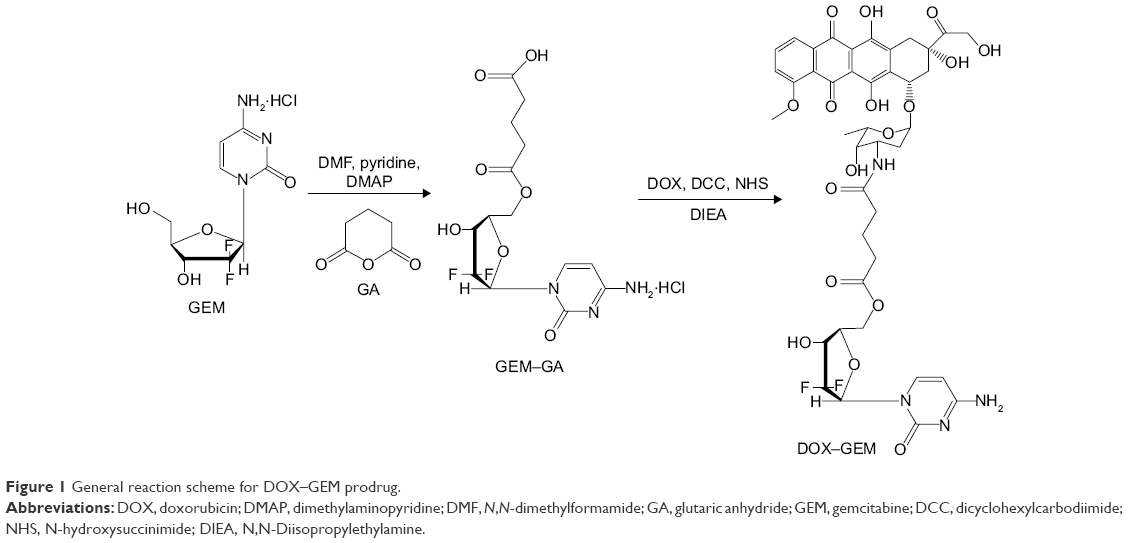 | Figure 1 General reaction scheme for DOX–GEM prodrug. |
GEM–GA and DOX (1:1, molar ratio) were dissolved in dimethyl sulfoxide (DMSO). Dicyclohexylcarbodiimide, N-hydroxysuccinimide, and N,N-Diisopropylethylamine (1:1:2, molar ratio) were then sequentially added into the DMSO solution.32 The reaction mixture was stirred at 600 rpm at room temperature for 4 h in the dark. Finally, DOX–GEM was obtained via rotary evaporation method and purification by HPLC.
Formulation of DOX–GEM VCR NLCs
DOX–GEM VCR NLCs were developed using the solvent diffusion method.33 Drug (1 g of DOX–GEM and 0.5 g of VCR), Precirol ATO 5 (5 g), and injectable soya lecithin (10 g) were mixed in a 50 mL solvent mixture of ethanol and acetone (1:1, v/v) followed by bath sonication for 10 min. The obtained mixture was kept on a water bath maintained at 60°C to make a clear solution of lipids and drug in an organic solvent system. The resultant organic mixture was hastily added into 100 mL of an aqueous phase comprising of polyvinyl acetate as stabilizer kept on water bath maintained at 70°C under mechanical agitation of 500 rpm for 10 min. The obtained DOX–GEM VCR NLC dispersion was cooled at room temperature for 4 h on magnetic stirrer for the liberation of organic solvent. The prepared DOX–GEM VCR NLC dispersion was transferred to centrifuge tubes equipped with cooling centrifuge, and centrifugation was carried out for 15,000 rpm and 20 min at −10°C to separate the precipitated DOX–GEM VCR NLCs.
DOX–GEM-loaded NLCs (DOX–GEM NLCs) were formulated using the same method described earlier without VCR.
VCR-loaded NLCs (VCR NLCs) were formulated using the same method described earlier without DOX–GEM.
Blank NLCs (NLCs) were formulated using the same method described earlier without DOX–GEM and VCR.
Characterization of DOX–GEM VCR NLCs
Morphology
To observe the morphology of DOX–GEM VCR NLCs, 2 μL of sample suspended in deionized water was placed on a carbon film coated on a copper grid and stained with 1% uranyl acetate for 30 s. The grid was allowed to dry for 10 min and then examined using a JEM-200 CX transmission electron microscope (TEM; JEOL, Tokyo, Japan).34
Particle size and surface charge
The particle size and zeta potential values of the NLCs were determined at pH 7.4 and at 25°C, using dynamic light scattering (DLS) in a Zetasizer NS (Malvern Instruments, Malvern, UK), by photon correlation spectroscopy (PCS) and by electrophoretic laser Doppler anemometry, respectively.35
DL efficiency and encapsulation efficiency (EE)
The DL efficiency and EE of drug-loaded NLCs were measured as follows: free DOX, GEM, and VCR were removed from NLCs by ultrafiltration (Millipore, Boston, MA, USA).
The amount of DOX loaded in the NLCs was determined by fluorescence at 480 nm with the maximum absorption at 590 nm.36
The amount of GEM incorporated in the NLCs was determined by HPLC.37 The NLCs’ suspension was diluted by adding water and acetonitrile (60:40) to a total volume of 0.5 mL. The mixture was then centrifuged for 15 min at 300 μg, after which 3.0 mL of the organic layer was transferred and evaporated to dryness under nitrogen stream and injected into a Symmetry C18 column. The detection was carried out by ultra violet adsorption measurement at 248 nm.
The amount of VCR loaded in the NLCs was determined by HPLC.38 Briefly, ethanol was added to disrupt the NLCs, and 20 μL of the resulting transparent solution was injected into an HPLC system (Agilent 1260; Agilent Technologies, Santa Clara, CA, USA). A Kromasil C18 reverse phase column (150×4.6 mm, 5 μm; AkzoNobel, Separation Products, Bohus, Sweden) and the mobile phase consisting of acetonitrile and 0.01 M NaH2PO4 (55/45, v/v, pH 7.0 adjusted with triethylamine) were used to separate the targeted component. The samples were eluted by the mobile phase at a flow rate of 1.0 mL/min at 35°C and monitored at 297 nm.
In vitro drug release
The drug release behavior of DOX–GEM VCR NLCs, DOX–GEM NLCs, and VCR NLCs was measured by the dialysis method.39 Samples were placed in the dialysis bag separately. Then, the bag was incubated with 50 mL of PBS (pH 7.4, containing 0.1% Tween 80). The medium (1 mL) was collected at predetermined time points and replaced with 50 mL of fresh medium. The concentrations of released drugs were determined by the method described in the “DL efficiency and encapsulation efficiency (EE)” section.
In vitro cytotoxicity
In vitro cytotoxicity was evaluated by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay.40 Raji cells were seeded at 1×105 cells/mL in 96-well plates using Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum and then incubated with DOX–GEM VCR NLCs, DOX–GEM NLCs, VCR NLCs, NLCs, free DOX–GEM, free VCR, and physical mixture of DOX–GEM and VCR (DOX–GEM/VCR, 2:1, w/w) for 48 h. Free drugs and physical mixture of drugs were dissolved with DMSO. Cells treated with Dulbecco’s Modified Eagle Medium served as controls. Then, 25 μL of MTT solution (5 mg/mL) was added to each well. The plate was incubated at 37°C for 3 h. After incubation, 100 μL of DMSO was added. The absorbance was measured at 570 nm using a microplate reader.
In vivo biodistribution
Biodistribution experiments were carried out in BALB/c nude mice, previously inoculated with Raji cells.41 Both DOX–GEM VCR NLCs and DOX–GEM/VCR (both contained 10 mg/kg of DOX–GEM and 5 mg/kg of VCR) were sterilized by filtering through 0.22 μm sterile filters and administered via the tail vein. The mice were sacrificed by cervical dislocation at predefined time periods (0.5, 2, 4, 8, and 12 h). The tumors, hearts, kidneys, livers, lungs, and spleens were harvested, washed, weighed, and homogenized. Tissue concentrations of drugs were determined by the method described in the “DL efficiency and encapsulation efficiency (EE)” section.
In vivo antitumor activity
Raji cells (1×107) in 100 μL were inoculated subcutaneously into the lateral flank of BALB/c nude mice.31 When the tumors reached ~500 mm3 in volume, mice were randomly assigned to seven groups with six each for the treatment of PBS, DOX–GEM VCR NLCs, DOX–GEM NLCs, VCR NLCs, DOX–GEM/VCR, free DOX–GEM, and free VCR via the tail vein every 3 days. At the determined time points, mice were sacrificed by cervical dislocation and the tumor size was measured in two perpendicular diameters with precision calipers and calculated in a range of 3 weeks. Tumor volume was measured according to the following formula:
|
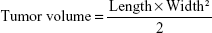
where length and width refer to the longest and the shortest diameters of tumors, respectively.
The antitumor efficacy of each formulation was evaluated by tumor inhibition efficiency (TIE), which was calculated by measuring the tumor weight using the following formula:
|

The body weight was measured simultaneously as an indicator of systemic toxicity.
In vivo pharmacokinetic studies
The pharmacokinetic studies of drug-loaded NLCs and free drugs were carried out in BALB/c mice. DOX–GEM VCR NLCs, free DOX, free GEM, and free VCR (containing 10 mg/kg of DOX–GEM, 6.9 mg/kg of DOX, 3.1 mg/kg of GEM, and 5 mg/kg of VCR, respectively) were sterilized by filtering through a 0.22 μm sterile filters and administered via the tail vein. Blood samples were collected at 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, 12, 16, 24, and 48 h. The blood samples were centrifuged at 15,000 rpm for 3 min at 4°C and then stored at −80°C prior to HPLC analysis.
Statistical analysis
Data were expressed as the mean ± SD. Statistical analysis was performed by Student’s unpaired t-test or one-way analysis of variance to identify significant differences unless otherwise indicated. Differences were considered significant at a P-value of <0.05.
Results
Synthesis and characterization of DOX–GEM prodrug
Figure 1 describes the synthesis of DOX–GEM prodrug. GEM–GA was first obtained by linking GEM and GA using ester bond. DOX–GEM was then prepared by the reaction between the carboxyl groups of DOX–GEM and amino groups of DOX. Chemical structures of DOX–GEM were confirmed using 1H nuclear magnetic resonance spectroscopy. As shown in Figure 2, 1H nuclear magnetic resonance (DMSO-d6, 300 mHz) δ (ppm): 1.27–1.79 (–CH2–), 1.98 (–OH), 2.26 (–CH2–C=O–O–), 2.46–2.53 (–CH2N–), 3.16–3.39 (–CH–), 3.77 (–C=OCH2–), 4.21 (O=C–O–CH2–), 4.39 (–NHC=O), 6.39 (–NHCO–CH=), and 8.08 (–NH). The production rate is 73.4%.
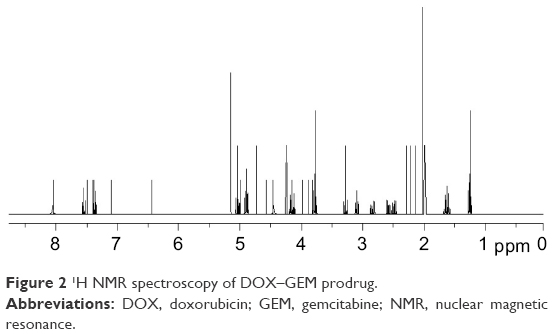 | Figure 2 1H NMR spectroscopy of DOX–GEM prodrug. |
Characterization of DOX–GEM VCR NLCs
TEM image shows that the DOX–GEM VCR NLCs were dispersed in the solution and the particle shape was uniform (Figure 3). The average particle size of the DOX–GEM VCR NLCs determined by DLS was 112.6 nm (Table 1). The size of blank NLCs, DOX–GEM NLCs, and VCR NLCs was ~110.9, 113.1, and 11.8 nm, respectively. The polydispersity index of four kinds of NLCs was between 0.1 and 0.2. The zeta potential of blank NLCs and DOX–GEM VCR NLCs was −26.4 and −39.7 mV, respectively. The EE of drugs loaded in NLCs was all over 85%. The DL of various drugs in different systems was between 4.6% and 10.1%.
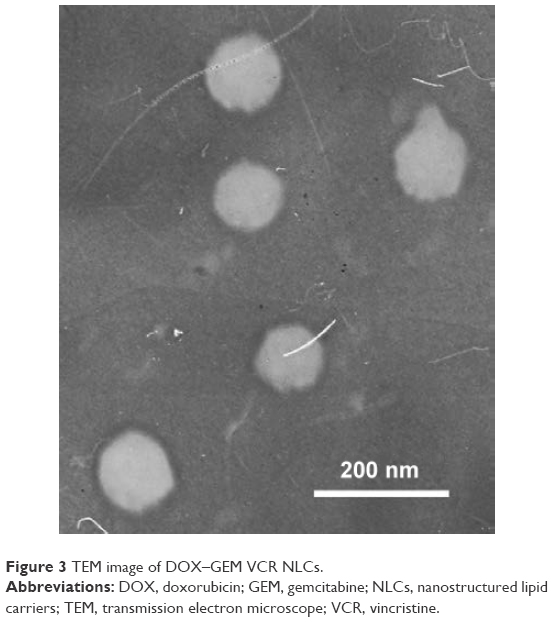 | Figure 3 TEM image of DOX–GEM VCR NLCs. |
In vitro drug release
In vitro release profile of DOX, GEM, and VCR from DOX–GEM VCR NLCs, DOX–GEM NLCs, and VCR NLCs showed a sustained release behavior (Figure 4). There was no significant difference in DOX release and GEM release between DOX–GEM VCR NLCs and DOX–GEM NLCs (Figure 4A and B). The in vitro release profiles of VCR from DOX–GEM VCR NLCs and VCR NLCs showed no obvious difference (Figure 4C).
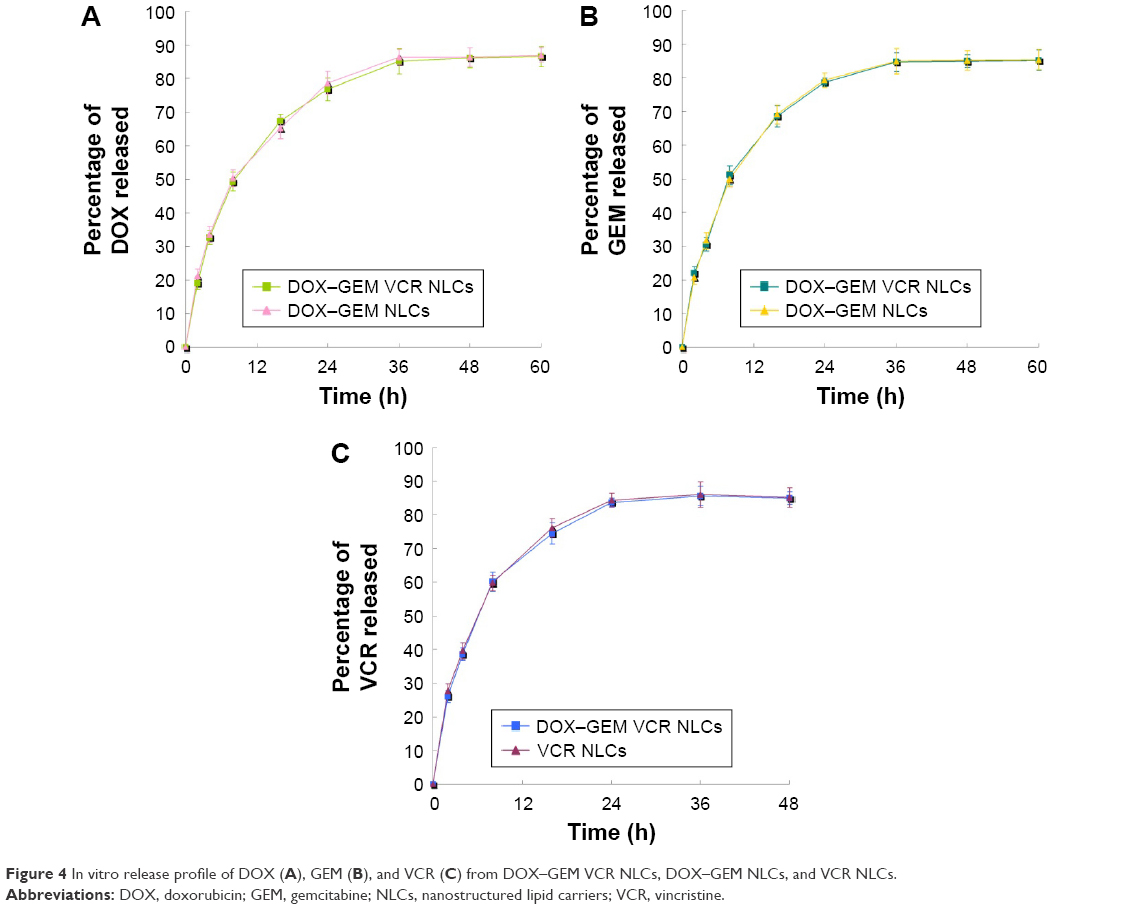 | Figure 4 In vitro release profile of DOX (A), GEM (B), and VCR (C) from DOX–GEM VCR NLCs, DOX–GEM NLCs, and VCR NLCs. |
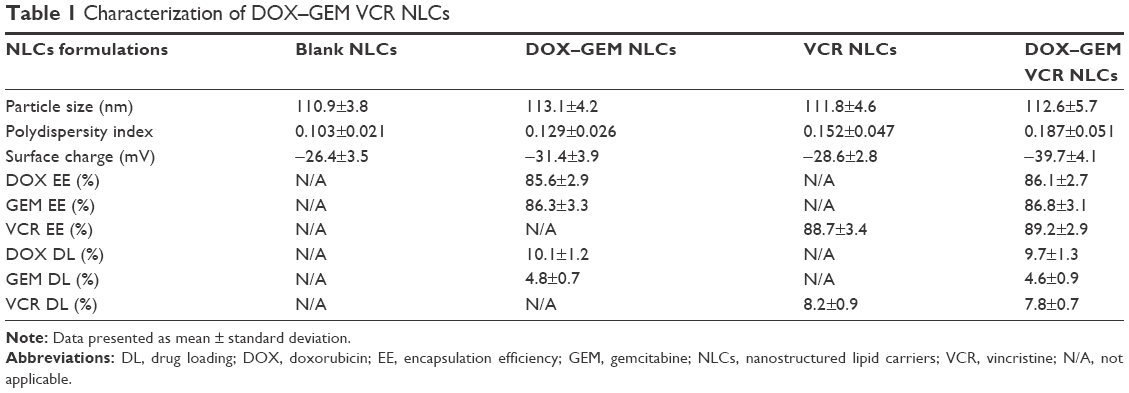 | Table 1 Characterization of DOX–GEM VCR NLCs |
In vitro cytotoxicity
In vitro viability of Raji cells treated with different formulations is illustrated in Figure 5. Blank NLCs without drugs showed high cell viability (88.1%). Significant inhibitory effects of drug solutions and drugs loaded in NLCs were observed at the concentrations of 1–10 μg/mL, and the toxicity conformed to a concentration-dependent pattern. Moreover, drugs loaded in NLCs showed significantly higher cytotoxicity than drug solutions (P<0.05). DOX–GEM VCR NLCs exhibited the highest cytotoxic effect among all samples tested. The half maximal inhibitory concentration values of DOX–GEM VCR NLCs, DOX–GEM NLCs, VCR NLCs, DOX–GEM/VCR, free DOX–GEM, and free VCR are 0.21, 0.52, 0.79, 4.85, 7.92, and 9.13 μg/mL, respectively.
 | Figure 5 In vitro viability of Raji cells treated with different formulations. |
In vivo biodistribution
In vivo tissue distributions of free drugs and drugs loaded in NLCs were investigated in lymph cancer-bearing mice (Figure 6). Drugs were widely distributed in most tissues following intravenous administration of DOX–GEM/VCR. By contrast, the drug concentration in the tumors, livers, spleens, and lungs was higher for the drugs loaded in the NLC group than for the free drugs group (P<0.05). On the opposite, the drug concentration in the heart and kidney for NLCs group was much lower (P<0.05).
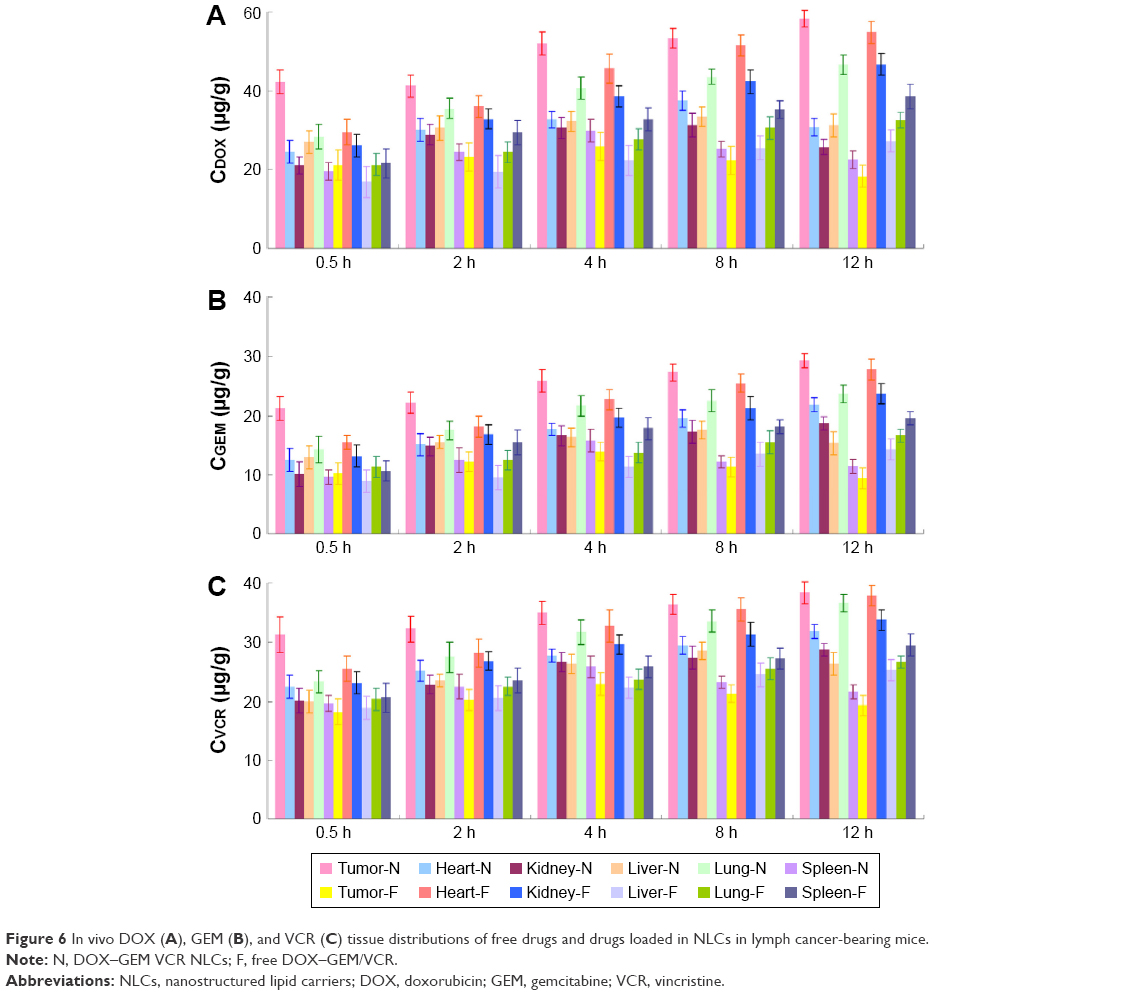 | Figure 6 In vivo DOX (A), GEM (B), and VCR (C) tissue distributions of free drugs and drugs loaded in NLCs in lymph cancer-bearing mice. |
In vivo antitumor activity
In vivo antitumor activity of free drugs and drugs in loaded NLCs was investigated in lymph cancer-bearing mice (Figure 7). Free drugs and drugs loaded in NLCs groups tested showed a significant tumor volume inhibition effect in tumor-bearing mice. At 3 weeks of administration, tumor weight and TIE of tumor-bearing mice were summarized (Table 2). DOX–GEM VCR NLCs exhibited the highest TIE (86.1%). The obvious emaciation could be observed in the free drug solutions groups and the PBS control group, while the DOX–GEM VCR NLC group did not cause a significant difference in body weight lost (Figure 8). During the treatment, reduction in food intake, energy sag, and inactive in moving were also observed in the free drug solution groups but not in the NLCs groups.
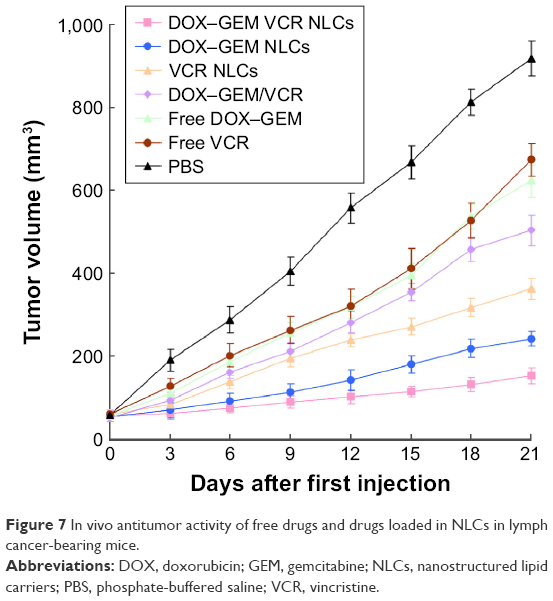 | Figure 7 In vivo antitumor activity of free drugs and drugs loaded in NLCs in lymph cancer-bearing mice. |
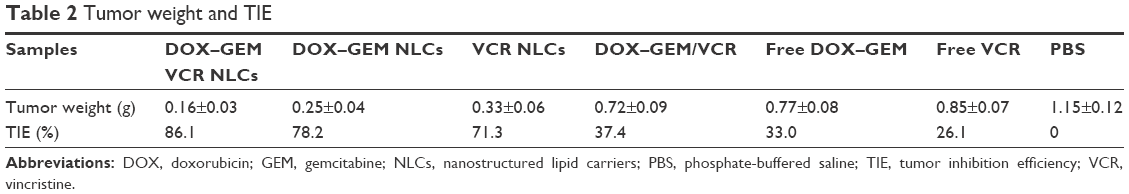 | Table 2 Tumor weight and TIE |
 | Figure 8 Body weight changes of lymph cancer bearing mice when injected with free drugs and drugs loaded in NLCs. |
In vivo pharmacokinetic studies
The pharmacokinetics of DOX, GEM, and VCR after the administration of drug-loaded NPs or free drugs is shown in Figure 9. Key pharmacokinetic parameters were calculated from these data and are presented in Tables 3–5. For DOX, the area under the curve (AUC), mean residence time (MRT), and t1/2 were significantly higher than free DOX. For instance, the AUC values of DOX–GEM VCR NLCs were 13.25 and 4.73 h μg/mL for DOX–GEM VCR NLCs and free DOX, respectively. For GEM, the AUC, MRT, and t1/2 were significantly higher than free GEM. Similarly, for VCR, the AUC, MRT, and t1/2 were significantly higher than free GEM.
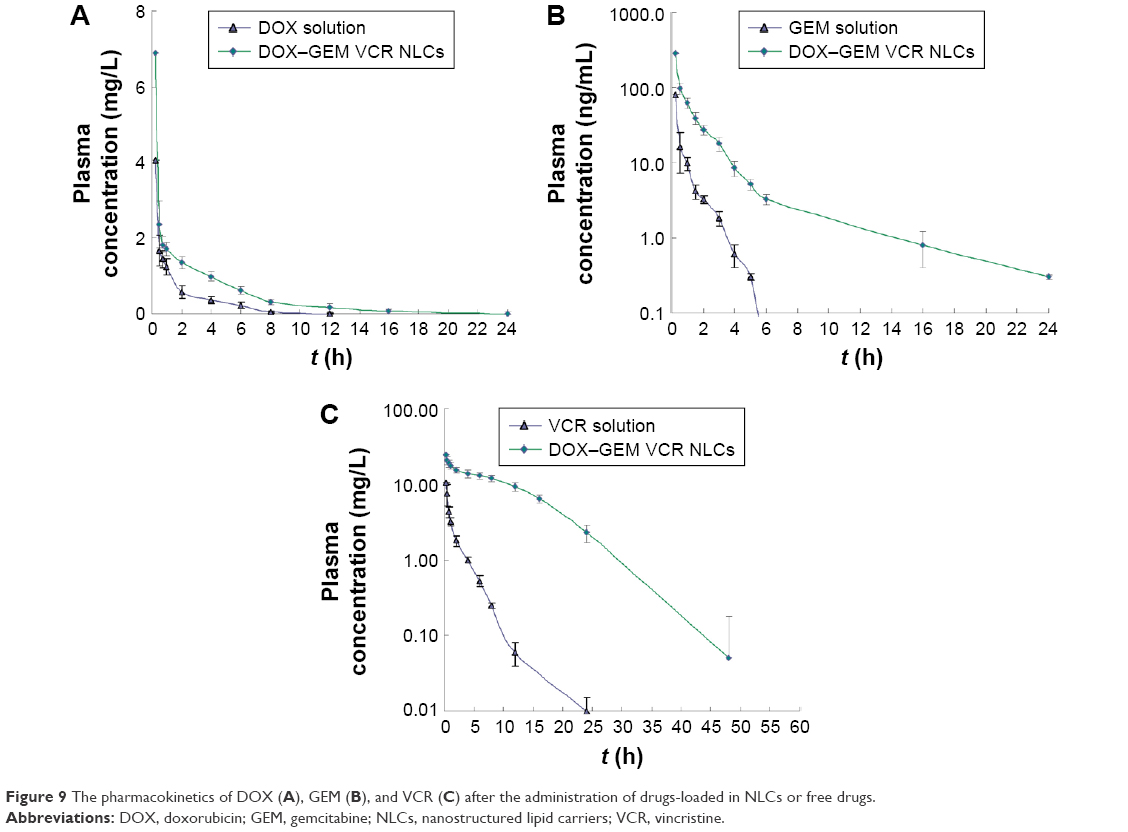 | Figure 9 The pharmacokinetics of DOX (A), GEM (B), and VCR (C) after the administration of drugs-loaded in NLCs or free drugs. |
 | Table 3 Pharmacokinetic parameters of DOX after intravenous administration of DOX–GEM VCR NLCs and DOX solution to mice (10 mg/kg DOX–GEM, 6.9 mg/kg DOX) |
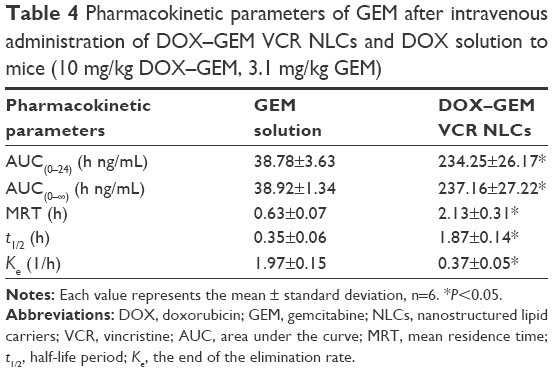 | Table 4 Pharmacokinetic parameters of GEM after intravenous administration of DOX–GEM VCR NLCs and DOX solution to mice (10 mg/kg DOX–GEM, 3.1 mg/kg GEM) |
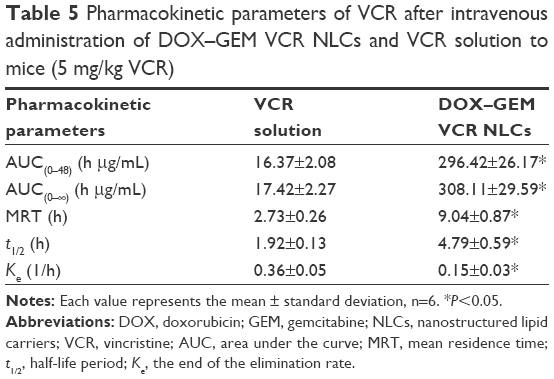 | Table 5 Pharmacokinetic parameters of VCR after intravenous administration of DOX–GEM VCR NLCs and VCR solution to mice (5 mg/kg VCR) |
Discussion
Currently, the front-line treatment of patients with B-cell lymphoma is CHOP, where the principal curative agents remain the DOX, GEM, or VCR. The aim of this study is to synthesize DOX–GEM prodrug and design DOX, GEM, and VCR coloaded nanocarriers. There are many nanosystems that can be used to deliver cancer drugs, including cross-linked self-assembly nanoparticles, polymersomes, liposomes, and others.42–45 Nanocarriers using lipids to load drugs form a carrier system with a number of desirable features, including a low toxicity, a biodegradable particulate matrix, nontoxic degradation products, a high capacity to incorporate lipophilic and hydrophilic drugs, a controlled release of the incorporated drug, and easy scale-up at low cost.46 For this purpose, different types of lipid carriers, including lipid–drug conjugates, SLNs, and NLCs, have been developed. NLCs represent a new generation of lipid nanoparticles, which are developed through the combination of advantages from different nanocarriers, including liposomes and SLN systems. So in this study, DOX–GEM VCR NLCs were constructed.
The size of blank NLCs, DOX–GEM NLCs, VCR NLCs, and DOX–GEM VCR NLCs was ~110 nm, indicating that drugs loading into NLCs did not increase the diameter of NLCs. The polydispersity index of four kinds of NLCs was <0.2, showing the narrow distributions. The zeta potential of DOX–GEM VCR NLCs was the lowest. Surface charge of DOX–GEM NLCs and VCR NLCs was lower than blank NLCs. The reduced zeta potential of DOX–GEM VCR NLCs was due to the incorporation of the negatively charged drugs.47 Over 85% EE of the drugs loaded in NLCs could be the evidence that the drugs were fully entrapped in the NLCs.48 DOX–GEM VCR NLCs have uniform spherical particle shape according to the TEM image. The obtained image indicates that the prepared drug-loaded NLCs were homogenous, and no aggregation was noticed. The later sustained release indicated that drugs might be stably retained in the lipid matrix before slow release by drug diffusion.49 Retardation of drug release in NLCs may be ascribed to the distribution of drugs in the lipid matrix.
Blank NLCs without drugs showed a high cell viability (88.1%). The low cytotoxic effect of blank NLCs is because they are formulated with glycerides consisting of fatty acids, which are safe and well tolerated by organisms and cells.50 Once they are delivered, they can adhere to the cell membrane, internalization, and degradation of byproducts in the cell culture medium or into the cells. The half maximal inhibitory concentration values of DOX–GEM VCR NLCs exhibited twofold dose advantage over DOX–GEM NLCs and many times over drug solutions. These results illustrate the best lymph tumor cell inhibition ability of the resulting NLCs formula, accounting for the highest antitumor activity in vitro.
In vivo tissue distributions of free drugs and drugs loaded in NLCs were investigated in lymph cancer-bearing mice. The drug concentration in the tumors, livers, spleens, and lungs was higher for the drugs loaded in the NLC group than for the free drugs group, while the drug concentration in the heart and kidney was much lower for the NLC group.
This may be because the nanocarriers had a relatively large mean diameter. As nanoparticles, NLCs could be recognized as foreign bodies in the blood circulation and rapidly cleared by mononuclear phagocyte system cells, which are abundant in special tissues and organs, such as the liver, lung, and spleen.51 The enhanced permeability and retention effects of tumors mean that the nanosized particles could passive targeted to the tumor, which resulted in the efficient drug accumulation in tumor tissue.52 Less drug distributions in heart and kidney may reduce the systemic toxicity; distribution mainly in tumor tissue than in the other tissues could decrease the side effects and lead to better antitumor therapeutic efficiency.
Excellent in vivo tumor inhibition activity of drug-loaded NLCs against lymph cancer was verified in tumor-bearing mice. The enhanced antitumor effect may be attributed to the reasons that, unlike free chemotherapeutics entering the cytoplasm by passive diffusion, drugs loaded in NLCs are endocytosed by tumor cells.31 The carriers with a regular spherical structure and proper size distribution can be easily uptaken by tumor cells and exert a stronger tumor-killing effect. In addition, sufficient accumulation and retention in tumor tissues via EFR effect play an important role. Moreover, controlled drug release can contribute to long circulation time and durable antilymphoma activity, which may be another important factor.
Conclusion
This study demonstrates the promising potential of a nanocarrier system to achieve a stable DL capacity, attractive anticancer therapeutic effects, and reduced toxicities in human Burkitt’s lymphoma cell line and mice bearing cancer model. The resulting DOX–GEM VCR NLCs could be an efficient antilymph cancer agent and could be developed further for the treatment of other tumors.
Disclosure
The authors report no conflicts of interest in this work.
References
Knapp CM, Whitehead KA. In pursuit of a moving target: nanotherapeutics for the treatment of non-Hodgkin B-cell lymphoma. Expert Opin Drug Deliv. 2014;11(12):1923–1937. | ||
Murawski N, Pfreundschuh M. New drugs for aggressive B-cell and T-cell lymphomas. Lancet Oncol. 2010;11(11):1074–1085. | ||
Alzouebi M, Goepel JR, Horsman JM, Hancock BW. Primary thyroid lymphoma: the 40 year experience of a UK lymphoma treatment centre. Int J Oncol. 2012;40(6):2075–2080. | ||
Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57(7):727–741. | ||
Emadi A, Jones RJ, Brodsky RA. Cyclophosphamide and cancer: golden anniversary. Nat Rev Clin Oncol. 2009;6(11):638–647. | ||
Gidding CE, Kellie SJ, Kamps WA, et al. Vincristine revisited. Crit Rev Oncol Hematol. 1999;29(3):267–287. | ||
Martelli M, Ferreri AJ, Agostinelli C, Di Rocco A, Pfreundschuh M, Pileri SA. Diffuse large B-cell lymphoma. Crit Rev Oncol Hematol. 2013;87(2):146–171. | ||
Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | ||
Toner LE, Vrhovac R, Smith EA, et al. The schedule-dependent effects of the novel antifolate pralatrexate and gemcitabine are superior to methotrexate and cytarabine in models of human non-Hodgkin’s lymphoma. Clin Cancer Res. 2006;12(3 pt1):924–932. | ||
Giovannetti E, Mey V, Loni L, et al. Cytotoxic activity of gemcitabine and correlation with expression profile of drug-related genes in human lymphoid cells. Pharmacol Res. 2007;55(4):343–349. | ||
Chiang CT, Yeh PY, Gao M, et al. Combinations of mTORC1 inhibitor RAD001 with gemcitabine and paclitaxel for treating non-Hodgkin lymphoma. Cancer Lett. 2010;298(2):195–203. | ||
Georgieva MC, Konstantinov SM, Topashka-Ancheva M, Berger MR. Combination effects of alkylphosphocholines and gemcitabine in malignant and normal hematopoietic cells. Cancer Lett. 2002;182(2):163–174. | ||
Zinzani PL, Venturini F, Stefoni V, et al. Gemcitabine as single agent in pretreated T-cell lymphoma patients: evaluation of the long-term outcome. Ann Oncol. 2010;21(4):860–863. | ||
Sivam V, Cook L, Hughes G, et al. Gemcitabine and vinorelbine chemotherapy for refractory or relapsing aggressive non-Hodgkin lymphoma. Hematol Oncol. 2012;30(4):214–215. | ||
Silverman JA, Deitcher SR. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol. 2013;71(3):555–564. | ||
Bai B, Huang HQ, Cai QQ, et al. Promising long-term outcome of gemcitabine, vinorelbine, liposomal doxorubicin (GVD) in 14-day schedule as salvage regimen for patients with previously heavily treated Hodgkin’s lymphoma and aggressive non-Hodgkin’s lymphoma. Med Oncol. 2013;30(1):350. | ||
Qian Z, Song Z, Zhang H, Wang X, Zhao J, Wang H. Gemcitabine, navelbine, and Doxorubicin as treatment for patients with refractory or relapsed T-cell lymphoma. Biomed Res Int. 2015;2015:606752. | ||
Straus DJ, Johnson JL, LaCasce AS, et al. Doxorubicin, vinblastine, and gemcitabine (CALGB50203) for stage I/II nonbulky Hodgkin lymphoma: pretreatment prognostic factors and interim PET. Blood. 2011;117(20):5314–5320. | ||
Gu J, Song ZP, Gui DM, Hu W, Chen YG, Zhang DD. Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in lymphoma nude mice by heme oxygenase-1 induction. Cardiovasc Toxicol. 2012;12(4):341–349. | ||
Wouters KA, Kremer LC, Miller TL, Herman EH, Lipshultz SE. Protecting against anthracycline-induced myocardial damage: a review of the most promising strategies. Br J Haematol. 2005;131(5):561–578. | ||
Limat S, Demesmay K, Voillat L, et al. Early cardiotoxicity of the CHOP regimen in aggressive non-Hodgkin’s lymphoma. Ann Oncol. 2003;14(2):277–281. | ||
Liu Y, He M, Niu M, et al. Delivery of vincristine sulfate-conjugated gold nanoparticles using liposomes: a light-responsive nanocarrier with enhanced antitumor efficiency. Int J Nanomedicine. 2015;10:3081–3095. | ||
Zhang P, Ling G, Sun J, et al. Multifunctional nanoassemblies for vincristine sulfate delivery to overcome multidrug resistance by escaping P-glycoprotein mediated efflux. Biomaterials. 2011;32(23):5524–5533. | ||
Aryal S, Hu CM, Zhang L. Combinatorial drug conjugation enables nanoparticle dual-drug delivery. Small. 2010;6(13):1442–1448. | ||
Thukral DK, Dumoga S, Mishra AK. Solid lipid nanoparticles: promising therapeutic nanocarriers for drug delivery. Curr Drug Deliv. 2014;11(6):771–791. | ||
Kang KW, Chun MK, Kim O, et al. Doxorubicin-loaded solid lipid nanoparticles to overcome multidrug resistance in cancer therapy. Nanomedicine. 2010;6(2):210–213. | ||
Parhi R, Suresh P. Preparation and characterization of solid lipid nanoparticles-a review. Curr Drug Discov Technol. 2012;9(1):2–16. | ||
Muller RH, Radtke M, Wissing SA. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv Drug Deliv Rev. 2002;54(suppl 1):S131–S155. | ||
Saupe A, Wissing SA, Lenk A, et al. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) – structural investigations on two different carrier systems. Biomed Mater Eng. 2005;15(5):393–402. | ||
Doktorovova S, Souto EB, Silva AM. Nanotoxicology applied to solid lipid nanoparticles and nanostructured lipid carriers – a systematic review of in vitro data. Eur J Pharm Biopharm. 2014;87(1):1–18. | ||
Li H, Guo K, Wu C, et al. Controlled and targeted drug delivery by a UV-responsive liposome for overcoming chemo-resistance in non-Hodgkin lymphoma. Chem Biol Drug Des. 2015;86(4):783–794. | ||
Huang S, Fang R, Xu J, et al. Evaluation of the tumor targeting of a FAPα-based doxorubicin prodrug. J Drug Target. 2011;19(7):487–496. | ||
Shah NV, Seth AK, Balaraman R, Aundhia CJ, Maheshwari RA, Parmar GR. Nanostructured lipid carriers for oral bioavailability enhancement of raloxifene: design and in vivo study. J Adv Res. 2016;7(3):423–434. | ||
Nam JP, Park SC, Kim TH, et al. Encapsulation of paclitaxel into lauric acid-O-carboxymethyl chitosan-transferrin micelles for hydrophobic drug delivery and site-specific targeted delivery. Int J Pharm. 2013;457(1):124–135. | ||
Loureiro A, Bernardes GJ, Shimanovich U, et al. Folic acid-tagged protein nanoemulsions loaded with CORM-2 enhance the survival of mice bearing subcutaneous A20 lymphoma tumors. Nanomedicine. 2015;11(5):1077–1083. | ||
Lv S, Tang Z, Li M, et al. Co-delivery of doxorubicin and paclitaxel by PEG-polypeptide nanovehicle for the treatment of non-small cell lung cancer. Biomaterials. 2014;35(23):6118–6129. | ||
Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Control Release. 2004;100(3):331–346. | ||
Wu M, Fan Y, Lv S, Xiao B, Ye M, Zhu X. Vincristine and temozolomide combined chemotherapy for the treatment of glioma: a comparison of solid lipid nanoparticles and nanostructured lipid carriers for dual drugs delivery. Drug Deliv. 2016;23(8):2720–2725. | ||
Shao Z, Shao J, Tan B, et al. Targeted lung cancer therapy: preparation and optimization of transferrin-decorated nanostructured lipid carriers as novel nanomedicine for co-delivery of anticancer drugs and DNA. Int J Nanomedicine. 2015;10:1223–1233. | ||
Zhao X, Chen Q, Li Y, Tang H, Liu W, Yang X. Doxorubicin and curcumin co-delivery by lipid nanoparticles for enhanced treatment of diethylnitrosamine-induced hepatocellular carcinoma in mice. Eur J Pharm Biopharm. 2015;93:27–36. | ||
Yang S, Zhang B, Gong X, Wang T, Liu Y, Zhang N. In vivo biodistribution, biocompatibility, and efficacy of sorafenib-loaded lipid-based nanosuspensions evaluated experimentally in cancer. Int J Nanomedicine. 2016;11:2329–2343. | ||
Liu X, Yaszemski MJ, Lu L. Expansile crosslinked polymersomes for pH sensitive delivery of doxorubicin. Biomater Sci. 2016;4(2):245–249. | ||
Yu Z, Xu Q, Dong C, et al. Self-assembling peptide nanofibrous hydrogel as a versatile drug delivery platform. Curr Pharm Des. 2015;21(29):4342–4354. | ||
Haeri A, Zalba S, Ten Hagen TL, Dadashzadeh S, Koning GA. EGFR targeted thermosensitive liposomes: a novel multifunctional platform for simultaneous tumor targeted and stimulus responsive drug delivery. Colloids Surf B Biointerfaces. 2016;146:657–669. | ||
Zhong T, Yao X, Zhang S, et al. A self-assembling nanomedicine of conjugated linoleic acid-paclitaxel conjugate (CLA-PTX) with higher drug loading and carrier-free characteristic. Sci Rep. 2016;6:36614. | ||
Liu Q, Li J, Pu G, Zhang F, Liu H, Zhang Y. Co-delivery of baicalein and doxorubicin by hyaluronic acid decorated nanostructured lipid carriers for breast cancer therapy. Drug Deliv. 2016;23(4):1364–1368. | ||
Zhang W, Peng F, Zhou T, et al. Targeted delivery of chemically modified anti-miR-221 to hepatocellular carcinoma with negatively charged liposomes. Int J Nanomedicine. 2015;10:4825–4836. | ||
Zhang Y, Zhou J, Yang C, et al. Folic acid-targeted disulfide-based cross-linking micelle for enhanced drug encapsulation stability and site-specific drug delivery against tumors. Int J Nanomedicine. 2016;11:1119–1130. | ||
Ahmed TA. Preparation of finasteride capsules-loaded drug nanoparticles: formulation, optimization, in vitro, and pharmacokinetic evaluation. Int J Nanomedicine. 2016;11:515–527. | ||
Bondì ML, Craparo EF, Giammona G, et al. Nanostructured lipid carriers-containing anticancer compounds: preparation, characterization, and cytotoxicity studies. Drug Deliv. 2007;14(2):61–67. | ||
Gao L, Zhang D, Chen M, et al. Studies on pharmacokinetics and tissue distribution of oridonin nanosuspensions. Int J Pharm. 2008;355(1–2):321–327. | ||
He X, Li L, Su H, et al. Poly(ethylene glycol)-block-poly(ε-caprolactone)-and phospholipid-based stealth nanoparticles with enhanced therapeutic efficacy on murine breast cancer by improved intracellular drug delivery. Int J Nanomedicine. 2015;10:1791–1804. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.