")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 15
Lumasiran in the Management of Patients with Primary Hyperoxaluria Type 1: From Bench to Bedside
Authors D'Ambrosio V, Ferraro PM
Received 12 March 2022
Accepted for publication 3 June 2022
Published 17 June 2022 Volume 2022:15 Pages 197—206
DOI https://doi.org/10.2147/IJNRD.S293682
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pravin Singhal
Viola D’Ambrosio,1,2 Pietro Manuel Ferraro1,2
1U.O.S. Terapia Conservativa della Malattia Renale Cronica, U.O.C. Nefrologia, Dipartimento di Scienze Mediche e Chirurgiche, Fondazione Policlinico Universitario A. Gemelli IRCCS, Roma, Italia; 2Dipartimento Universitario di Medicina e Chirurgia Traslazionale, Università Cattolica del Sacro Cuore, Roma, Italia
Correspondence: Pietro Manuel Ferraro, U.O.S. Terapia Conservativa della Malattia Renale Cronica, U.O.C. Nefrologia, Dipartimento di Scienze Mediche e Chirurgiche, Fondazione Policlinico Universitario A. Gemelli IRCCS, Largo Agostino Gemelli 8, Roma, 00168, Italia, Tel +39-06-3015-7819, Fax +39-06-3015-942, Email [email protected]
Abstract: Primary hyperoxaluria (PH) is a rare genetic disease caused by excessive hepatic production and elevated urinary excretion of oxalate that leads to recurrent nephrolithiasis, nephrocalcinosis and, eventually, kidney failure. As glomerular filtration rate declines, oxalate accumulates leading to systemic oxalosis, a debilitating condition with high morbidity and mortality. Although PH is usually diagnosed during infancy, it can present at any age with different phenotypes, ranging from mild symptoms to extremely debilitating manifestations. PH is an autosomal recessive disorder and, to date, three types have been identified: PH1, PH2 and PH3. PH1 is the most common and most aggressive type, accounting for almost 80% of primary hyperoxaluria diagnoses. Until 2020, general treatment for PH1 consisted mainly in high fluid intake, urine alkalization, surgical management of recurrent nephrolithiasis and eventually, if and when kidney failure occurred, intensive dialysis regimens and transplantation strategies (simultaneous or sequential liver-kidney transplant or isolated liver/kidney transplant in carefully selected patients). Specific treatment did and still consists in administration of pyridoxine hydrochloride, although it is only effective in a subset of PH1 patients. Lumasiran, a novel biological drug based on mRNA interference that has been recently approved in the US and European Union, showed promising results and is set to be a turning point in the management of PH1. This literature review aims to summarize the available evidence on PH1 treatment with lumasiran, in order to provide both pediatric and adult nephrologists and clinicians with the knowledge for the identification and management of PH1 patients suitable for treatment.
Keywords: primary hyperoxaluria, oxalate, lumasiran, iRNA, nephrolithiasis
Introduction: Primary Hyperoxaluria Type 1 – Disease Overview
The term hyperoxaluria refers to an elevated urinary excretion of oxalate, generally above 45 mg/1.73 m2/24h (0.50 mmol/1.73 m2/24h, conversion factor 0.011 mmol/mg) with normal values in healthy individuals below this threshold.1
Oxalate Metabolism
Oxalate is the ionic form of oxalic acid and is obtained from exogenous sources (eg, nuts, chocolate, beetroot, rhubarb and spinach) and endogenous synthesis.2 The endogenous synthesis of oxalate takes place in the liver where glyoxylate acts as an intermediate molecule generated either by the oxidation of glycolate (via the glycolate oxidase) or from metabolism of hydroxyproline found in collagen or dietary sources.3 Defects in the metabolism of glyoxylate – as seen in primary hyperoxaluria – lead to accumulation of glyoxylate, conversion of glyoxylate to oxalate via a lactate dehydrogenase type A (liver-specific) and ultimately to oxalate overproduction. Under physiological conditions, oxalate is both absorbed daily by the gut from dietary sources and endogenously produced by the liver; oxalate is then handled by the kidney via glomerular filtration – that depends on plasma oxalate levels – and tubular secretion and reabsorption4 mediated by SLC26A6 and SLC26A1, respectively.5
Classification of Hyperoxaluria
Hyperoxaluria can be classified as: primary, secondary and unclassified. While secondary hyperoxalurias depend on different causes such as increased dietary intake of oxalate enriched food,6 intestinal fat malabsorption (also called enteric hyperoxaluria seen in bariatric surgery and inflammatory bowel diseases7), excessive intake of oxalate precursors such as vitamin C8–10 and alteration in the intestinal microflora,11 primary hyperoxalurias (PHs) are a group of genetic diseases that lead to excessive hepatic production and consequent elevated urinary excretion of oxalate.12
Epidemiology of Primary Hyperoxaluria
The estimated prevalence of PHs is approximately 1 to 3 cases/1,000,000,13,14 although population analysis conducted by Hopp et al demonstrated that the inferred prevalence of PH may be higher (1:58.000).15 PHs are also responsible for 1% of end-stage kidney disease (ESKD) in the pediatric population in some European studies,16 although prevalence may vary in different endemic countries. PH is classified into three different groups: type 1 (PH1), type 2 (PH2) and type 3 (PH3). PH1 is the most common and severe form, accounting for approximately 80% of all cases and it is caused by a genetic defect of the AGXT gene that encodes for a liver-specific peroxisomal enzyme called alanine-glyoxylate aminotransferase (AGT),17 PH2 (8–9% of the cases in a European cohort18) is caused by a genetic defect of the GRHPR gene that encodes for the glyoxylate reductase/hydroxypyruvate reductase (GR/HPR)19 and PH3 (7–8% of the cases in a European cohort20) is caused by mutations in the HOGA1 gene that encodes for the mitochondrial 4-hydroxy 2-oxoglutarate aldolase 1 (HOGA1);21 all enzymes involved in the hepatic metabolism of oxalate.3
Clinical Manifestations of Primary Hyperoxaluria
All three conditions are characterized by recurrent nephrolithiasis, nephrocalcinosis and eventually kidney failure. Based on that, as glomerular filtration rate (GFR) declines, oxalate accumulates and when it exceeds its plasmatic supersaturation threshold (currently set at 30 mcmol/L) it starts to accumulate as calcium oxalate deposits in extrarenal organs and soft tissues leading to a condition called systemic oxalosis. Involvement of the myocardium, cardiac conduction system, bones, bone marrow, vessels, eyes, skin and central nervous system have been described, which is not only extremely debilitating, but also more resistant to treatment,22,23 although cases of improved organ function after specific treatment are present in the literature.24 Recent evidence has suggested a non-linear correlation between eGFR and plasma oxalate levels, with no clear cut-off point, but rather an exponential increase of plasma oxalate levels for eGFR values below 60 mL/min/1.73 m2.25 Moreover, a recent analysis conducted by Milliner et al demonstrated a statistically significant inverse correlation between eGFR and plasma oxalate levels even at early stages of chronic kidney disease (1–3b),26 however, these results need further validation as there are controversial data in the literature.27 PH is usually diagnosed during adolescence or adulthood, however, it can present at any age and under-recognized symptoms may be present since infancy. It is characterized by inter-individual clinical variability with different phenotypes, ranging from mild symptoms to extremely debilitating manifestations. As already stated above, PH1 is the most aggressive form and in 10% of the cases (at least in Europe and North America28) it can present as infantile oxalosis. This form is characterized by a very early onset of symptoms (usually before the age of 1) such as failure to thrive, vomiting, oligo-anuria and bilateral nephrocalcinosis alongside a quick progression towards ESKD (by 3 years of age in 80% of the cases) needing intensive renal replacement regimens.29 This form represents a life-threatening condition and therefore should be promptly diagnosed and treated.
Treatment Options for PH1
Before the approval of specific treatments, PH1 management consisted mainly in a general approach that could be applied to all subtypes of primary hyperoxaluria, with the exception of pyridoxine hydrochloride, recommended only for PH1 patients – pyridoxine hydrochloride or vitamin B6 is metabolised to pyridoxal phosphate, an essential cofactor of AGT;30 it is considered a first-line therapy in PH1 patients with B6-responsive mutations. Conservative measures can be proposed in case of preserved kidney function and they should be promptly initiated once the diagnosis is established. These measures consist mainly in massive hydration and urine alkalinization. The recommended volume of fluid intake is above 3 L/m2 per day equally distributed throughout the 24 h.31 Ensuring an adequate fluid intake allows the reduction of urinary supersaturation for calcium oxalate (SS CaOx), therefore preventing crystals aggregation, stone formation and growth. While in adults this goal is more easily achievable (except in cases of severe fluid losses such as diarrhea, vomiting and fever that require intravenous administration of fluids), in infants and small children a feeding tube or a gastrostomy may be required. Urinary alkalinization can be achieved through the administration of potassium citrate (or sodium citrate in patients with chronic kidney disease and/or hyperkalemia) that reduces SS CaOx32 by increasing citrate excretion and urinary pH. The recommended dose is 0.10–0.15 g/kg/day (0.5–0.8 mmol/kg/day).31 Restriction of dietary oxalate intake has been proven to be of limited efficacy, since the main source of oxalate in PH1 patients is endogenous and intestinal oxalate absorption seems to be lower compared to the general population,33 although an adequate dietary calcium intake is usually recommended to increase oxalate binding in the gastrointestinal tract.34,35 Generally, surgical procedures to remove uncomplicated renal calculi are not recommended; however, in case of obstruction with or without acute kidney injury (AKI), infection or recurrent nephrolithiasis endoscopic procedures are preferred.31 When kidney failure occurs, intensive dialysis regimens such as high efficacy dialysis, daily hemodialysis (HD), nocturnal dialysis, combined HD and peritoneal dialysis should be promptly initiated. Conventional intermittent dialysis alone is unable to remove plasma oxalate properly; plasma oxalate is generated at a rate of 4–7 mmol/1.73 m2, while conventional dialysis removes oxalate at a rate of 1–2 mmol/1.73 m2 per day in adults and 3–4 mmol/1.73 m2 per day in children.36–38
For decades, the only available option to correct the defective metabolism of glyoxylate in PH1 has been liver transplantation, possibly in combination with kidney transplantation (both simultaneous and sequential) in case of kidney failure. Pre-emptive liver transplantation at chronic kidney disease (CKD) stage G3b KDIGO is usually recommended to avoid systemic oxalosis,31 although it raises ethical concerns and could burden patients with a still intact organ function with long-term immunosuppression. Isolated kidney transplantation is currently not an option, with the exception of selected B6-responsive cases. However, this treatment is burdened with elevated morbidity and mortality39–41 and obviously, life-long immunosuppressive therapy.
Lumasiran: Mechanism of Action (Pharmacodynamics and Pharmacokinetics)
Lumasiran is a subcutaneously administered drug based on the mechanism of RNA interference. RNA interference is a biological process designated to regulate – more specifically, to silence – cellular gene expression via small interfering RNAs (siRNAs).42,43 siRNAs are short double-stranded RNAs (dsRNAs) that interfere with cellular translation and prevent messenger RNAs (mRNAs), produced by cellular transcription, from being decoded into proteins by ribosomes.44 Lumasiran is a synthetic double-stranded siRNA conjugated with the carbohydrate N-acetylgalactosamine (GalNAc) that targets the hydroxyacid oxidase 1 (HAO1) gene in hepatocytes, more specifically, it prevents HAO1 mRNA translation to the enzyme glycolate oxidase (GO). In the hepatocyte peroxisomes, GO catalyzes the conversion of glycolate to glyoxylate, which is the direct precursor of oxalate. Therefore, a reduction in GO expression results in a reduction of oxalate production and an increase in glycolate levels.45,46 Proof of GO’s inhibition efficacy in reducing oxalate production came from a study conducted by Martin-Higueras et al in double knockout mice Hao1-/-Agxt1-/-.47 As a consequence of GO’s inhibition glycolate can reach elevated plasma and urinary levels;48,49 glycolate is an acid (glycolic acid) and therefore can potentially cause metabolic acidosis. Further studies are needed to evaluate this effect and its long-term outcomes. Hepatic selectivity is guaranteed by the interaction of GalNAc to the asialoglycoprotein receptor 1, highly expressed by the hepatocytes.50 This receptor facilitates the hepatic uptake of the siRNA via endocytosis, and, once in the acidic endosome, the siRNA is released from GalNAc42,45 and can exert its gene-silencing function.
Because GO is upstream of the defective AGT enzyme, lumasiran acts independently of the specific AGXT mutation and it is only used for PH1.
First pre-clinical studies conducted on wild-type mice demostrated lumasiran’s efficacy in silencing HAO1 mRNA, even after a single dose. Lumasiran’s efficacy was subsequently proved in a murine model of the disease (Agxt1-/-), in which a single dose (3 mg/kg) of subcutaneously administered lumasiran proved to reduce urinary oxalate by 50% in 2–3 weeks, with an effect lasting up to 7 weeks. On the other hand, multiple administrations of different doses four days per week (respectively, 0.3, 1 and 3 mg/kg) induce more than 95% of HAO1 mRNA silencing and an almost complete suppression of urinary oxalate excretion.45
With regard to pharmacokinetics, once administered, lumasiran is rapidly absorbed with a maximum plasma concentration reached after 4 hours and a plasma protein binding of 85%, and distributes primarily to the liver. As a consequence, lumasiran plasma half-life of elimination is very short (5.2 hours). Plasma clearance in adults was 26.5 L/h, while urinary clearance ranged from 2.0 to 3.4 L/h in both affected adults and children. Although lumasiran is primarily metabolized in the liver in a non-CYP450-dependent manner, a relatively small percentage (7% to 26%) is excreted by the kidneys.51
Lumasiran: Current Clinical Evidence
Preclinical and clinical evidence of lumasiran efficacy comes from several studies (Table 1), most of which are currently active. In 2016, the first Phase I/II study was initiated to assess safety, tolerability, pharmacokinetics and pharmacodynamics of subcutaneously administered lumasiran in healthy adult subjects and patients with PH1 (clinicaltrials.gov identifier: NCT02706886). This was a randomized, single-blind, placebo-controlled trial with single and multiple ascending doses of lumasiran.50 The study was composed of two parts: part A evaluated single doses in ascending dose groups (0.3, 1, 3 or 6 mg/kg) in 32 healthy adult participants; part B evaluated two lumasiran doses (1.0 mg/kg once a month or 3.0 mg/kg once a month) in 20 adults and children (> 6 years of age) affected by PH1. In both part A and B, participants were randomly assigned 3:1 to lumasiran or placebo. The primary outcome of this study was the incidence of adverse events, with secondary exploratory outcomes being parameters such as pharmacokinetics and pharmacodynamics. In part A, adverse events were reported in 83% and 63% of participants receiving lumasiran and placebo, respectively. The most frequent adverse events in the lumasiran-treated group were upper respiratory tract infections (eg nasopharyngitis and rhinitis), headache and injection site reactions. Only one lumasiran-treated healthy subject developed a serious adverse event, that was assessed as not drug-related. In part B, adverse events were reported in 59% of lumasiran-treated PH1 patients compared to 67% of the placebo group. The most common adverse events were abdominal pain, headache, rhinitis, nephrolithiasis and cough. Serious adverse events were reported in 1 placebo-treated patient and 2 lumasiran-treated patients, although they were assessed as not related to the drug. In all the 20 PH1 patients that received lumasiran in part B, a rapid and sustained reduction in 24-hour urinary oxalate was observed post last dosing (mean maximum reduction 75%) as well as reduction in plasma oxalate levels, with all patients achieving normal or near-normal 24-hour urinary oxalate levels. All 20 patients entered an extension study (clinicaltrials.gov identifier: NCT03350451) where they received lumasiran 3 mg/kg once every three months. The positive results regarding the lowering of urinary and plasma oxalate levels, already seen in the phase I/II study, were confirmed. Three phase-III studies investigating lumasiran in PH1 patients have been designed. In the randomized, double-blind, placebo-controlled clinical trial ILLUMINATE-A (clinicaltrials.gov identifier: NCT0368118452), patients affected by PH1 who were >6 years of age and had an eGFR >30 mL/min/1.73 m2 were randomly assigned to subcutaneous lumasiran (3 mg/kg given at baseline, at month 1, 2, 3 and 6) or placebo in a 2:1 ratio for six months.53 39 patients were enrolled (26 receiving lumasiran and 13 receiving placebo), with only one withdrawing from the trial. The primary outcome regarding the percentage decrease in urinary oxalate excretion was expressed as least-square mean (LSM) difference between the lumasiran and the placebo group. LSM from baseline to month 6 was −53.5% (−65.4% in the lumasiran group and −11.8% in the placebo group). This reduction in urinary oxalate excretion was confirmed in every subgroup of patients adjusted for baseline oxalate excretion, baseline pyridoxine use and baseline eGFR. At month six, 84% of patients who received lumasiran had a near-normalization (defined as not higher than 1.5 times the upper limit of normal) of urinary oxalate levels vs 0% in the placebo group, with 52% reaching normalization of urinary oxalate excretion. After month six, all patients entered an extension period of up to 54 months, where lumasiran-receiving patients continued on lumasiran while placebo-receiving patients switched to lumasiran: the beneficial effects of lumasiran on urinary oxalate levels were confirmed. Two clinical exploratory end points were also investigated: kidney-stone event rate and ultrasound-assessed nephrocalcinosis. Kidney-stone event rate was reduced after six and twelve months (1.09 and 0.85 events per person-year, respectively) of lumasiran in the lumasiran/lumasiran group compared to the twelve months prior randomization (3.19 events per person-year), while in the placebo/lumasiran group the kidney-stone event rate remained the same after 6 months of placebo compared to the twelve months prior randomization (0.66 vs 0.54 events per person-year) and was reduced after 6 months of lumasiran (0.17 events per person-year). In patients who had two ultrasound evaluations at month 1 and month 6, 3 out of 22 lumasiran-receiving patients showed an improvement in nephrocalcinosis, compared to none in the placebo group; nephrocalcinosis worsened in 1 out of 12 placebo-receiving patients compared to none in the lumasiran group. Given the exclusion of children <6 years of age in ILLUMINATE-A, a second open-label, phase-III trial (ILLUMINATE-B, clinicaltrials.gov identifier: NCT03905694) assessed lumasiran efficacy in reducing urinary oxalate excretion in this population with an eGFR >45 mL/min/1.73 m2,54 if aged ≥12 months, or normal serum creatinine, if aged < 12 months. 18 patients were enrolled and lumasiran was administered in different doses according to weight: patients who weighed < 10 kg received 6 mg/kg once a month for three months, subsequently tapered at 3 mg/kg monthly; patients who weighed 10–20 kg received 6 mg/kg once a month for the first three months followed by 6 mg/kg every 3 months; patients who weighed > 20 kg received 3 mg/kg once a month for the first three months followed by 3 mg/kg every three months. In this trial, lumasiran was confirmed to be able to reduce spot urinary oxalate: creatinine ratio at month six compared to baseline (LSM percent reduction in spot UOx: Cr was 72.0%) as well as plasma oxalate levels (31.7%). Change in eGFR (evaluated only in patients >12 months of age) and change in kidney-stone event rate compared to the twelve months prior randomization were respectively minimal and absent. Of the 14 (78%) patients with baseline nephrocalcinosis, 3 had bilateral improvement, 5 had unilateral improvement, and no patients worsened after 6 months of treatment. The third non-comparative Phase III trial, ILLUMINATE-C (clinicaltrials.gov identifier: NCT04152200) intends to evaluate lumasiran efficacy and safety in PH1 patients of all ages with impaired kidney function (eGFR <45 mL/min/1.73 m2) including patients undergoing stable hemodialysis51,55 and patients on stable doses of vitamin B6. 6 patients with advanced PH1 were enrolled in cohort A and 15 patients undergoing hemodialysis were enrolled in cohort B, lumasiran was administered following the same regimens as in ILLUMINATE-B, previously reported in the paper. Data from the 6-month primary analysis period showed that patients in both cohorts had significant reduction in plasma oxalate (POx). In cohort A, percent change in POx from baseline to months 6 led to a LSM reduction of 33.3% (95% CI, −15.16 to 81.82). In cohort B, percent change in predialysis POx from baseline to month 6 led to a LSM reduction of 42.43% (95% CI, 34.15 to 50.71). Reduction in POx was evident by Month 1 and persisted through the end of the 6-month primary analysis period. BONAPH1DE (clinicaltrials.gov identifier: NCT04982393) is a prospective observational study that aims to describe the natural history and progression of patients diagnosed with PH1 and to characterize the long-term real-world safety and effectiveness of lumasiran. The trials’ most relevant results are summarized in Table 2. In November 2020, lumasiran was approved for the treatment of PH1 in all age groups in the European Union (EU) and in pediatric and adult patients in the United States (US). Current recommended regimens of subcutaneous lumasiran consist of a loading dose and a maintenance dose based on body weight (Table 3). For affected patients with an eGFR between 30 and 90 mL/min/1.73 m2, dose adjustment is not required. However, limited data are available for patients with an eGFR <30 mL/min/1.73 m2, on dialysis and/or under 1 year of age; therefore, lumasiran should be cautiously administered and closely monitored in these patients. The role of lumasiran in the treatment of the infantile form of PH1 still needs to be completely clarified and widely evaluated, however, there are some case reports in the literature that show evidence of lumasiran’s efficacy in improving clinical manifestations and quality of life in infantile forms.56
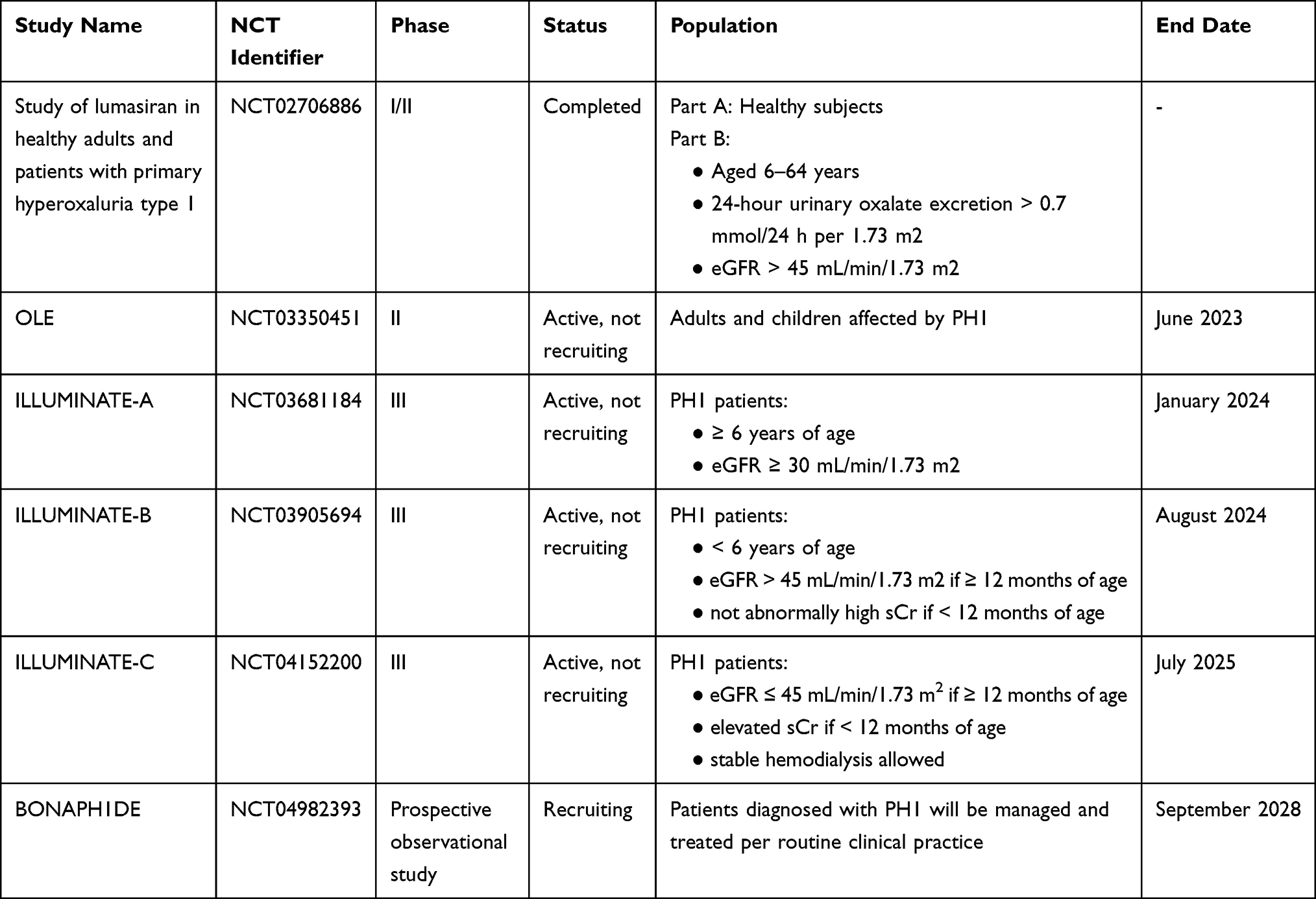 |
Table 1 Registered Preclinical and Clinical Trials Involving Lumasiran |
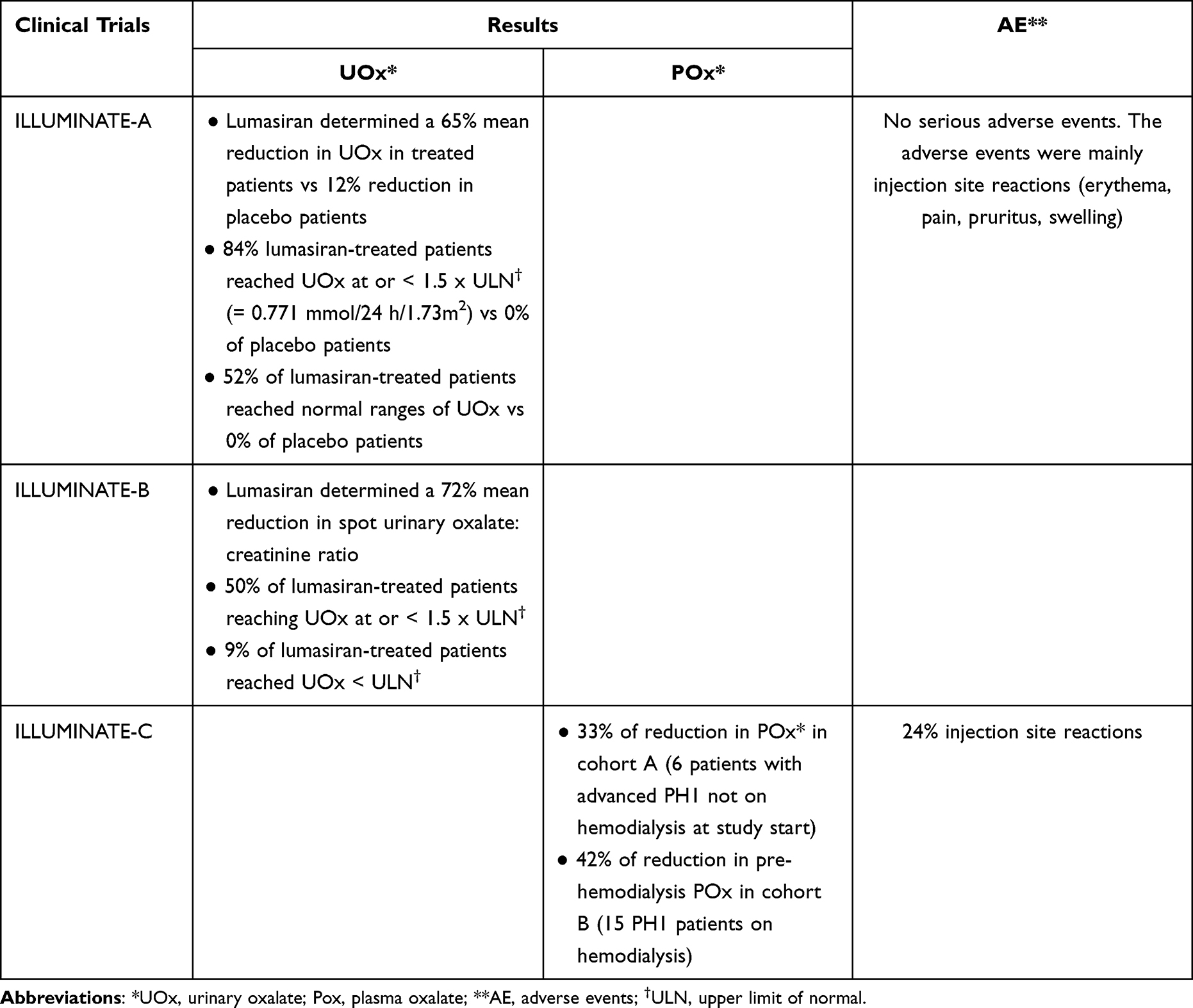 |
Table 2 Clinical Trials’ Most Relevant Results for Lumasiran |
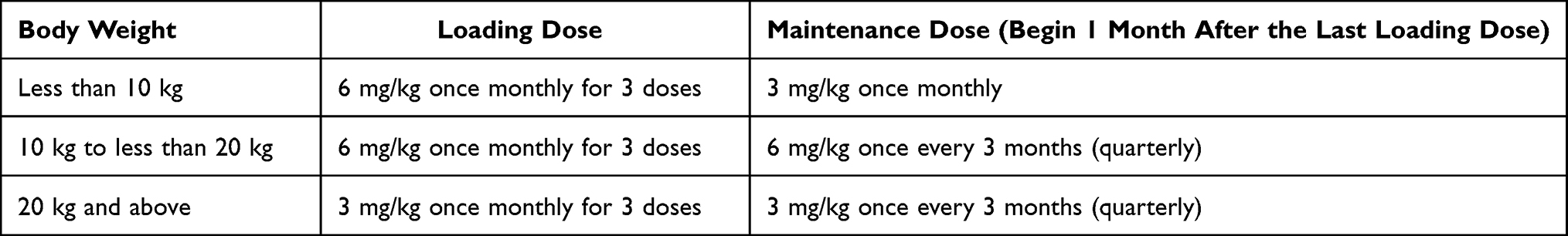 |
Table 3 Recommended Lumasiran Regimens Based on Body Weight, Currently Approved in the US and EU |
Patient Selection and Place in Therapy
Lumasiran is currently approved for patients affected by PH1. Although evidence from the above-mentioned clinical trials supports the safety and efficacy in adult and pediatric patients, data on pregnant women and patients >65 years of age are less clear. More specifically, EMA indications declare that there is no need for dose adjustment in patients ≥65 years old, while FDA states that data in the literature are not sufficient to determine whether this population responds differently from younger patients. With regard to patients with liver disease, since lumasiran has not been studied in patients with severe hepatic impairment, EMA states that no dose adjustment is required in patients that present with transient elevation of total bilirubin (total bilirubin >1.0 to 1.5x upper limit of normal [ULN]), however it should be cautiously administered in patients with moderate or severe hepatic impairment. Similarly, FDA does not recommend any dose adjustment for mild and moderate hepatic impairment (respectively, defined as total bilirubin >1.0 to 1.5x ULN or AST >1.0x ULN and total bilirubin >1.5 to 3.0x ULN with any AST). To date, lumasiran is only available for patients with a definite diagnosis of PH1.
Novel Therapies
Despite the fact that primary hyperoxaluria is an extremely rare condition, the interest in developing novel therapeutic strategies has been growing over the last few years. Recently, novel technological approaches have been proposed. Nedosiran, a novel therapy based on a siRNA technology similar to lumasiran but targeting hepatic lactate dehydrogenase (LDH) activity, holds promising results. Nedosiran is a GalNac-conjugated siRNA that targets the LDH subtype A (LDHA, primarily expressed in the liver), the enzyme that catalyzes the final cytosolic conversion of glyoxylate to oxalate, a common step in all 3 subtypes of PH. In murine models of both PH1 and PH2, nedosiran showed sustained reduction of 24-hour urinary oxalate excretion.57 A Phase 1 study was conducted on healthy volunteers and patients with PH1 or PH2 (clinicaltrials.gov identifier: NCT03392896) in which a single dose of nedosiran showed acceptable safety and a urinary oxalate-lowering effect in both type 1 and type 2 of PH.58 A phase III study aiming at evaluating the safety and efficacy of nedosiran in children and adults with PH1 and PH2 was also conducted, but results are yet to be published (clinicaltrials.gov identifier: NCT03847909). Another strategy based on LDHA inhibition, is the one of stiripentol, an anti-convulsant usually administered to children affected by Dravet syndrome. Stiripentol shares the mechanism of action with nedosiran, hepatic selectivity and ability to reduce urinary oxalate excretion. A clinical trial evaluating stiripentol efficacy in the treatment of PH1, 2 and 3 was concluded last year, although results are still to be published (clinicaltrials.gov identifier: NCT03819647), and 5 cases are currently recorded in the literature.59–62 Other strategies that target the underlying metabolic defect of PH currently under development and/or investigation are: substrate reduction therapies as such as the one using CRISPR/Cas9 technologies targeting either glycolate oxidase or lactate dehydrogenase, replacement therapies such as gene therapy, cell therapy and enzyme replacement therapy; pharmacological chaperone therapy.63 On the other hand, since the deposition of calcium oxalate (CaOx) crystals in the kidney leads to the activation of several pathways that mediate kidney damage, strategies that aim to treat renal manifestations of PH are also under development (anti-inflammatory, anti-fibrotic and crystallization preventing therapies).3,63
Conclusions
In conclusion, despite being a rare disease, PH has witnessed incredible progresses in its treatment over the last few years. Results from the ILLUMINATE trials led the way to the very recent approval by FDA and European Union of lumasiran for the treatment of PH1, a pivotal milestone for an ultra-rare yet potentially life-threatening disease. Simultaneously, research is making important progress in unravelling the underlying pathophysiology of PH, allowing not only a better understanding of the disease, but also the identification of novel potential therapeutic targets. Further studies are nevertheless needed to assess lumasiran’s long-term safety and ability to prevent and possibly reverse systemic oxalosis, an extremely debilitating condition that, to date, has only liver transplantation as specific therapeutic option.
Author Contributions
Both authors made a significant contribution to the work reported, whether that is in the conception, study design, execution; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was not funded.
Disclosure
PMF received consultant fees and grant support from Allena Pharmaceuticals, Alnylam, AstraZeneca, BioHealth Italia, Otsuka Pharmaceuticals, Vifor Fresenius, and royalties as an author for UpToDate. VDA received consultant fees from Allena Pharmaceuticals. PMF and VDA are members of the European Reference Network for Rare Kidney Diseases (ERKNet) – Project ID No 739532.
References
1. Bhasin B, Ürekli HM, Atta MG. Primary and secondary hyperoxaluria: understanding the enigma. World J Nephrol. 2015;4(2):235–244. doi:10.5527/wjn.v4.i2.235
2. Asplin JR. Hyperoxaluric calcium nephrolithiasis. Endocrinol Metab Clin North Am. 2002;31(4):927–949. doi:10.1016/s0889-8529(02)00030-0
3. Belostotsky R, Frishberg Y. Novel therapeutic approaches for the primary hyperoxalurias. Pediatr Nephrol. 2021;36(9):2593–2606. doi:10.1007/s00467-020-04817-8
4. Osswald H, Hautmann R. Renal elimination kinetics and plasma half-life of oxalate in man. Urol Int. 1979;34(6):440–450. doi:10.1159/000280294
5. Mount DB, Romero MF. The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch. 2004;447(5):710–721. doi:10.1007/s00424-003-1090-3
6. Holmes RP, Kennedy M. Estimation of the oxalate content of foods and daily oxalate intake. Kidney Int. 2000;57(4):1662–1667. doi:10.1046/j.1523-1755.2000.00010.x
7. Karaolanis G, Lionaki S, Moris D, Palla VV, Vernadakis S. Secondary hyperoxaluria: a risk factor for kidney stone formation and renal failure in native kidneys and renal grafts. Transplant Rev. 2014;28(4):182–187. doi:10.1016/j.trre.2014.05.004
8. Ferraro PM, Curhan GC, Gambaro G, Taylor EN. Total, dietary, and supplemental vitamin c intake and risk of incident kidney stones. Am J Kidney Dis. 2016;67(3):400–407. doi:10.1053/j.ajkd.2015.09.005
9. Canavese C, Petrarulo M, Massarenti P, et al. Long-term, low-dose, intravenous vitamin C leads to plasma calcium oxalate supersaturation in hemodialysis patients. Am J Kidney Dis. 2005;45(3):540–549. doi:10.1053/j.ajkd.2004.10.025
10. Nasr SH, Kashtanova Y, Levchuk V, Markowitz GS. Secondary oxalosis due to excess vitamin C intake. Kidney Int. 2006;70(10):1672. doi:10.1038/sj.ki.5001724
11. Stewart CS, Duncan SH, Cave DR. Oxalobacter formigenes and its role in oxalate metabolism in the human gut. FEMS Microbiol Lett. 2004;230(1):1–7. doi:10.1016/S0378-1097(03)00864-4
12. Cochat P, Rumsby G, Ingelfinger JR. Primary hyperoxaluria. N Engl J Med. 2013;369(7):649–658. doi:10.1056/NEJMra1301564
13. Cochat P, Deloraine A, Rotily M, Olive F, Liponski I, Deries N. Epidemiology of primary hyperoxaluria type 1. Société de Néphrologie and the Société de Néphrologie Pédiatrique. Nephrol Dial Transplant. 1995;10(Suppl 8):3–7. doi:10.1093/ndt/10.supp8.3
14. Primary hyperoxaluria type 1 in the Netherlands: prevalence and outcome - PubMed. Available from: https://pubmed.ncbi.nlm.nih.gov/12543880/.
15. Hopp K, Cogal AG, Bergstralh EJ, et al. Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol. 2015;26(10):2559–2570. doi:10.1681/ASN.2014070698
16. Harambat J, van Stralen KJ, Espinosa L, et al. Characteristics and outcomes of children with primary oxalosis requiring renal replacement therapy. Clin J Am Soc Nephrol. 2012;7(3):458–465. doi:10.2215/CJN.07430711
17. Purdue PE, Takada Y, Danpure CJ. Identification of mutations associated with peroxisome-to-mitochondrion mistargeting of alanine/glyoxylate aminotransferase in primary hyperoxaluria type 1. J Cell Biol. 1990;111(6 Pt 1):2341–2351. doi:10.1083/jcb.111.6.2341
18. Garrelfs SF, Rumsby G, Peters-Sengers H, et al. Patients with primary hyperoxaluria type 2 have significant morbidity and require careful follow-up. Kidney Int. 2019;96(6):1389–1399. doi:10.1016/j.kint.2019.08.018
19. Molecular analysis of the glyoxylate reductase (GRHPR) gene and description of mutations underlying primary hyperoxaluria type 2 - PubMed. Available from: https://pubmed.ncbi.nlm.nih.gov/14635115/.
20. Martin-Higueras C, Garrelfs SF, Groothoff JW, et al. A report from the European Hyperoxaluria Consortium (OxalEurope) Registry on a large cohort of patients with primary hyperoxaluria type 3. Kidney Int. 2021;100(3):621–635. doi:10.1016/j.kint.2021.03.031
21. Belostotsky R, Seboun E, Idelson GH, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet. 2010;87(3):392–399. doi:10.1016/j.ajhg.2010.07.023
22. Leumann E, Hoppe B. The primary hyperoxalurias. J Am Soc Nephrol. 2001;12(9):1986–1993. doi:10.1681/ASN.V1291986
23. Bobrowski AE, Langman CB. The primary hyperoxalurias. Semin Nephrol. 2008;28(2):152–162. doi:10.1016/j.semnephrol.2008.01.008
24. Hoppe B, Pellikka PA, Dehmel B, Banos A, Lindner E, Herberg U. Effects of Oxalobacter formigenes in subjects with primary hyperoxaluria Type 1 and end-stage renal disease: a Phase II study. Nephrol Dial Transplant. 2021;36(8):1464–1473. doi:10.1093/ndt/gfaa135
25. Milliner DS, McGregor TL, Thompson A, et al. End points for clinical trials in primary hyperoxaluria. Clin J Am Soc Nephrol. 2020;15(7):1056–1065. doi:10.2215/CJN.13821119
26. Milliner DS, Cochat P, Hulton SA, et al. Plasma oxalate and eGFR are correlated in primary hyperoxaluria patients with maintained kidney function-data from three placebo-controlled studies. Pediatr Nephrol. 2021;36(7):1785–1793. doi:10.1007/s00467-020-04894-9
27. Hillebrand P, Hoppe B. Plasma oxalate levels in primary hyperoxaluria type I show significant intra-individual variation and do not correlate with kidney function. Pediatr Nephrol. 2020;35(7):1227–1233. doi:10.1007/s00467-020-04531-5
28. Milliner DS, Harris PC, Sas DJ, Cogal AG, Lieske JC. Primary hyperoxaluria type 1. 2002 Jun 19 [updated 2022 Feb 10]. In: Adam MP, Ardinger H, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 2022:1993–2022. PMID: 20301460.
29. Guillaume A, Chiodini B, Adams B, Dahan K, Deschênes G, Ismaili K. The struggling odyssey of infantile primary hyperoxaluria. Front Pediatr. 2021;9:317. doi:10.3389/fped.2021.615183
30. Fargue S, Rumsby G, Danpure CJ. Multiple mechanisms of action of pyridoxine in primary hyperoxaluria type 1. Biochim Biophys Acta. 2013;1832(10):1776–1783. doi:10.1016/j.bbadis.2013.04.010
31. Cochat P, Hulton SA, Acquaviva C, et al. Primary hyperoxaluria Type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant. 2012;27(5):1729–1736. doi:10.1093/ndt/gfs078
32. Management of primary hyperoxaluria: efficacy of oral citrate administration - PubMed. Available from: https://pubmed.ncbi.nlm.nih.gov/8476722/.
33. Sikora P, von Unruh GE, Beck B, et al. [13C2]oxalate absorption in children with idiopathic calcium oxalate urolithiasis or primary hyperoxaluria. Kidney Int. 2008;73(10):1181–1186. doi:10.1038/ki.2008.63
34. von Unruh GE, Voss S, Sauerbruch T, Hesse A. Dependence of oxalate absorption on the daily calcium intake. J Am Soc Nephrol. 2004;15(6):1567–1573. doi:10.1097/01.asn.0000127864.26968.7f
35. Holmes RP, Goodman HO, Assimos DG. Contribution of dietary oxalate to urinary oxalate excretion. Kidney Int. 2001;59(1):270–276. doi:10.1046/j.1523-1755.2001.00488.x
36. Marangella M, Petrarulo M, Cosseddu D, Vitale C, Linari F. Oxalate balance studies in patients on hemodialysis for type I primary hyperoxaluria. Am J Kidney Dis. 1992;19(6):546–553. doi:10.1016/s0272-6386(12)80833-x
37. Marangella M, Petrarulo M, Vitale C, Cosseddu D, Linari F. Plasma and urine glycolate assays for differentiating the hyperoxaluria syndromes. J Urol. 1992;148(3 Pt 2):986–989. doi:10.1016/s0022-5347(17)36796-4
38. Illies F, Bonzel KE, Wingen AM, Latta K, Hoyer PF. Clearance and removal of oxalate in children on intensified dialysis for primary hyperoxaluria type 1. Kidney Int. 2006;70(9):1642–1648. doi:10.1038/sj.ki.5001806
39. Jamieson NV; European PHI Transplantation Study Group. A 20-year experience of combined liver/kidney transplantation for primary hyperoxaluria (PH1): the European PH1 transplant registry experience 1984–2004. Am J Nephrol. 2005;25(3):282–289. doi:10.1159/000086359
40. Khorsandi SE, Samyn M, Hassan A, et al. An institutional experience of pre-emptive liver transplantation for pediatric primary hyperoxaluria type 1. Pediatr Transplant. 2016;20(4):523–529. doi:10.1111/petr.12705
41. Shapiro R, Weismann I, Mandel H, et al. Primary hyperoxaluria type 1: improved outcome with timely liver transplantation: a single-center report of 36 children. Transplantation. 2001;72(3):428–432. doi:10.1097/00007890-200108150-00012
42. Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19(10):673–694. doi:10.1038/s41573-020-0075-7
43. Debacker AJ, Voutila J, Catley M, Blakey D, Habib N. Delivery of oligonucleotides to the liver with GalNAc: from research to registered therapeutic drug. Mol Ther. 2020;28(8):1759–1771. doi:10.1016/j.ymthe.2020.06.015
44. Dana H, Chalbatani GM, Mahmoodzadeh H, et al. Molecular mechanisms and biological functions of siRNA. Int J Biomed Sci. 2017;13(2):48–57.
45. Liebow A, Li X, Racie T, et al. An investigational RNAi therapeutic targeting glycolate oxidase reduces oxalate production in models of primary hyperoxaluria. J Am Soc Nephrol. 2017;28(2):494–503. doi:10.1681/ASN.2016030338
46. Dutta C, Avitahl-Curtis N, Pursell N, et al. Inhibition of glycolate oxidase with dicer-substrate siRNA reduces calcium oxalate deposition in a mouse model of primary hyperoxaluria type 1. Mol Ther. 2016;24(4):770–778. doi:10.1038/mt.2016.4
47. Martin-Higueras C, Luis-Lima S, Salido E. Glycolate oxidase is a safe and efficient target for substrate reduction therapy in a mouse model of primary hyperoxaluria type I. Mol Ther. 2016;24(4):719–725. doi:10.1038/mt.2015.224
48. Clifford-Mobley O, Rumsby G, Kanodia S, Didi M, Holt R, Senniappan S. Glycolate oxidase deficiency in a patient with congenital hyperinsulinism and unexplained hyperoxaluria. Pediatr Nephrol. 2017;32(11):2159–2163. doi:10.1007/s00467-017-3741-1
49. Frishberg Y, Zeharia A, Lyakhovetsky R, Bargal R, Belostotsky R. Mutations in HAO1 encoding glycolate oxidase cause isolated glycolic aciduria. J Med Genet. 2014;51(8):526–529. doi:10.1136/jmedgenet-2014-102529
50. Frishberg Y, Deschênes G, Groothoff JW, et al. Phase 1/2 Study of lumasiran for treatment of primary hyperoxaluria type 1: a placebo-controlled randomized clinical trial. Clin J Am Soc Nephrol. 2021;16(7):1025–1036. doi:10.2215/CJN.14730920
51. Scott LJ, Keam SJ. Lumasiran: first approval. Drugs. 2021;81(2):277–282. doi:10.1007/s40265-020-01463-0
52. Alnylam Pharmaceuticals. ILLUMINATE-A: a Phase 3 Randomized, Double-Blind, Placebo-Controlled Study with an extended dosing period to evaluate the efficacy and safety of lumasiran in children and adults with primary hyperoxaluria type 1. clinicaltrials.gov; 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT03681184.
53. Garrelfs SF, Frishberg Y, Hulton SA, et al. Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N Engl J Med. 2021;384(13):1216–1226. doi:10.1056/NEJMoa2021712
54. Deschênes G, Cochat P, Magen D, et al. ILLUMINATE-B, a Phase 3 open-label study to evaluate lumasiran, an RNAi therapeutic, in young children with Primary Hyperoxaluria Type 1 (PH1); 2020:10.
55. Michael M, Groothoff JW, Shasha-Lavsky H, et al. ILLUMINATE-C, a single-arm, Phase 3 study of lumasiran in patients with primary hyperoxaluria type 1 and CKD3b-5, including those on hemodialysis; 2022:10.
56. Aldabek K, Grossman OK, AL-Omar O, Fox JA, Moritz ML. Infantile primary hyperoxaluria type 1 treated with lumasiran in twin males. Cureus. 2022;14(1):e21673. doi:10.7759/cureus.21673
57. Lai C, Pursell N, Gierut J, et al. Specific inhibition of hepatic lactate dehydrogenase reduces oxalate production in mouse models of primary hyperoxaluria. Mol Ther. 2018;26(8):1983–1995. doi:10.1016/j.ymthe.2018.05.016
58. Hoppe B, Koch A, Cochat P, et al. Safety, pharmacodynamics, and exposure-response modeling results from a first-in-human phase 1 study of nedosiran (PHYOX1) in primary hyperoxaluria. Kidney Int. 2022;101(3):626–634. doi:10.1016/j.kint.2021.08.015
59. Kempf C, Pfau A, Holle J, et al. Stiripentol fails to lower plasma oxalate in a dialysis-dependent PH1 patient. Pediatr Nephrol. 2020;35(9):1787–1789. doi:10.1007/s00467-020-04585-5
60. Le Dudal M, Huguet L, Perez J, et al. Stiripentol protects against calcium oxalate nephrolithiasis and ethylene glycol poisoning. J Clin Invest. 2019;129(6):2571–2577. doi:10.1172/JCI99822
61. Violier P, Boyer O, Berthaud R, Dorval G. Treatment with stiripentol in a patient with primary hyperoxaluria type 1: lesson for the clinical nephrologist. J Nephrol. 2022;35(3):1049–1051. doi:10.1007/s40620-021-01116-9
62. Martin-Higueras C, Feldkötter M, Hoppe B. Is stiripentol truly effective for treating primary hyperoxaluria? Clin Kidney J. 2021;14(1):442–444. doi:10.1093/ckj/sfaa068
63. Moya-Garzon MD, Gomez-Vidal JA, Alejo-Armijo A, et al. Small molecule-based enzyme inhibitors in the treatment of primary hyperoxalurias. J Pers Med. 2021;11(2):74. doi:10.3390/jpm11020074
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.