")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Lumacaftor-ivacaftor in the treatment of cystic fibrosis: design, development and place in therapy
Authors Connett GJ
Received 28 March 2019
Accepted for publication 1 July 2019
Published 19 July 2019 Volume 2019:13 Pages 2405—2412
DOI https://doi.org/10.2147/DDDT.S153719
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
GJ Connett
National Institute for Health Research, Southampton Respiratory Biomedical Research Centre, University Hospital Southampton NHS Foundation Trust, Southampton, SO16 6YD, UK
Abstract: Lumacaftor-ivacaftor is a combination of two small molecule therapies targeting the basic defect in cystic fibrosis (CF) at a cellular level. It is a precision medicine and its effects are specific to individuals with two copies of the p.Phe508del gene mutation. The drug combination works by restoring functioning CF transmembrane conductance regulator (CFTR) protein in cell surface membranes and was the first CFTR modulator licensed for the homozygous p.Phe508del genotype. The drug is a combination of a CFTR corrector and potentiator. Lumacaftor, the corrector, works by increasing the trafficking of CFTR proteins to the outer cell membrane. Ivacaftor, the potentiator, works by enabling the opening of what would otherwise be a dysfunctional chloride channel. In vivo lumacaftor-ivacaftor improves Phe508del-CFTR activity in airways, sweat ducts and intestine to approximately 10–20% of normal CFTR function with greater reductions in sweat chloride levels in children versus adults. Its use results in a modest improvement in lung function and a decreased rate of subsequent decline. Perhaps more importantly, those treated report increased levels of well-being and their rate of respiratory exacerbations is significantly improved. This review traces the development and use of this combination of CFTR modulators, the first licensed drug for treating the homozygous p.Phe508del CF genotype at the intracellular level by correcting the protein defect.
Keywords: corrector, modifier, modulator, Phe508del, DF508, Orkambi
Introduction
Cystic Fibrosis (CF) is an autosomal recessive disease caused by mutations in a gene on the long arm of chromosome 7 that encodes for the CF transmembrane conductance regulator (CFTR) protein. This protein is a cyclic adenosine monophosphate-regulated channel that facilitates the passive movement of chloride ions across the surface membranes of epithelial cells according to concentration gradients. It might also have important interactions with adjacent sodium channels and the movement of bicarbonate, but these interactions are incompletely understood. It is extraordinary that the loss of function of this protein channel results in such a severe life-shortening disease.
The cystic fibrosis clinical phenotype
The most important complication for the vast majority of CF sufferers is a predilection to respiratory infection and particularly with the bacteria Pseudomonas aeruginosa that results in progressive lung damage, irreversible respiratory failure and early mortality.1 CFTR function is also of critical importance to the health of other organs including the intestines, pancreas, liver, vas deferens and sweat glands.2,3 Up to 15% of those who are homozygous for CFTR gene mutations resulting in minimal CFTR protein function, present at birth with intestinal obstruction due to the retention of abnormally viscid meconium throughout their bowel. Recurrent intestinal symptoms are common amongst all patients and typically are increasingly problematic in adult life. The vast majority of patients, and in particular those with gene mutations that result in little or no residual CFTR function, are pancreatic insufficient. Such patients need to take pancreatic enzyme supplements before meals to compensate for their loss of exocrine function and are at risk of significant malnutrition including deficiencies of fat-soluble vitamins. Many go on to also suffer a loss of pancreatic endocrine function and develop diabetes mellitus for which they have to take insulin. Elevation of liver enzymes is common but small numbers of CF sufferers additionally go on to develop a focal biliary cirrhosis that can result in portal hypertension, variceal bleeding and eventually hepatic decompensation. Male CF sufferers are invariably sterile. The sweat glands of CF patients produce sweat with a markedly increased salt concentration. Whilst measuring this is a useful diagnostic confirmatory test of the functional abnormality of CFTR, in hot weather, excessive salt loss can result in metabolic decompensation and heat prostration.
The CF gene and CFTR gene therapy
The gene abnormality causing CF was discovered in 19854 and its 1480 amino acid sequence was determined 4 years later in 1989.5–7 The three base pair deletion known as p.Phe508del was also identified shortly thereafter and elucidated how gene mutations of this protein were disease-causing. The p.Phe508del mutation has since been characterized as the most common and particularly amongst Caucasian populations. Fifty percent of UK CF patients are homozygous for this gene defect and approximately 40% are compound heterozygotes for p.Phe508del plus a second disease-causing mutation.8 Since the discovery of the CFTR gene, 1900 sequence variations have been reported.9 A huge research effort has lead to a detailed understanding of a small percentage of these which account for the most common disease-causing variants. The functional consequences of many of the less common sequence variations are either unknown or might be associated with so-called CFTR-related disorders such as pancreatitis and congenital absence of the vas deferens.2
The breakthrough understanding about how CFTR mutations were disease-causing led to early hope for the prospect of gene therapy as a CF treatment modality. Research over subsequent years in the US and the UK resulted in a double-blind randomized controlled trial conducted by the Cystic Fibrosis Gene Therapy Consortium sponsored by the CF Trust in the UK. In this study, the CF gene was delivered directly to the airway by nebulizer.10 The study established the potential and proof of principle for this treatment approach, but clinical benefits were small compared to the placebo group. Future research collaborations are investigating the potential for modified lentivirus gene delivery to the airway which has the potential to restore gene transcription in the basal cells of the respiratory epithelium and thus obviate the need for repeat dosing.11 Whilst still promising, this direction of research has more recently been eclipsed by the prospect of small molecule therapies which can be taken orally called CFTR modulators. These drugs have the far greater potential of restoring CFTR protein function throughout the body as a result of their systemic mode of delivery and are a step change from the more limited conventional approach of treating the downstream secondary consequences of the biological defect.
Small molecule discovery and CFTR modulator therapy
Research to discover compounds that might act on dysfunctional CFTR protein was financed by the Cystic Fibrosis Foundation in the US in the 1990s through a company called Aurora Sciences. The company was subsequently acquired by Vertex in 2001. Vertex went on to develop and continues to manufacture Lumacaftor-ivacaftor alongside other CFTR modulator compounds. The researchers developed this new class of drugs through harvesting explanted lung cells from CF patients who had undergone lung transplantation. These cells were cultured in microplates to create an in-vitro pharmacology model that was subsequently utilized to robotically test hundreds of thousands of compounds for their cellular effects on CFTR function.12
Although the research focus was initially on patients with the most common gene mutation, the first major discovery using this approach was a compound that worked as a potentiator. This molecule, ivacaftor, proved highly effective in restoring the function of CFTR proteins with so-called gating defects.13,14 Gene mutations resulting in gating defects make CFTR proteins that are effectively transported to the outer cell membrane, but their ion channel is blocked. Ivacaftor is able to “wedge open” the channel and restore CFTR function. The most common CFTR gating mutation is called p.Gly551Asp. The prevalence of this mutation varies between countries, but approximately 5% of the UK CF population is heterozygous for this defect and this is the second most common gene defect after p.Phe508del.8 There are many other gating defects and ivacaftor is similarly effective across this mutation class.15 Ivacaftor has also been shown to be of benefit in adults with the R117H residual function mutation who tend to have less severe disease during childhood.16
To treat p.Phe508del patients, researchers needed to discover an additional “corrector” compound. Using ivacaftor alone to treat patients homozygous for the p.Phe508del mutation was shown to be ineffective.17 An additional molecule was needed to correct the shape of misfolded p.Phe508del CFTR proteins thus helping them to reach the cell membranes where they could be further improved through the secondary effect of ivacaftor correction of their additional gating defect (Figure 1). The first corrector drug discovered for p.Phe508del patients was lumacaftor. It is in current use in combination with ivacaftor and is licensed as the lumacaftor-ivacaftor drug combination Orkambi. The safety and effectiveness of the drug has been evaluated through global studies conducted through the CF Therapeutics Development Network and it is currently licensed by the FDA for use in children down to the age of two years.18,19 There are ongoing studies evaluating its efficacy in infants to determine whether it might prevent early CF complications including the development of pancreatic insufficiency which invariably occurs in p.Phe508del homozygote patients.
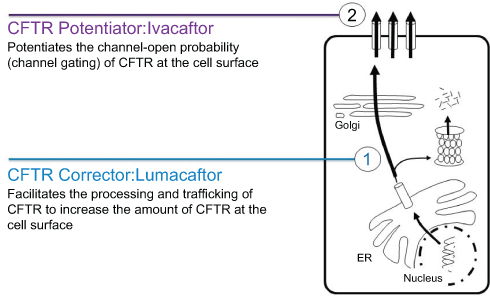 |
Figure 1 CFTR corrector (1) and potentiator (2) modulators work together to improve CFTR Function on the cell surface. |
Lumacaftor-ivacaftor key studies
The key trials leading to market authorization for the use of lumacaftor-ivacaftor were two Phase III multinational, double-blind, placebo-controlled studies published in 2015 called TRAFFIC and TRANSPORT.18 The studies were included in a recent Cochrane review of CFTR correctors for Class II mutations.20 The studies demonstrated similar efficacy for Lumacaftor 600 mg once daily and 400 mg twice daily used in combination with ivacaftor 250 mg twice daily and the primary outcome measure of significant improvement in percent predicted forced expiratory volume in 1 second (FEV1%) was achieved. The marketed formulation included the twice daily dosing regimen of lumacaftor for simplicity of use given that the ivacaftor component of the drug combination had to be given twice daily to be optimally effective.
More recently, a formulation of deuterated ivacaftor has been developed.21 The deuteration of small molecules, whereby one or more of their hydrogen atoms are replaced by the heavier, more stable isotope deuterium, results in significantly lower rates of metabolism and therefore a longer half-life. This opens up the possibility of lumacaftor-ivacaftor being relaunched as a once-daily treatment which might result in better adherence to treatment over the long term.
The Phase III studies reported a modest, but highly significant (P<0.001) mean absolute improvement in FEV1% of 2.6–4.0% compared to placebo at 24 weeks. The study thus achieved its primary outcome measure albeit with a mean improvement in lung function that was less than the generally accepted minimally important clinical difference of 5% (Figure 2).18 Perhaps more importantly, the rate of pulmonary exacerbations was 30–39% lower in the lumacaftor-ivacaftor groups compared with placebo (Figure 3) and it has been subsequently shown in a post hoc analysis that such benefits occurred irrespective of whether there were initial improvements in lung function.22 This is a particularly important finding given the increasing realization about the importance of pulmonary exacerbations in causing long-term lung function decline and poor respiratory health.23 The study was followed by a 96-week open-label trial extension called PROGRESS.19 In this study, an attempt was made to compare long-term outcomes with the natural history of lung function decline in untreated patients with the same genotype using CF foundation US-registry-matched controls. The study reported a 42% slower rate of FEV1% decline over the 96-week study period in support of sustained long-term benefits from treatment. However, these data were subject to several confounders. These included the possibility of systematic differences between the participants in this clinical trial compared to those who were not recruited, the calculation of each individual’s rate of decline being based on differing observation periods and the use of US control data that might not be comparable to that of other countries in which the open-label extension period took place. Quality of life, as measured using the CFQ-R respiratory domain score, was significantly improved by 4.1 points from baseline after 24 weeks of treatment in the TRAFFIC and TRANSPORT studies. This change was above the 4 points cutoff for a minimal clinically important difference from baseline for assessing interventions in stable patients.24 However, the between-group difference from baseline compared to placebo, although significant, was only 2.2. The trend toward improved CFQ-R scores was sustained in the open trial extension period but improvements were not significant in those who had transitioned from the placebo arm of the controlled trial period. Nutritional indices were also significantly improved and benefits were sustained across the double-blind and open-label trial periods.
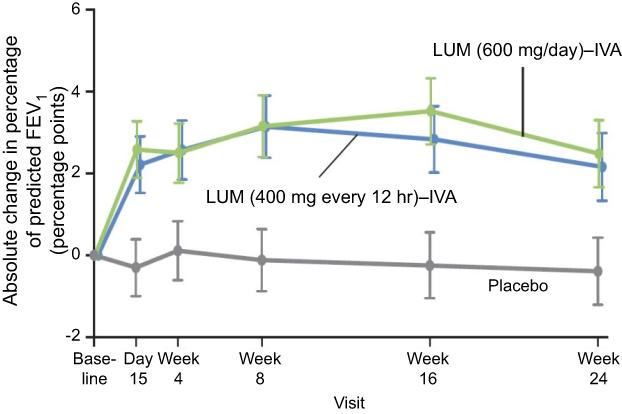 |
Figure 2 Change from baseline in percentage of predicted FEV1: Lumacaftor-ivacaftor (LUM-IVA) versus placebo.Note: Reprinted from Lancet Respir Med, 5(2), Konstan MW, McKone EF, Moss RB, et al, Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study, 107–118, Copyright 2017, with permission from Elsevier.19). |
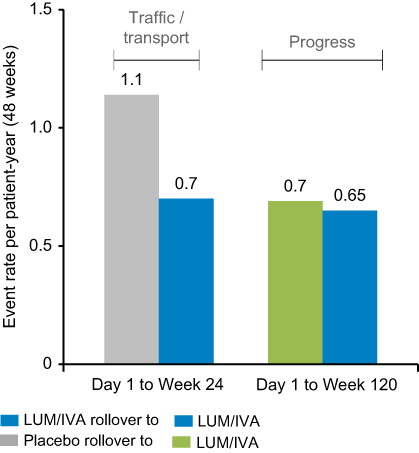 |
Figure 3 Lumacaftor-ivacaftor (LUM/IVA) treatment effects on projected annual rate of pulmonary exacerbations.Note: Reprinted from Lancet Respir Med, 5(2), Konstan MW, McKone EF, Moss RB, et al, Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study, 107–118, Copyright 2017, with permission from Elsevier.19 |
Lumacaftor-ivacaftor treatment for adults heterozygous for the p.Phe508del mutation has shown a modest restitution of CFTR function with an 11 mmol/L difference in sweat chloride levels from placebo after 56 days treatment. However, despite significant between-group minimal clinically important differences in mean CFQ-R scores of 6.5, there was no effect on the primary outcome measure FEV1%.25
The above studies were carried out in patients aged 12 years and above. Subsequent to this, an open label 24-week study in children aged 6–11 years reported that lumacaftor-ivacaftor was well tolerated with a similar safety profile to that seen in adults.26 There were no significant changes in spirometry, which was perhaps expected given the relatively well-preserved mean FEV1% of 91.4 in the study cohort as is typical for CF children in this age group. The study did however show significant improvements in lung clearance index, a lung function measure perhaps more indicative of more subtle changes in small airways disease which are less easily identified with spirometry. This lead the way for a subsequent double-blind, randomized controlled study in a similarly aged study cohort which demonstrated statistically significant improvements in lung clearance index, the studies primary outcome measure, as well as FEV1% when compared with placebo over a 24-week study period.27 Similar to the adult trials, CFQ-R scores were significantly improved (5.5 versus 3.0) but the treatment difference versus placebo was only 2.5 and thus below that of a minimal clinically important difference.
In both these studies, there were significant improvements in sweat chloride levels of around 20 mmol/L which were greater than the improvements seen in a Phase II dose-ranging study of adults in which improvements in sweat chloride were around 10 mmol/L.28 These improvements were similar to those reported in a post-market authorization study including adults and children aged >12 years in which sweat chloride levels improved by a mean of 17.8 mmol/L after 8–16 weeks treatment.29 Taken together, these studies suggest perhaps greater efficacy in children versus adults in relation to the level of CFTR correction.
More recently, Vertex funded a 24-week open-label study assessing the safety, tolerability and pharmacokinetics of lumacaftor-ivacaftor in children aged 2–5 years.2,30 The drug was generally well tolerated in this age group although 3 out of 60 patients discontinued lumacaftor/ivacaftor because of significant elevations in liver enzymes. Such elevations in liver enzymes have occasionally limited the use of this drug in clinical practice in all age groups and important safety information advises about the need to monitor for such adverse reactions. The results of ongoing studies are awaited into the use of lumacaftor-ivacaftor in 1–2-year-olds. Such early use of these treatments offers the greater potential for the prevention of CF-related complications and in particular the development of bronchiectasis. Whether the early use of lumacaftor-ivacaftor will significantly impact on the occurrence of pancreatic damage and pancreatic insufficiency as has occurred with the early use of ivacaftor is as yet unknown.31 In contrast to patients with a gating mutation, patients homozygous for the p.Phe508del genotype typically have more rapid onset of pancreatic failure. Lumacaftor-ivacaftor is less effective for p.Phe508del homozygotes than ivacaftor is for patients with G551D and other gating mutations. Whilst early use of lumacaftor-ivacaftor might lead to a reduction in the doses of enzymes needed to correct malabsorption, the effects are likely to be less than that achieved with ivacaftor for gating mutations.
Lumacaftor-ivacaftor has been studied in a stratified analysis of patients including adults with relatively severe lung disease as demonstrated by FEV1% of less than 40 at study commencement, albeit that lung function was greater than 40 at their screening visits. This patient sub-group achieved similar benefits in the reduction in the frequency of acute exacerbations as in those with less severe lung pathology.32 Use of lumacaftor-ivacaftor has also been reported in a young adult with end-stage lung disease.33 Used in this scenario, lumacaftor-ivacaftor was associated with improvements including a decrease in inflammatory markers and a greater level of stability. This enabled “bridging” as part of the preparation in advance of successful lung transplantation. A group of 20 patients awaiting heart lung transplant in Switzerland have also been recently reported.34 Lumacaftor-ivacaftor was introduced according to a step-wise protocol to overcome initial issues with tolerance and was associated with clinical improvements including stabilization of lung function and a decreased number of exacerbations compared with prior to treatment.
There is limited experience of lumacaftor-ivacaftor in children with severe CF lung disease, but the drug has been made available for compassionate use in individuals with either a sustained, rapid decline in lung function or those with persistently reduced lung function to an FEV1% of less than 40 despite optimal use of currently available treatments. This has happened in countries where there has been limited access to this medication. Sustained recovery of lung function has been achieved when children have been treated with lumacaftor-ivacaftor in this scenario although these benefits have not been consistent across all patients.35 Such differential responses might be due to differences in the pathophysiology of lung disease in CF adults versus some children in whom reduced lung function might be due in a greater part to small airways disease. It is possible that airways obstruction at this level is more reversible than extensive bronchiectasis through restoration of CFTR function.36
Side effects and drug interactions
Feelings of breathlessness and chest tightness are relatively common at the start of treatment and particularly in adults and those with more severe lung disease. Such problems can be overcome where there is concern by starting treatment at a lower dose.37 Some adult centers initiate lumacaftor-ivacaftor in hospital with close monitoring for such initial effects. This has not been an issue for the majority of pediatric patients. Gastrointestinal side effects such as diarrhea, nausea and abdominal pain in addition to breathlessness have been reasons for discontinuation. In a real-world setting, 14% of patients discontinued medication within a year of starting treatment and in two-thirds of cases, this was reported as being due to adverse drug reactions.38
Ivacaftor undergoes extensive liver metabolism through the cytochrome P450/CPY3A system and it has been suggested that liver injury could occur through toxic or immunogenic breakdown products. Lumacaftor is excreted largely unchanged in feces39
Elevations of alanine aminotransferase and aspartate aminotransferase liver enzymes have been reported in relation to lumacaftor-ivacaftor treatment, but most elevations are mild and transient.19 It is difficult to determine the relevance of these changes because similar changes occur in CF patients not on treatment as a result of CF-related liver dysfunction. Thus far there is no convincing evidence that lumacaftor-ivacaftor causes clinically significant liver injury, but it is recommended that liver function is checked 3-monthly for the first year of treatment and annually thereafter. Treatment should be interrupted if enzyme levels rise to greater than five times the upper limit of normal. Closer monitoring and dose reductions are recommended if the drug is used in patients with pre-existing moderate or severe CF-related liver disease.
Small elevations in blood pressure have been noted in clinical trials and so regular monitoring has been recommended whilst on treatment.18,19 Also, non-congenital lens opacities have been reported in children treated with drugs containing ivacaftor. Although these have not affected vision and their relevance is uncertain, ivacaftor-related cataracts were observed in newborn rats in pre-clinical studies and so baseline and follow-up ophthalmological examinations are recommended in pediatric patients.40
Given ivacaftor’s breakdown by CPY3A, concomitant use with strong inducers and inhibitors of this enzyme can affect the efficacy of lumacaftor-ivacaftor treatment through alterations in drug levels. Antifungal agents such as itraconazole, which are commonly used in CF patients, are strong CPY3A inhibitors and so lumacaftor-ivacaftor dosing schedules should be reduced when such drugs are used. Paradoxically, strong CPY3A inducers such as rifampicin and the herbal treatment St John’s wort, will reduce the efficacy of lumacaftor-ivacaftor and should therefore be avoided.
Complicating drug interactions further, lumacaftor is itself a strong inducer of CYP3A and can therefore limit the efficacy of drugs that are cleared through this metabolic pathway. In particular, the efficacy of hormonal contraceptives is impaired and alternative methods of contraception should be used to avoid pregnancy when using this drug. The list of drug interactions is extensive and a comprehensive drug history including the use of complementary therapies needs to be taken before starting treatment.
Unexpected benefits
Quite apart from its effects as a CFTR modulator, lumacaftor-ivacaftor has also been reported as exhibiting unexpected synergistic activity against highly polymyxin-resistant P. aeruginosa CF isolates.41 Such anti-microbial-resistant bacteria are an evolving problem in individuals with chronic lung infection. Whilst CFTR modulators are unlikely to eradicate these bacteria in patients with significant bronchiectasis, they might prove to be useful as part of anti-microbial treatment regimes to minimize their impact on disease progression.
The future
Whilst lumacaftor-ivacaftor has been a land mark treatment for CF, it will be succeeded by the next generation of CFTR modulators. Lumacaftor has already been substituted with tezacaftor in combination with ivacaftor in the drug Symdeko/Symkevi. This drug currently has approval for use in CF patients aged 12 years and older and studies are ongoing in younger age groups. Whilst this corrector-promoter drug combination is of similar efficacy to lumacaftor-ivacaftor, it appears to be better tolerated and has fewer problematic drug interactions.42 The tezacaftor-ivacaftor drug combination is currently being used alongside additional candidate corrector molecules as part of novel triple therapies. Phase III trials have recently been completed and published Phase II data show far greater efficacy for this triple therapy.42 Importantly, this combination of two correctors and a promoter is effective in patients heterozygous for p.Phe508del. The best mean FEV1% improvements in this dose-ranging study were 13.8% in those with p.Phe508del plus a second minimal function mutation and 11% in those who were homozygous. These improvements were in addition to tezacaftor-ivacaftor treatment at baseline and comparable to that achieved with ivacaftor used to treat gating mutations. Elexacaftor-tezacaftor-ivacaftor is the triple combination being taken forward for market approval.
More recently, the concept of theratyping has been developed whereby mutations are characterized according to their response to CFTR modulators using functional and biochemical modeling systems in-vitro. These include the use of cells from rectal biopsies to create organoid cell cultures to determine modulator drug efficacy to treat rarer genotypes.43
Conclusions
Lumacaftor-ivacaftor represents a significant advancement in treatment for CF patients homozygous for the p.Phe508del gene mutation. Its use has been associated with modest improvements in sweat chloride levels and lung function and the significant prevention of respiratory exacerbations. Its future use however is likely to be time limited by the availability of more effective combinations of CFTR modulators.
Disclosure
GJ Connett has been an investigator in Vertex, Flateley, Proteostasis, Zambon, Mylan and Corbus-sponsored cystic fibrosis clinical trials. The Southampton Children’s Hospital has received support for educational meetings from Vertex, Chiesi, Mylan, Novartis, PARI GmbH and Teva pharmaceuticals. GJ Connett reports personal fees from Vertex during the conduct of the study. The author reports no other conflicts of interest in this work.
References
1. Bhagirath AY, Li Y, Somayajula D, Dadashi M, Badr S, Duan K. Cystic fibrosis lung environment and pseudomonas aeruginosa infection. BMC Pulm Med. 2016;16(1):174. doi:10.1186/s12890-016-0339-5
2. Ferec C, Cutting GR. Assessing the disease-liability of mutations in CFTR. Cold Spring Harb Perspect Med. 2012;2(12):a009480. doi:10.1101/cshperspect.a009480
3. Shwachman H, Holsclaw DS. Complications of cystic fibrosis. N Engl J Med. 1969;281:500–501. doi:10.1056/NEJM196908282810910
4. Tsui LC, Buchwald M, Barker D, et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science. 1985;230:1054–1057. doi:10.1126/science.2997931
5. Kerem B-S, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. doi:10.1126/science.2570460
6. Rommens JM, Iannuzzi MC, Kerem B-S, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi:10.1126/science.2772657
7. Riordan JR, Rommens JM, Kerem B-S, et al. Identification of the cystic fibrosis gene: cloning and characterization of the complementary DNA. Science. 1989;245:1066–1073. doi:10.1126/science.2475911
8. UK Cystic Fibrosis. Registry annual data report 2017. Available from: https://www.cysticfibrosis.org.uk/the-work-we-do/uk-cf-registry/reporting-and-resources.
9. Cystic Fibrosis mutation database. Available from: http://www.genet.sickkids.on.ca.
10. Alton EW, Armstrong DK, Ashby D, et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med. 2015;3:684–691. doi:10.1016/S2213-2600(15)00245-3
11. Alton EW, Beekman JM, Boyd AC, et al. Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax. 2017;72(2):137–147. doi:10.1136/thoraxjnl-2016-208406
12. Pedemonte NGL, Lukacs K, Du E, et al. Small-molecule correctors of defective {Delta}F508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115(9):2564–2571. doi:10.1172/JCI24898
13. Ramsey BW, Davies J, McElvaney G. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672. doi:10.1056/NEJMoa1105185
14. De Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674–680. doi:10.1016/j.jcf.2014.09.005
15. Guimbellot J, Solomon GM, Baines A, et al. Effectiveness of ivacaftor in cystic fibrosis patients with non-G551D gating mutations. J Cyst Fibros. 2019;18(1):102–109. doi:10.1016/j.jcf.2018.04.004
16. Moss RB, Flume PA, Elborn JS, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015;3(7):524–533. doi:10.1016/S2213-2600(15)00201-5
17. Flume PA, Liou TG, Borowitz DS. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest. 2012;142(3):718–724. doi:10.1378/chest.11-2672
18. Wainwright CE, Elborn JS, Ramsey BW, et al. (TRAFFIC study group) lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–231. doi:10.1056/NEJMoa1409547
19. Konstan MW, McKone EF, Moss RB, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med. 2017;5(2):107–118. doi:10.1016/S2213-2600(16)30427-1
20. Southern KW, Patel S, Sinha IP, Nevitt SJ,. Correctors (specific therapies for class II CFTR mutations) for cystic fibrosis. Cochrane Database Syst Rev. 2018;8: Art. No.: CD010966. doi:10.1002/14651858.CD010966.pub2
21. Harbeson SL, Morgan AJ, Liu JF, et al. Altering metabolic profiles of drugs by precision deuteration 2: discovery of a deuterated analog of ivacaftor with differentiated pharmacokinetics for clinical development. J Pharmacol Exp Ther. 2017;362(2):359–367. doi:10.1124/jpet.117.241497
22. McColley SA, Konstan MW, Ramsey BW, et al. Lumacaftor/Ivacaftor reduces pulmonary exacerbations in patients irrespective of initial changes in FEV1. J Cyst Fibros. 2019;18(1):94–101. doi:10.1016/j.jcf.2018.07.011
23. Sanders DB, Bittner RC, Rosenfeld M, Redding GJ, Goss CH. Pulmonary exacerbations are associated with subsequent FEV1 decline in both adults and children with cystic fibrosis. Pediatr Pulmonol. 2011;46(4):393. doi:10.1002/ppul.v46.4
24. Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the cystic fibrosis questionnaire-revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic pseudomonas aeruginosa airway infection. Chest. 2009;135(6):1610–1618. doi:10.1378/chest.08-1190
25. Rowe SM, McColley SA, Rietschel E, et al. Lumacaftor/ivacaftor treatment of patients with cystic fibrosis heterozygous for F508del-CFTR. Ann Am Thorac Soc. 2017;14(2):213–219. doi:10.1513/AnnalsATS.201609-689OC
26. Milla CE, Ratjen F, Marigowda G, et al. Lumacaftor/ivacaftor in patients aged 6–11 years with cystic fibrosis and homozygous for F508del-CFTR. Am J Respir Crit Care Med. 2017;195(7):912–920. doi:10.1164/rccm.201701-0150WS
27. Ratjen F, Hug C, Marigowda G, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017;5(7):557–567. doi:10.1016/S2213-2600(17)30215-1
28. Boyle MP, Bell SC, Konstan MW, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014;2(7):527–538. Epub 2014 Jun 24. doi:10.1016/S2213-2600(14)70132-8
29. Graeber SY, Dopfer C, Naehrlich L, et al. Effects of lumacaftor-ivacaftor therapy on cystic fibrosis transmembrane conductance regulator function in Phe508del homozygous patients with cystic fibrosis. Am J Respir Crit Care Med. 2018;197(11):1433–1442. doi:10.1164/rccm.201710-1983OC
30. McNamara JJ, McColley SA, Marigowda G, et al. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2–5 years with cystic fibrosis homozygous for F508del-CFTR: an open-label phase 3 study. Lancet Respir Med. 2019. doi:10.1016/S2213-2600(18)30460-0
31. Davies JC, Cunningham S, Harris WT, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. 2016;(2):107–115. doi:10.1016/S2213-2600(15)00545-7
32. Elborn S
33. Pedersen SS, Jensen-Fangel S, Jeppesen M. Bridging for lung transplantation with lumacaftor/ivacaftor. Bridging for lung transplantation with lumacaftor/ivacaftor. Breathe. 2018;14:e68–e71. doi:10.1183/20734735.019318
34. Murer C, Huber LC, Kurowski T, et al. First experience in Switzerland in Phe508del homozygous cystic fibrosis patients with end-stage pulmonary disease enrolled in a lumacaftor-ivacaftor therapy trial – preliminary results. Swiss Med Wkly. 2018;148:w14593. doi:10.4414/smw.2018.14575
35. Hammond JA, Connett GJ. The use of lumacaftor/ivacaftor to treat acute deterioration in paediatric cystic fibrosis. Paediatr Respir Rev. 2018;27:16–17. doi:10.1016/j.prrv.2018.05.008
36. Tiddens HA, Donaldson SH, Rosenfeld M, Paré PD. Cystic fibrosis lung disease starts in the small airways: can we treat it more effectively? Pediatr Pulmonol. 2010;45(2):107–117. doi:10.1002/ppul.v45:2
37. Taylor-Cousar JL, Jain M, Barto TL, et al. Lumacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease homozygous for F508del-CFTR. J Cyst Fibros. 2018;17(2):228–235. doi:10.1016/j.jcf.2017.09.012
38. Brokaar E, van Leeuwen M, Leegwater E, et al. Adverse drug reactions and discontinuation rate during the first year on orkambi – the earliest results of the STORM study. J Cyst Fibros. 2019;18(Suppl 1):S130. doi:10.1016/S1569-1993(19)30553-3
39. Whiting P, Al M, Burgers L, et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health Technol Assess. 2014;18:18. doi:10.3310/hta18180
40. McColley SA. A safety evaluation of ivacaftor for the treatment of cystic fibrosis. Expert Opin Drug Saf. 2016;15(5):709–715. doi:10.1517/14740338.2016.1165666
41. Schneider EK, Azad MA, Han ML, et al. An unlikely pair: the antimicrobial synergy of polymyxin b in combination with the cystic fibrosis transmembrane conductance regulator drugs KALYDECO and ORKAMBI. ACS Infect Dis. 2016;2(7):478–488. doi:10.1021/acsinfecdis.6b00082
42. Keating D, Marigowda G, Burr L, et al. VX-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379(17):1612–1620. doi:10.1056/NEJMoa1807120
43. Berkers G, van Mourik P, Vonk AM, et al. Rectal organoids enable personalized treatment of cystic fibrosis. Cell Rep. 2019;26(7):1701–1708.e3. doi:10.1016/j.celrep.2019.01.068
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.