")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 8
LRRK2 inhibitors and their potential in the treatment of Parkinson’s disease: current perspectives
Authors Atashrazm F, Dzamko N
Received 16 August 2016
Accepted for publication 13 September 2016
Published 20 October 2016 Volume 2016:8 Pages 177—189
DOI https://doi.org/10.2147/CPAA.S102191
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Arthur E. Frankel
Farzaneh Atashrazm,1 Nicolas Dzamko2
1Neuroscience Research Australia, Randwick; 2School of Medical Sciences, Faculty of Medicine, University of New South Wales, Kensington, NSW, Australia
Abstract: Major advances in understanding how genetics underlies Parkinson’s disease (PD) have provided new opportunities for understanding disease pathogenesis and potential new targets for therapeutic intervention. One such target is leucine-rich repeat kinase 2 (LRRK2), an enigmatic enzyme implicated in both familial and idiopathic PD risk. Both academia and industry have promoted the development of potent and selective inhibitors of LRRK2, and these are currently being employed to assess the safety and efficacy of such compounds in preclinical models of PD. This review examines the evidence that LRRK2 kinase activity contributes to the pathogenesis of PD and outlines recent progress on inhibitor development and early results from preclinical safety and efficacy testing. This review also looks at some of the challenges remaining for translation of LRRK2 inhibitors to the clinic, if indeed this is ultimately warranted. As a disease with no current cure that is increasing in prevalence in line with an aging population, there is much need for developing new treatments for PD, and targeting LRRK2 is currently a promising option.
Keywords: synuclein, inflammation, lysosome, Rab, inhibitor, autophagy
Introduction
Parkinson’s disease
As global populations age, neurodegenerative diseases are rapidly emerging as a major human health burden. As the most common neurodegenerative movement disorder, Parkinson’s disease (PD) contributes substantially to this burden. Defined pathologically by the accumulation of proteinaceous inclusions in the brain, termed Lewy bodies, PD results in neuronal dysfunction and progressive loss of dopamine-producing neurons located in the substantia nigra pars compacta region of the midbrain. Declining dopamine levels are responsible for the classical PD motor symptoms of resting tremor, postural instability, rigidity, and slow movement. To a certain extent, dopamine replacement medication can improve motor function in PD patients, but treatment remains purely symptomatic with no currently available cures or disease-modifying therapies. Moreover, current therapies do not adequately address the nonmotor symptoms that manifest with PD such as sleep disorders, hallucinations, gastrointestinal dysfunction, depression, cognitive decline leading to dementia, and loss of smell. Collectively, the motor and nonmotor symptoms combine over decades to produce a progressively debilitating disease, the exact cause(s) of which have remained elusive for almost 200 years. A wealth of data, however, has implicated key biological processes that may contribute to neuronal dysfunction. Chief among these are autophagy/lysosomal dysfunction,1,2 mitochondrial dysfunction,3,4 and inflammation,5,6 all of which are highly integrated and complexly regulated. Unfortunately, due to a lack of disease biomarkers, studying the early phase of PD is not currently possible. Consequently, delineating which biological processes are causes, or indeed consequences, of up to decades of disease is exceedingly difficult. Knowing that in some instances PD can be caused by missense mutations in a number of proteins may give further clues to help unravel the complex etiology of PD.
PD genetics
Over the past two decades, new information describing how genetics may influence PD susceptibility has been uncovered. Inherited mutations that cause familial PD have been identified in ~18 genes, collectively accounting for ~10% of all PD.7–9 Familial PD mutations can be inherited in either a recessive or autosomal dominant manner and are predominantly missense mutations altering the function(s) of the proteins they encode. There is consequently much interest in delineating the biological function of these PD-implicated proteins and identifying converging disease-causing pathways. This is not trivial, however, as a number of familial PD genes cause disease in a manner atypical from the common idiopathic form. For example, mutations in Parkin are the most common cause of recessively inherited PD, but Parkin-associated PD presents with a much earlier onset and slower disease progression than idiopathic PD.10,11 By way of contrast, mutations in α-synuclein are often associated with a more aggressive form of PD.12 Indeed, clinical studies have long suggested heterogeneity in PD, suggesting caution in extrapolating from familial PD to the more common idiopathic form of PD. In the case of leucine-rich repeat kinase 2 (LRRK2), however, where the LRRK2 mutations are the most common cause of autosomal-dominant PD, there is substantial overlap with idiopathic PD in terms of clinical symptoms.13,14 Thus, it is plausible that understanding LRRK2 at least may provide insight into both familial and idiopathic PD.
The connection between LRRK2 and idiopathic PD is further strengthened by results from large-scale genome wide association studies and subsequent meta-analyses, suggesting that LRRK2 polymorphisms are linked to increased risk of idiopathic PD.15–17 Indeed, of the ~18 familial PD genes, only those encoding LRRK2 and α-synuclein are consistently associated with idiopathic PD risk. The polymorphisms identified by genome wide association studies are largely in the noncoding regions of the LRRK2 gene and are thus different from the missense mutations that cause familial PD. How these polymorphisms contribute to PD risk is unknown, but it is possible they may affect the transcriptional regulation of the LRRK2 gene and thus protein production. Being genetically linked to both familial and idiopathic forms of PD has elevated interest in LRRK2, but perhaps even more so, is that as a protein kinase, LRRK2 may present a readily druggable target. The majority of LRRK2 research has therefore focused on the enzyme’s catalytic kinase activity. Pubmed was used to search the literature with a focus on the past 5 years of LRRK2 research.
Evidence linking LRRK2 kinase activity to PD
Pathogenic LRRK2 mutations and kinase activity
By far, the most common pathogenic LRRK2 mutation is the substitution of glycine at position 2019 to serine (G2019S mutation). This mutation lies in a conserved part of the protein kinase domain that commences the activation loop, which, as the name implies, regulates catalytic enzyme activity (Figure 1). The neighboring I2020T mutation also lies in the kinase domain, while the other pathogenic LRRK2 mutations lie in either the Ras-like GTPase domain (R1441C/G/H), or the C-terminal of Ras domain (Y1699C) that links the GTPase and kinase domains of LRRK2.18 Studies using autophosphorylation as a surrogate readout of LRRK2 activity suggest a common mechanism across pathogenic mutations to increase kinase activity.19,20 In vitro kinase assays with model LRRK2 peptide substrates confirm that the G2019S mutation results in a direct two–threefold increase in kinase activity.21 The recent identification of Rab family GTPases as LRRK2 substrates has also allowed for in vivo assessment of LRRK2 activity by measuring Rab phosphorylation. Consistent with autophosphorylation results, pathogenic LRRK2 mutations increased Rab phosphorylation in cells.22 However, in vitro, only G2019S LRRK2 can increase Rab phosphorylation, a result consistent across in vitro kinase assay peptide substrates.22,23 This suggests distinct mechanisms by which the G2019S mutation directly increases LRRK2 kinase activity, presumably due to its location in the kinase domain, but other pathogenic mutations indirectly increase LRRK2 kinase activity. Exactly how mutations outside of G2019S increase kinase activity remains to be elucidated, but likely involves the complex interplay between the GTPase and kinase domains that has been observed. For example, a number of autophosphorylation sites in the GTPase domain may affect GTPase activity, and conversely, modulating GTPase activity has effects on LRRK2 kinase activity.24,25 Understanding this complex domain cross-talk will likely be important for understanding LRRK2 function, but nonetheless, biochemical evidence from mutation studies suggests a key role for LRRK2 kinase activity in mediating PD pathogenesis.
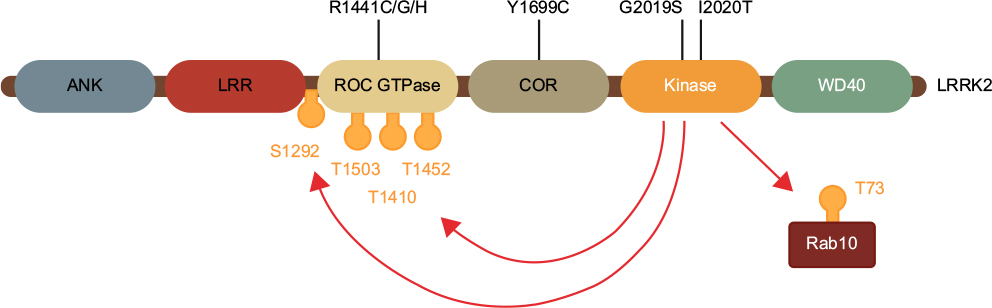 | Figure 1 Pathogenic LRRK2 mutations increase LRRK2 autophosphorylation and downstream substrate phosphorylation. Notes: The most common pathogenic LRRK2 missense mutations are located in the catalytic kinase domain or the ROC-COR tandem GTPase domain. All mutations increase the low levels of autophosphorylation on LRRK2, particularly Ser1292, and all mutations increase the phosphorylation of downstream Rab GTPase substrates such as Rab10. Abbreviation: LRRK2, leucine-rich repeat kinase 2. |
Increased kinase activity and LRRK2 pathobiology
Early studies in cells,26,27 flies,28 worms,29,30 and transgenic animal models31–33 demonstrated that overexpression of G2019S LRRK2 could induce or potentiate PD-like neurodegenerative phenotypes, particularly in regard to neuronal toxicity, dopamine transmission, and neurite morphology. In most instances, overexpression of G2019S LRRK2 was compared to wild-type (WT) LRRK2, the overexpression of which induced little to no phenotype in most models studied. This led to a concept that the kinase activity increasing LRRK2 G2019S mutation at least, may impart a new toxic gain of function. However, any mechanism by which increased LRRK2 kinase activity actually mediates PD-associated toxicity remains unclear. Indeed, even the normal function of LRRK2 remains unclear. The majority of reports though suggest roles for LRRK2 in the highly integrated pathways of inflammation, autophagy/lysosomal function and mitochondrial function, all of which are also highly implicated in PD pathogenesis.34–37 Overexpression models are not perfect, as there is no current evidence that LRRK2 is overexpressed in human PD; however, their use has predominated as mice with PD-causing knockin mutations in endogenous LRRK2 show almost no constitutive nigral neurodegenerative phenotype, even with aging.38–40 It is also noteworthy that the majority of overexpression-based studies do not directly demonstrate a pathogenic role for LRRK2 kinase activity, but rather provide models that could be used for testing LRRK2 inhibitors to better prove this hypothesis. Indeed, substantial resources have subsequently gone into LRRK2 inhibitor development.
The development of potent and selective LRRK2 inhibitors
The discovery of pathogenic LRRK2 mutations in 200441,42 opened a new opportunity for PD therapy, with a focus on developing LRRK2 inhibitors. However, drug development has proven to be highly challenging with a lack of knowledge regarding LRRK2 biology, blood–brain barrier (BBB) permeability limitation, and a lack of preclinical models that faithfully recapitulate PD phenotypes, among many challenging factors that need to be overcome to develop drugs for potential first-in-human trials.
Early nonselective compounds
The earliest compounds tested for blocking LRRK2 activity were largely nonselective pan-kinase inhibitors, mostly derived from cancer research. In 2009, Covy and Giasson examined the effect of 120 kinase inhibitors on WT and G2019S LRRK2, five of which were reported to be favorable candidates for LRRK2 inhibition; JAK3 inhibitor VI, K252A, staurosporine, Su-11248, and Ro-31-8220.43 Staurosporine is a natural indolinone component derived from a bacterium and inhibits a large number of protein kinases through prevention of adenosine triphosphate binding to the kinase.44 Among the five best targets, staurosporine was the most potent compound with an IC50 of 2 and 1.8 nM for WT and G2019S LRRK2, respectively. Despites high potency, its nonselectivity limits its usage. In the following studies, many such pan-kinase inhibitors were analyzed for their inhibitory activity against LRRK2, among which a wide range of nonselective LRRK2 inhibitors were described, including the tyrosine kinase inhibitor imatinib, mitogen-activated protein kinase kinase inhibitors PD98059 and U0126, Z8205, sunitinib, the Raf kinase inhibitors sorafenib and GW5074, and the anaplastic lymphoma kinase inhibitor, TAE684.45–49 Although many of these nonselective kinase inhibitors have a significant effect on LRRK2 activity in cells and rodents, their relatively poor potency, multiple kinase targets, and poor pharmacokinetic profiles limited their clinical application, necessitating further development of more favorable compounds.
First-generation selective compounds
LRRK2-IN-1 was first among a number of compounds with an improved potency and selectivity profile (Table 1).50 The selectivity of LRRK2-IN-1 was robustly characterized using a high-throughput kinase profiling screening of 442 kinases. Having a strong binding affinity to both WT and G2019S mutant LRRK2 in addition to a good pharmacokinetic exposure in mice with a half-life of 4.5 hours, LRRK2-IN-1 appeared to be a promising agent for PD therapeutic studies. However, its bulky size and impermeability through the BBB limited its use in vivo.50 Moreover, LRRK2-IN-1 appears to cause a number of off-target effects relating to inflammatory pathways,51 which confounds its use in this emerging area of LRRK2 biology.
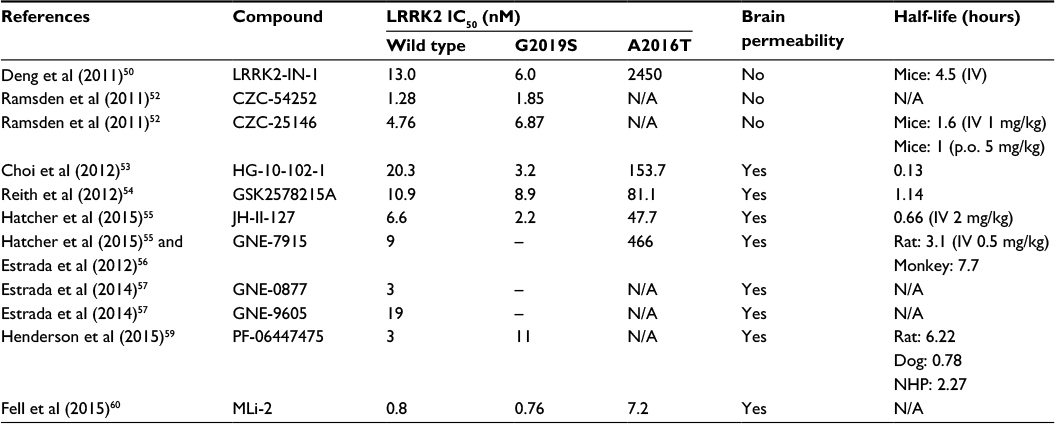 | Table 1 Characteristics and potency of selective LRRK2 inhibitors Abbreviations: LRRK2, leucine-rich repeat kinase 2; IV, intravenous; N/A, not available; NHP, non-human primate. |
CZC-54252 and CZC-25146 are other early “tool” inhibitors with improved specificity and potency against LRRK2.52 Analysis of the inhibitory effects of these two components on 185 diverse kinases showed that CZC-54252 inhibited only ten kinases whereas CZC-25146 showed a cleaner profile, inhibiting only five kinases. In vivo, CZC-25146 had relatively good pharmacokinetic properties and an extensive distribution throughout animal body following intravenous injection into mice. Unfortunately, similar to LRRK2-IN-1, both CZC compounds failed to cross the BBB, displaying only 4% BBB penetration.52 Thus, although providing excellent tool compounds for assay development, further improvements were still required to generate compounds for in vivo testing.
Second-generation highly potent and selective compounds with BBB permeability
HG-10-102-1 was the first selective LRRK2 inhibitor reported to penetrate the BBB and inhibit LRRK2 activity in mouse brain.53 It has an IC50 close to LRRK2-IN-1 and completely inhibits LRRK2 in brain following intraperitoneal administration at a dose of 100 mg/kg. However, with lower doses of 10 and 30 mg/kg, HG-10-102-1 could only partially inhibit LRRK2 in brain. Another highly potent and selective LRRK2 inhibitor that achieved high exposure to brain was GSK2578215A.54 Having good BBB permeability and a half-life of 1.14 hours, it seemed to be a reasonable candidate. However, despite the high exposure, GSK2578215A failed to inhibit LRRK2 in brain when it was injected intraperitoneal into mice. The pyrrolopyrimidine JH-II-127 is another selective LRRK2 inhibitor, which like HG-10-102-1, caused complete inhibition of LRRK2 in mouse brain at 100 mg/kg, but only partial inhibition at 30 and 10 mg/kg doses.55
Next, the small molecule GNE-7915 was reported following a high throughput screening system, revealing promising in vitro and in vivo profiles.56 This highly potent and selective LRRK2 inhibitor displayed low clearance values by rat hepatocytes, and a long half-life, in addition to high exposures in the periphery and brain throughout a 7-day dosing regimen in both rats and monkeys. Further development led to two more potent LRRK2 inhibitors, GNE-0877 and GNE-9605, both with enhanced brain penetration and cellular potency.57 These compounds demonstrated robust, dose-dependent inhibition of LRRK2 in the brain of transgenic mice expressing human LRRK2 G2019S protein.57 Both compounds also inhibited LRRK2 in the brain of nonhuman primates following oral administration.58
PF-06447475 is another second-generation LRRK2 inhibitor with high potency, selectivity, and good BBB permeability properties.59 Although good brain permeability and availability, and moderate clearance were observed in rats, administration of 5 mg/kg of this compound to rat, dog, and nonhuman primates showed a poor oral pharmacokinetic profile in three species. Treatment of bacterial artificial chromosome transgenic WT LRRK2 and G2019S LRRK2 mice with PF-06447475, however, decreased the LRRK2 activity in brain and kidney after 90 minutes, even at a low dose of 3 mg/kg. Moreover, in a safety study, oral administration of PF-06447475 over 2 weeks was well tolerated at doses of 65 mg/kg.59
Most recently, another novel potent, selective, brain penetrant LRRK2 inhibitor known as MLi-2 was reported.60 In vitro, MLi-2 presents a marked improvement in potency for G2019S LRRK2. In pharmacokinetic profile studies, MLi-2 had a mean maximum plasma concentration at 0.75 hours after 10 mg/kg oral administration and unbound fraction of 0.008 and 0.009 in plasma and brain, respectively. LRRK2 activity was decreased in brain following oral doses of MLi-2 in WT mice at 1 hour post-dose, with maximal suppression (>90% reduction) at 10 mg/kg. The development of LRRK2 blocking drugs has thus advanced to an extent that highly potent and selective compounds are now available to interrogate preclinical models of PD.
Preclinical efficacy testing
Key to advancing the translation of LRRK2 inhibitors to clinic is demonstrating proof-of principle of LRRK2 inhibitor efficacy in preclinical PD-models. This is challenging, as unfortunately for PD research, preclinical models are rarely ideal. Recent efforts have focused on generating improved overexpression-based models using viral vectors, and also human-induced pluripotent stem cell models to determine any protective effect of the newer, more potent, and selective LRRK2 inhibitors.
Viral vector rodent models
The use of recombinant viral vectors to induce regional overexpression in adult rodent brain holds promise for developing quicker and more controlled models for studying potential PD therapeutics.61 The use of viral vectors also allows a ready comparison between pathogenic mutation and kinase inactive forms of LRRK2. Two studies, one using mice and the other rats, have demonstrated that viral mediated overexpression of G2019S LRRK2 via unilateral injection into the striatum can result in the progressive loss of substantia nigra dopaminergic neurons.48,62 In contrast, in mice, the overexpression of kinase inactive LRRK2 did not cause neuronal loss. Moreover, evidence was presented that the relatively nonselective LRRK2 inhibitor GW5074 could attenuate the LRRK2 G2019S-induced neuronal loss in mice.48 It would be important to see if such studies can be repeated with the more selective inhibitors now available. The use of LRRK2 viral vector models is not without challenge, however. The retrograde transport of viral particles from the striatum to the substantia nigra can be variable and indeed the vectors themselves can induce an inflammatory response with variable toxicity.63 Additionally there is no aggregation of α-synuclein in these LRRK2 overexpression models,62,63 potentially due to the short timeframes involved. Also the extent to which any motor phenotype develops has not been robustly explored.
In addition to direct overexpression of LRRK2, there is also much interest in determining the efficacy of LRRK2 inhibitors in more established models of α-synuclein overexpression. Of particular note are the experiments by Daher et al that show that the LRRK2 inhibitor, PF-06447475, effectively attenuates α-synuclein-induced dopaminergic neurodegeneration in WT and transgenic LRRK2 G2019S expressing rats.64 These experiments involved adeno-associated viral mediated α-synuclein overexpression via unilateral injection directly into the substantia nigra pars compacta. Daily oral dosing of PF-06447475 over 4 weeks markedly prevented the loss of dopaminergic neurons in both the substantia nigra and striatum and also reduced α-synuclein-mediated neurodegeneration.64 The results in WT rats are potentially exciting, as they suggest a protective role for LRRK2 inhibitors in idiopathic PD; however, the relationship between LRRK2 and α-synuclein is complex and the extent to which LRRK2 mediates α-synuclein pathology and if/how this relates to neural toxicity is very unclear.65,66
Pluripotent stem cell models
The relatively new and rapidly accelerating field of induced pluripotent stem cells holds much promise for PD research. Such models allow for endogenous pathogenic variants to be studied in a neuronal context using primary human cells. The first step in the differentiation of induced pluripotent stem cells (IPSCs) to neurons is most often via neural stem cells, and a number of phenotypes have been uncovered for these cells that harbour LRRK2 mutations. Using a polymerase chain reaction-based assay to quantify mitochondrial DNA damage, Sanders et al could demonstrate increased damage in neural stem cells with either R1441C or G2019S mutations compared to nonmutation cells.67 Importantly, increased mitochondrial DNA damage could be reversed with zinc-finger nuclease-mediated correction of the G2019S mutation back to WT. This result is suggestive of mitochondrial dysfunction, and indeed, another study using G2019S and R1441C induced pluripotent stem cells-derived neural stem cells further reported reduced basal oxygen consumption, impaired mitochondrial dynamics, and increased sensitivity to valinomycin-induced stress in mutant cells.68 In addition to mitochondrial dysfunction, the LRRK2 G2019S mutation has also been associated with impaired nuclear envelope morphology, a phenotype often associated with cellular aging, and also impaired neural differentiation ability.69 Neural stem cells with the LRRK2 G2019S mutation have also been differentiated to dopamine neurons and show greater sensitivity to oxidative stress,70 as well as reduced neurite outgrowth71 and impaired autophagy.71,72 Moreover, a number of studies report abnormal and/or increased α-synuclein in G2019S neuronal cell lines compared to control.70–72 A number of these phenotypes have been reversed using early-generation inhibitors of LRRK2 such as LRRK2-IN1 and GW5074, and thus may serve as excellent models for testing the latest generation of specific and potent compounds.
Preclinical safety testing
To be efficacious for the treatment of PD, it is generally regarded that LRRK2 inhibitors will need to cross the BBB; however, it is established that other tissues, particularly lung, kidney, and certain peripheral immune cells robustly express LRRK2, potentially resulting in peripheral side effects (Figure 2). Indeed, brain tissue has far lower expression of LRRK2 than other tissues (~6.2-fold less than that of kidney). It is noteworthy that LRRK2 knockout mice have a normal life span; however, a number of studies have reported pathology in peripheral organs. Whether such pathologies are safety liabilities or indeed, are even observed in pharmacological studies, where the kinase activity of LRRK2 is only partially inhibited rather than genetically ablated, is currently under investigation.
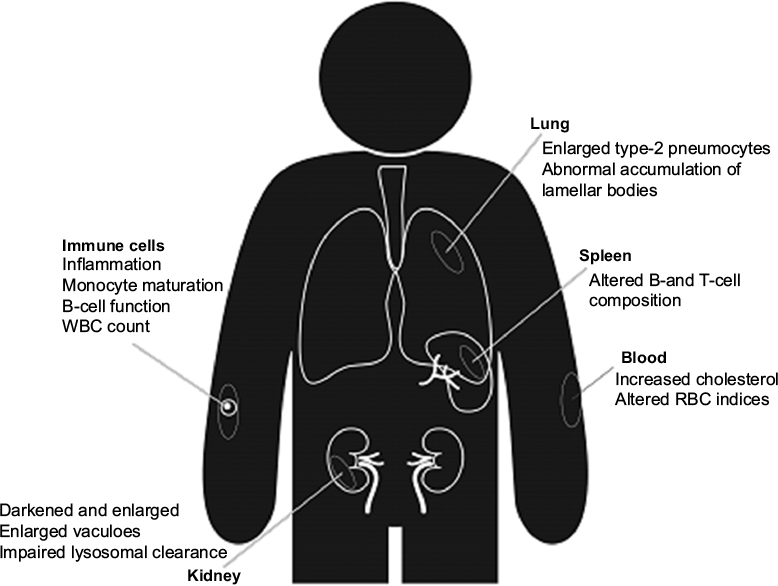 | Figure 2 Peripheral actions of LRRK2 inhibitors. Notes: LRRK2 has a particularly high expression in kidney, lung, spleen, and peripheral immune cells. A number of studies using knockout, kinase inactive knockin, and LRRK2 inhibitor-treated rodents have reported potential peripheral side effects of LRRK2 manipulation. To what extent these relate to a safety liability in humans is still unknown; however, compared to many drugs, LRRK2 inhibitors appear to be well tolerated. Abbreviations: LRRK2, leucine-rich repeat kinase 2; WBC, white blood cells; RBC, red blood cells. |
Kidney function
Among different tissues, renal cells express the highest level of LRRK2 and therefore, not surprisingly, LRRK2 loss has the most striking effect on kidney. Macroscopically, darkening of kidney and its altered weight are the main reported features in genetically modified rats and mice with knockout of LRRK2 and less consistently, in kinase-dead LRRK2 mice (animals with kinase-inactivating point mutations).73,74 These morphological alterations seem to be gender biased as they are mainly seen in male animals and not females.75,76 Histological examination of LRRK2 knockout rats and mice shows hyaline droplets, brown pigment accumulation, and cytoplasmic vacuolation in renal tubes of the cortex and outer medulla,73,74 as well as microvacuoles in proximal tubules, which become larger when animals age. Herzig et al characterized the observed enlarged vacuoles to be of lysosomal structures, which were redistributed from an apical distribution in WT kidney to a perinuclear distribution in knockout and kinase-dead kidney of mice.73 Indeed, there is some evidence of impaired autophagy-lysosomal function with aggregation of α-synuclein reported in the kidney of 20-month-old LRRK2 knockout mice, whereas it is normally present at very low levels (∼1/200 compared to the brain).74 Despite the marked macroscopic and immunohistochemical differences, there is little evidence that accumulation of proteins and altered lysosomal/autophagy causes kidney dysfunction. Proteinuria was observed in old, but not younger LRRK2 knockout rodents,73 however, this was perhaps due to reduced tubular function in old mice rather than glomerular function. In another study, no differences were observed in blood urea nitrogen, serum creatinine, and their ratio between WT and LRRK2 mice, suggesting that the renal filtration function is not altered in LRRK2 knockout mice.76 Perhaps most importantly, however, is that the described kidney phenotype is not observed in heterozygous animals73 or indeed rodents or nonhuman primates treated with LRRK2 inhibitors, including MLi-2.60 This suggests a chronic loss of LRRK2 protein or activity caused genetically, rather than a partial or acute loss of kinase activity caused by pharmacological inhibition, is required before kidney phenotypes will present.
Phospholipidosis
Another reported side effect of loss/inhibited LRRK2 is phospholipidosis (PLD). In a study by Fuji et al, administration of LRRK2 inhibitors GNE-7915 and GNE-0877 in monkeys resulted in decreased urine di-22:6-BMP.58 Di-22:6-BMP is a phospholipid that is normally localized in the internal membrane of lysosomes and late endosomes, and is responsible for lysosomal degradation. Enlarged and increased numbers of lysosomes with stacked, whorled membranes and lipid were also observed in proximal tubules of LRRK2 knockout mice kidney,73 suggestive of accumulated phospholipid membranes in lysosomes. Drug-induced PLD is an acquired lysosomal storage disorder characterized by excessive accumulation of phospholipids and drugs in lysosomes in different tissues such as kidney, heart, and lungs.77 Since 1970, when the term PLD started to be used, more than 500 publications have reported PLD findings.78,79 It should be noted that PLD findings mainly include situations during preclinical studies in animals, and its significance in humans is unclear. Based on a FDA report, only a small number of PLD have induced toxicity in humans, because, first, it could be an adaptive response of the host to a drug, rather than a toxic manifestation, and second, the PLD seen in animals in preclinical studies is not predictive of similar findings in clinics.79 Thus, it is unclear if the observed PLD in animals with knockout or pharmacological inhibition of LRRK2 is a significant safety concern.
Lamellar bodies in lung
Lung is another organ in which LRRK2 has been implicated in pathological features. Unlike kidney tissue, pathological features in lung are observed with both knockout and pharmacological kinase inhibition of LRRK2, although with variable consistency across different models. First observed in knockout mice and rats, the major change in lung is abnormal accumulation of lamellar bodies in type II pneumocytes.73,80 Lamellar bodies are secretory lysosomal-related organelles measuring 0.2–0.3 μM in diameter. In pneumocytes, they secrete their stored contents (mainly phospholipids) known as pulmonary surfactants into the extracellular space. Enlargement of type II pneumocytes (defined as very slight) was observed in lungs of mice treated with MLi-2.60 Monkeys treated with GNE-7915 over a 7-day period also displayed increased size and number of lamellar bodies in lungs, similar to those seen in knockout animals, suggesting it to be an on-target side effect of LRRK2 inhibition.58 Apart from the accumulated lamellar bodies, which did not increase following a longer period of treatment (29 days), no other effect on the lungs was seen in monkeys. Interestingly, in contrast to monkeys, no pathological feature, including accumulation of lamellar bodies was seen in the lungs of mice and rats that were treated with GNE-7915 and GNE-0877,58 or mice treated with PF-06447475.64 The enlarged type II pneumocyte phenotype was also not observed in the lungs of LRRK2 mice with a kinase activating point mutation.73 Importantly, no changes have been reported in the size or weight of lungs in LRRK2 knockout, kinase inactive, or inhibitor-treated animals.73,75 As for kidney, it is therefore unclear if such a phenotype is a safety liability for human trials. Regardless, lung function could be readily monitored in early safety trials.
Inhibitor-induced degradation of LRRK2 protein
Another potential concern for LRRK2 inhibitors stems from observations that chronic LRRK2 inhibition often results in reduced expression of LRRK2 protein. Reduced levels of LRRK2 have been observed in mice with a kinase inactivating mutation73 and also in inhibitor-treated primates,58 mice,81 and primary human peripheral blood mononuclear cells.82 Mechanistically, LRRK2 inhibitor treatment appears to promote the ubiquitination of LRRK2 and consequently its proteosomal degradation.81 While quite high doses are required to achieve 20%–40% loss of LRRK2 protein, the consequences of continued pharmacological inhibition are still unclear. A reduction in LRRK2 levels with inhibitor treatment could explain the overlap in pathology seen in the lungs of knockout and inhibitor-treated animals. Moreover, loss of LRRK2 has been associated with increased susceptibility to inflammatory bowel disease83 and infection84 in mice. Thus, what happens to total LRRK2 levels under therapeutic dosing conditions would be useful to monitor.
Challenges for advancing to clinical trials
Despite more than 10 years of research since LRRK2 was first identified as a genetic cause of PD, many aspects of LRRK2 biology still remain unclear. How LRRK2 mutations lead to PD is unknown, and consequently what should/could be used as readouts of target engagement or inhibitor efficacy. Moreover, who should potentially be treated with LRRK2 kinase inhibitors and when?
Identifying the PD-relevant pathobiology
As major regulators of signal transduction pathways it is not unusual for protein kinases to mediate a number of biological responses. In this regard, LRRK2 may be no exception, having been linked to, among others, innate immunity, mitochondrial function, lysosomal trafficking and autophagy, WNT signaling, neurogenesis, synaptogenesis, transcriptional regulation and protein synthesis, cell death, and dopamine release (Figure 3). Trying to identify which aspects of LRRK2 biology are important for mediating PD is a major challenge. The recent identification of Rab GTPase proteins as bona fide LRRK2 substrates22 is a potentially exciting advance; however, the ~70-member family of Rab proteins themselves regulate many aspects of cell biology.85 Intriguingly, however, Rab7L1, also known as Rab29, has been genetically implicated in PD risk associated with the PARK16 locus.86,87 Studies in model organisms suggest that both LRRK2 and Rab7L1 work in the same biological pathway to regulate lysosomal function and cargo trafficking, particularly between the trans Golgi network and early/late endosomes.88–90 However, in vitro, LRRK2 has a marked preference for phosphorylating threonine residues over serine,91 and thus LRRK2 could only very weakly phosphorylate the serine containing Rab7L1 isoform.22 It still remains possible that LRRK2 could phosphorylate and regulate Rab7L1 in vivo, but under what conditions remains to be determined. Interestingly, Rab7L1 has also been implicated in innate immunity and pathogen clearance,92 another biological function involving LRRK2.93 Moreover, LRRK2 was found to interact with Rab10 and Rab2a, with LRRK2 and Rab2a in particular, being required for proper lysosome sorting in Paneth cells and protection from intestinal infection.84 Rab10, the most robustly phosphorylated of the identified LRRK2 substrates,22 is further involved in innate immunity through trafficking of toll-like receptors,94 the activation of which in turn leads to phosphorylation of LRRK2.95 Thus, lysosomal function and protein trafficking are likely to be key mediators of LRRK2 pathobiology with relevance for PD and also other LRRK2-associated diseases such as Crohn’s inflammatory bowel disease.83 However, understanding the specific context in which dysfunction leads to disease is still a major challenge.
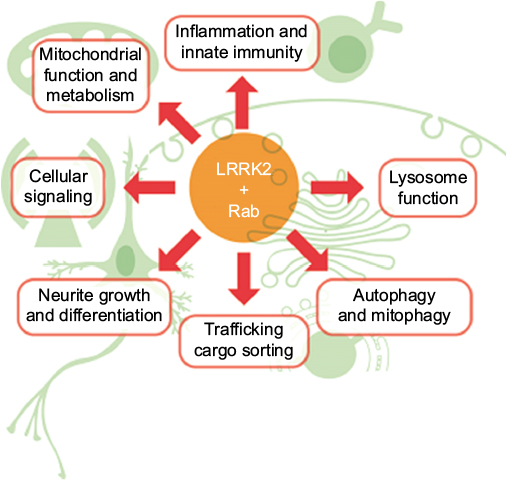 | Figure 3 Potential mediators of LRRK2 pathobiology. Notes: LRRK2 has been implicated in a variety of biological processes known to be perturbed in Parkinson’s disease. Rab GTPases, a number of which were recently identified as LRRK2 substrates, also contribute to the complex regulation of many PD-implicated biological processes. Abbreviations: LRRK2, leucine-rich repeat kinase 2; PD, Parkinson’s disease. |
Better direct measures of target engagement
The current gold standard for demonstrating LRRK2 inhibitor target engagement is measuring the loss of phosphorylation on serine residues 910, 935, 955, and 973 (Figure 4). These residues are located just before the leucine-repeat domains in LRRK2 and, at least in the case of Ser910 and Ser935, serve to bind 14-3-3 adaptor proteins and potentially regulate the cellular localization of LRRK2.23 It has been unequivocally demonstrated in many cell and animal models that LRRK2 inhibitors cause a dose-dependent loss of phosphorylation at these sites,50,96,97 but exactly how this occurs is unknown. It is evident that these serine residues are not autophosphorylation sites, rather they are constitutively phosphorylated by other kinases whose identity has remained elusive.96,97 Therefore, rather than a direct effect, following inhibitor treatment LRRK2 must either negatively regulate the upstream kinase(s), or engage phosphatases. In regard to the latter, LRRK2 inhibitor treatment increases binding of protein phosphatase 1α to LRRK2 and promotes dephosphorylation of serines 910, 935, 955, and 973.98 However, what phosphorylates LRRK2 at these residues in the first place is largely unknown. Protein kinase A,99 casein kinase 1α,100 and the IkappaB kinases95 can all phosphorylate some or all of these LRRK2 serine residues when stimulated, but it is unclear if one or any of these are the kinase responsible for the constitutive LRRK2 serine phosphorylation. Thus, although effective, the complicated nature of using serines 910, 935, 955, and/or 973 to infer target engagement requires careful validation. To some extent this has been demonstrated for peripheral blood mononuclear cells from idiopathic PD patients, where LRRK2 Ser910 and Ser935 phosphorylation were found to be invariant to age, gender, PD severity or duration, and L-Dopa medication,101 and were dephosphorylated to a similar extent with ex vivo LRRK2 inhibitor treatment.82 Nonetheless, it would be prudent to employ a second readout of target engagement for validation.
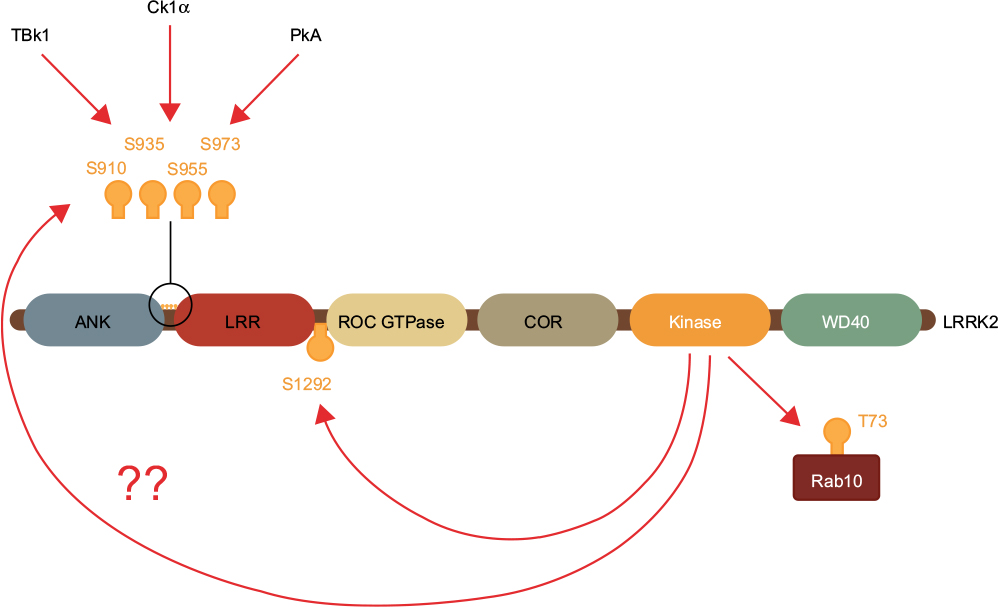 | Figure 4 Measuring LRRK2 inhibitor target engagement. Notes: LRRK2 inhibitors cause the loss of constitutive phosphorylation on serine residues 910 and 935. However, these are not autophosphorylation sites and only serve as indirect measures of target engagement. Unlike S910 and S935, S1292 is a bona fide LRRK2 autophosphorylation site, but its low stoichiometry of phosphorylation makes it difficult to measure. LRRK2 can also directly phosphorylate certain Rab GTPase proteins such as Rab10, and these may serve as good direct measures of target engagement if robust assays can be developed. Abbreviation: LRRK2, leucine-rich repeat kinase 2. |
One possibility for a second readout is measuring LRRK2 Ser1292 phosphorylation, which unlike serines 910, 935, 955, and 973 is a bona fide autophosphorylation site.19 While there has been some success in measuring Ser1292 phosphorylation in exosomes purified from urine,102 the very low stoichiometry of phosphorylation at this residue has complicated its measurement in other biospecimens and highly sensitive assay platforms will be required. As well as potentially providing much needed insight into LRRK2 pathobiology, the recent identification of Rab GTPases as direct LRRK2 substrates may also hold promise for developing measures of target engagement.
Finally, it is also noteworthy that no central measures of inhibitor target engagement have yet been developed that would be suitable for human drug trials. Although LRRK2 is highly expressed in peripheral immune cells, its expression in cerebrospinal fluid is minimal and thus measuring LRRK2 phosphorylation problematic. Whether LRRK2 phosphorylated Rab proteins can be detected in cerebrospinal fluid remains to be determined, likewise whether imaging agents can be used to measure LRRK2 or its substrates in living brain. A central readout of target engagement will be important as it is generally regarded that brain penetration will be required to treat PD.
When to treat and who
If LRRK2 blocking drugs are ultimately developed that are safe and efficacious in preclinical models then trials in humans would seemingly be warranted. Based on evidence to date, it would seem prudent to first trial such drugs in patients with activating LRRK2 mutations, particularly G2019S as this is by far the most common. A role for LRRK2 in neurogenesis has been proposed;103 however, there is no strong evidence that LRRK2 inhibitors will restore lost dopaminergic neurons and reverse PD. It will thus be important to determine if LRRK2 inhibitors should be used to try to slow down / halt the disease in already clinically diagnosed subjects (ie. already substantial loss of dopamine neurons). Or indeed whether LRRK2 inhibitors could / should be administered to asymptomatic mutation carriers to delay or even prevent the onset of neurodegeneration. The latter scenario is complicated by the incomplete penetrance of the G2019S mutation, which is estimated to be ~20%–50%,104–106 and therefore the need for earlier disease measures or predictors of which mutation carriers are likely to convert to clinical PD. A number of such measures have been proposed for asymptomatic LRRK2 mutation carriers, including increased peripheral inflammation,107 asymmetrical arm swing108 or gait changes when challenged,109 reduced nigrostriatal denervation,110 and subtle differences in nonmotor symptoms.111 However, lengthy longitudinal assessments are required to determine if any markers can be identified that robustly predict conversion to PD.
Another interesting prospect is whether LRRK2 inhibitors could also be used to treat more common idiopathic PD. Potential mediators of LRRK2 pathobiology, including lysosomal clearance defects, inflammation, and mitochondrial dysfunction, also clearly underlie idiopathic PD. PD by nature is heterogeneous, but LRRK2 G2019S-associated PD at least is largely similar to idiopathic PD in terms of age-of-onset, disease progression, and neuropathology. As outlined, LRRK2 is also genetically implicated in the increased risk of idiopathic PD. Thus, it is worth considering if LRRK2 inhibitors may have a broader utility; however, there is no evidence yet that LRRK2 activity is increased in idiopathic PD patients. This is largely due to the challenge of measuring LRRK2 activity in vivo, which will likely become feasible with the identification of LRRK2 substrates.
Conclusion
Increasing evidence suggests that LRRK2 kinase activity contributes to its pathogenic role in PD. Recent studies suggest a converging theme of pathogenic LRRK2 mutations to increase both LRRK2 autophosphorylation and Rab GTPase substrate phosphorylation. Early preclinical studies also suggest a protective role of LRRK2 inhibitors in some models, but replication with the newer generation of highly potent and selective inhibitors is required. Moreover, preclinical models of PD themselves are not ideal and advances in developing models that recapitulate key PD features would enable better testing of LRRK2 inhibitors. Any mechanism of action of how LRRK2 inhibitors may work to protect neurons in preclinical models is also largely unknown. Emerging trends, however, suggest LRRK2 functions in the highly integrated pathways of autophagy/lysosomal function, mitochondrial function, and inflammation. LRRK2 inhibitors also perturb some of these functions in peripheral organs, particularly lung tissue, resulting in potentially unwanted side effects. Compared to many drugs though, LRRK2 inhibitors appear to be well tolerated. Further research to understand LRRK2 function and build evidence of LRRK2 inhibitor efficacy will aid in the translation of these drugs to potential clinical trials.
Acknowledgments
This research into PD is funded by the Michael J Fox Foundation for PD research, the Shake It Up Australia Foundation, Parkinson’s NSW, the University of New South Wales, and the National Health and Medical Research Council. We thank Heidi Cartwright for making the figures.
Disclosure
The authors report no conflicts of interest in this work.
References
Xilouri M, Brekk OR, Stefanis L. Autophagy and alpha-synuclein: relevance to Parkinson’s disease and related synucleinopathies. Mov Disord. 2016;31(2):178–192. | ||
Rivero-Rios P, Madero-Perez J, Fernandez B, Hilfiker S. Targeting the autophagy/lysosomal degradation pathway in Parkinson’s disease. Curr Neuropharmacol. 2016;14(3):238–249. | ||
Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. Epub 2016 Aug 21. | ||
Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40(4):200–210. | ||
Chao Y, Wong SC, Tan EK. Evidence of inflammatory system involvement in Parkinson’s disease. Biomed Res Int. 2014;2014:308654. | ||
Su X, Federoff HJ. Immune responses in Parkinson’s disease: interplay between central and peripheral immune systems. Biomed Res Int. 2014;2014:275178. | ||
Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson’s disease: progress and therapeutic implications. Mov Disord. 2013;28(1):14–23. | ||
Ferreira M, Massano J. An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol Scand. Epub 2016 Jun 8. | ||
Gasser T, Hardy J, Mizuno Y. Milestones in PD genetics. Mov Disord. 2011;26(6):1042–1048. | ||
Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–608. | ||
Lucking CB, Durr A, Bonifati V, et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342(21):1560–1567. | ||
Devine MJ, Gwinn K, Singleton A, Hardy J. Parkinson’s disease and alpha-synuclein expression. Mov Disord. 2011;26(12):2160–2168. | ||
Haugarvoll K, Rademakers R, Kachergus JM, et al. Lrrk2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology. 2008;70(16 Pt 2):1456–1460. | ||
Healy DG, Falchi M, O’Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7(7):583–590. | ||
Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308–1312. | ||
Nalls MA, Pankratz N, Lill CM, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 2014;46(9):989–993. | ||
Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41(12):1303–1307. | ||
Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci. 2010;11(12):791–797. | ||
Sheng Z, Zhang S, Bustos D, et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med. 2012;4(164):164ra161. | ||
West AB, Moore DJ, Biskup S, et al. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102(46):16842–16847. | ||
Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem J. 2007;405(2):307–317. | ||
Steger M, Tonelli F, Ito G, et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016;5:e12813 | ||
Nichols RJ, Dzamko N, Morrice NA, et al. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem J. 2010;430(3):393–404. | ||
Taymans JM. The GTPase function of LRRK2. Biochem Soc Trans. 2012;40(5):1063–1069. | ||
Gilsbach BK, Kortholt A. Structural biology of the LRRK2 GTPase and kinase domains: implications for regulation. Front Mol Neurosci. 2014;7:32. | ||
Greggio E, Jain S, Kingsbury A, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23(2):329–341. | ||
Heo HY, Park JM, Kim CH, Han BS, Kim KS, Seol W. LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp Cell Res. 2010;316(4):649–656. | ||
Liu Z, Wang X, Yu Y, et al. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A. 2008;105(7):2693–2698. | ||
Saha S, Guillily MD, Ferree A, et al. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J Neurosci. 2009;29(29):9210–9218. | ||
Yao C, El Khoury R, Wang W, et al. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol Dis. 2010;40(1):73–81. | ||
Li X, Patel JC, Wang J, et al. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J Neurosci. 2010;30(5):1788–1797. | ||
Melrose HL, Dachsel JC, Behrouz B, et al. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol Dis. 2010;40(3):503–517. | ||
Ramonet D, Daher JP, Lin BM, et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One. 2011;6(4):e18568. | ||
Kumaran R, Cookson MR. Pathways to Parkinsonism Redux: convergent pathobiological mechanisms in genetics of Parkinson’s disease. Hum Mol Genet. 2015;24(R1):R32–R44. | ||
Wallings R, Manzoni C, Bandopadhyay R. Cellular processes associated with LRRK2 function and dysfunction. FEBS J. 2015;282(15):2806–2826. | ||
Martin I, Kim JW, Dawson VL, Dawson TM. LRRK2 pathobiology in Parkinson’s disease. J Neurochem. 2014;131(5):554–565. | ||
Esteves AR, Swerdlow RH, Cardoso SM. LRRK2, a puzzling protein: insights into Parkinson’s disease pathogenesis. Exp Neurol. 2014;261:206–216. | ||
Tong Y, Pisani A, Martella G, et al. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci U S A. 2009;106(34):14622–14627. | ||
Liu HF, Lu S, Ho PW, et al. LRRK2 R1441G mice are more liable to dopamine depletion and locomotor inactivity. Ann Clin Transl Neurol. 2014;1(3):199–208. | ||
Yue M, Hinkle KM, Davies P, et al. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol Dis. 2015;78:172–195. | ||
Paisan-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600. | ||
Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–607. | ||
Covy JP, Giasson BI. Identification of compounds that inhibit the kinase activity of leucine-rich repeat kinase 2. Biochem Biophys Res Commun. 2009;378(3):473–477. | ||
Galkina SI, Stadnichuk VI, Molotkovsky JG, Romanova JM, Sud’ina GF, Klein T. Microbial alkaloid staurosporine induces formation of nanometer-wide membrane tubular extensions (cytonemes, membrane tethers) in human neutrophils. Cell Adh Migr. 2010;4(1):32–38. | ||
Ray S, Liu M. Current understanding of LRRK2 in Parkinson’s disease: biochemical and structural features and inhibitor design. Future Med Chem. 2012;4(13):1701–1713. | ||
Lang CA, Ray SS, Liu M, Singh AK, Cuny GD. Discovery of LRRK2 inhibitors using sequential in silico joint pharmacophore space (JPS) and ensemble docking. Bioorg Med Chem Lett. 2015;25(13):2713–2719. | ||
Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270(46):27489–27494. | ||
Lee BD, Shin JH, VanKampen J, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med. 2010;16(9):998–1000. | ||
Zhang J, Deng X, Choi HG, Alessi DR, Gray NS. Characterization of TAE684 as a potent LRRK2 kinase inhibitor. Bioorg Med Chem Lett. 2012;22(5):1864–1869. | ||
Deng X, Dzamko N, Prescott A, et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat Chem Biol. 2011;7(4):203–205. | ||
Luerman GC, Nguyen C, Samaroo H, et al. Phosphoproteomic evaluation of pharmacological inhibition of leucine-rich repeat kinase 2 reveals significant off-target effects of LRRK-2-IN-1. J Neurochem. 2014;128(4):561–576. | ||
Ramsden N, Perrin J, Ren Z, et al. Chemoproteomics-based design of potent LRRK2-selective lead compounds that attenuate Parkinson’s disease-related toxicity in human neurons. ACS Chem Biol. 2011;6(10):1021–1028. | ||
Choi HG, Zhang J, Deng X, et al. Brain penetrant LRRK2 inhibitor. ACS Med Chem Lett. 2012;3(8):658–662. | ||
Reith AD, Bamborough P, Jandu K, et al. GSK2578215A; a potent and highly selective 2-arylmethyloxy-5-substitutent-N-arylbenzamide LRRK2 kinase inhibitor. Bioorg Med Chem Lett. 2012;22(17):5625–5629. | ||
Hatcher JM, Zhang J, Choi HG, Ito G, Alessi DR, Gray NS. Discovery of a pyrrolopyrimidine (JH-II-127), a highly potent, selective, and brain penetrant LRRK2 inhibitor. ACS Med Chem Lett. 2015;6(5):584–589. | ||
Estrada AA, Liu X, Baker-Glenn C, et al. Discovery of highly potent, selective, and brain-penetrable leucine-rich repeat kinase 2 (LRRK2) small molecule inhibitors. J Med Chem. 2012;55(22):9416–9433. | ||
Estrada AA, Chan BK, Baker-Glenn C, et al. Discovery of highly potent, selective, and brain-penetrant aminopyrazole leucine-rich repeat kinase 2 (LRRK2) small molecule inhibitors. J Med Chem. 2014;57(3):921–936. | ||
Fuji RN, Flagella M, Baca M, et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci Transl Med. 2015;7(273):273ra215. | ||
Henderson JL, Kormos BL, Hayward MM, et al. Discovery and preclinical profiling of 3-[4-(morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl]benzonitrile (PF-06447475), a highly potent, selective, brain penetrant, and in vivo active LRRK2 kinase inhibitor. J Med Chem. 2015;58(1):419–432. | ||
Fell MJ, Mirescu C, Basu K, et al. MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J Pharmacol Exp Ther. 2015;355(3):397–409. | ||
Low K, Aebischer P. Use of viral vectors to create animal models for Parkinson’s disease. Neurobiol Dis. 2012;48(2):189–201. | ||
Dusonchet J, Kochubey O, Stafa K, et al. A rat model of progressive nigral neurodegeneration induced by the Parkinson’s disease-associated G2019S mutation in LRRK2. J Neurosci. 2011;31(3):907–912. | ||
Tsika E, Nguyen AP, Dusonchet J, Colin P, Schneider BL, Moore DJ. Adenoviral-mediated expression of G2019S LRRK2 induces striatal pathology in a kinase-dependent manner in a rat model of Parkinson’s disease. Neurobiol Dis. 2015;77:49–61. | ||
Daher JP, Abdelmotilib HA, Hu X, et al. Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates alpha-synuclein gene-induced neurodegeneration. J Biol Chem. 2015;290(32):19433–19444. | ||
Schapansky J, Nardozzi JD, LaVoie MJ. The complex relationships between microglia, alpha-synuclein, and LRRK2 in Parkinson’s disease. Neuroscience. 2015;302:74–88. | ||
Hyun CH, Yoon CY, Lee HJ, Lee SJ. LRRK2 as a potential genetic modifier of synucleinopathies: interlacing the two major genetic factors of Parkinson’s disease. Exp Neurobiol. 2013;22(4):249–257. | ||
Sanders LH, Laganiere J, Cooper O, et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol Dis. 2014;62:381–386. | ||
Cooper O, Seo H, Andrabi S, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. 2012;4(141):141ra190. | ||
Liu GH, Qu J, Suzuki K, et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature. 2012;491(7425):603–607. | ||
Nguyen HN, Byers B, Cord B, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8(3):267–280. | ||
Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med. 2012;4(5):380–395. | ||
Orenstein SJ, Kuo SH, Tasset I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16(4):394–406. | ||
Herzig MC, Kolly C, Persohn E, et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. 2011;20(21):4209–4223. | ||
Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ 3rd, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010;107(21):9879–9884. | ||
Ness D, Ren Z, Gardai S, et al. Leucine-rich repeat kinase 2 (LRRK2)-deficient rats exhibit renal tubule injury and perturbations in metabolic and immunological homeostasis. PLoS One. 2013;8(6):e66164. | ||
Tong Y, Giaime E, Yamaguchi H, et al. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol Neurodegener. 2012;7:2. | ||
Shayman JA, Abe A. Drug induced phospholipidosis: an acquired lysosomal storage disorder. Biochim Biophys Acta. 2013;1831(3):602–611. | ||
Kruhlak NL, Choi SS, Contrera JF, Weaver JL, Willard JM, Hastings KL, Sancilio LF. Development of a phospholipidosis database and predictive quantitative structure-activity relationship (QSAR) models. Toxicol Mech Methods. 2008;18(2–3):217–227. | ||
The Regulatory Challenges of Drug-induced Phospholipidosis. U.S. Food and Drug Administration; 2010. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM210798.pdf. Accessed July 1, 2016. | ||
Baptista MA, Dave KD, Frasier MA, et al. Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS One. 2013;8(11):e80705. | ||
Zhao J, Molitor TP, Langston JW, Nichols RJ. LRRK2 dephosphorylation increases its ubiquitination. Biochem J. 2015;469(1):107–120. | ||
Perera G, Ranola M, Rowe DB, Halliday GM, Dzamko N. Inhibitor treatment of peripheral mononuclear cells from Parkinson’s disease patients further validates LRRK2 dephosphorylation as a pharmacodynamic biomarker. Sci Rep. 2016;6:31391. | ||
Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol. 2011;12(11):1063–1070. | ||
Zhang Q, Pan Y, Yan R, et al. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat Immunol. 2015;16(9):918–926. | ||
Pfeffer SR. Structural clues to Rab GTPase functional diversity. J Biol Chem. 2005;280(16):15485–15488. | ||
Tucci A, Nalls MA, Houlden H, et al. Genetic variability at the PARK16 locus. Eur J Hum Genet. 2010;18(12):1356–1359. | ||
Pihlstrom L, Rengmark A, Bjornara KA, et al. Fine mapping and resequencing of the PARK16 locus in Parkinson’s disease. J Hum Genet. 2015;60(7):357–362. | ||
MacLeod DA, Rhinn H, Kuwahara T, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77(3):425–439. | ||
Dodson MW, Zhang T, Jiang C, Chen S, Guo M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet. 2012;21(6):1350–1363. | ||
Kuwahara T, Inoue K, D’Agati VD, et al. LRRK2 and RAB7L1 coordinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci Rep. 2016;6:29945. | ||
Nichols RJ, Dzamko N, Hutti JE, et al. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson’s disease. Biochem J. 2009;424(1):47–60. | ||
Spano S, Liu X, Galan JE. Proteolytic targeting of Rab29 by an effector protein distinguishes the intracellular compartments of human-adapted and broad-host Salmonella. Proc Natl Acad Sci U S A. 2011;108(45):18418–18423. | ||
Dzamko N, Geczy CL, Halliday GM. Inflammation is genetically implicated in Parkinson’s disease. Neuroscience. 2015;302:89–102. | ||
Wang D, Lou J, Ouyang C, et al. Ras-related protein Rab10 facilitates TLR4 signaling by promoting replenishment of TLR4 onto the plasma membrane. Proc Natl Acad Sci U S A. 2010;107(31):13806–13811. | ||
Dzamko N, Inesta-Vaquera F, Zhang J, et al. The IkappaB kinase family phosphorylates the Parkinson’s disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS One. 2012;7(6):e39132. | ||
Dzamko N, Deak M, Hentati F, Reith AD, Prescott AR, Alessi DR, Nichols RJ. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem J. 2010;430(3):405–413. | ||
Doggett EA, Zhao J, Mork CN, Hu D, Nichols RJ. Phosphorylation of LRRK2 serines 955 and 973 is disrupted by Parkinson’s disease mutations and LRRK2 pharmacological inhibition. J Neurochem. 2012;120(1):37–45. | ||
Lobbestael E, Zhao J, Rudenko IN, et al. Identification of protein phosphatase 1 as a regulator of the LRRK2 phosphorylation cycle. Biochem J. 2013;456(1):119–128. | ||
Muda K, Bertinetti D, Gesellchen F, et al. Parkinson-related LRRK2 mutation R1441C/G/H impairs PKA phosphorylation of LRRK2 and disrupts its interaction with 14-3-3. Proc Natl Acad Sci U S A. 2014;111(1):E34–E43. | ||
Chia R, Haddock S, Beilina A, et al. Phosphorylation of LRRK2 by casein kinase 1alpha regulates trans-Golgi clustering via differential interaction with ARHGEF7. Nat Commun. 2014;5:5827. | ||
Dzamko N, Chua G, Ranola M, Rowe DB, Halliday GM. Measurement of LRRK2 and Ser910/935 phosphorylated LRRK2 in peripheral blood mononuclear cells from idiopathic Parkinson’s disease patients. J Parkinsons Dis. 2013;3(2):145–152. | ||
Fraser KB, Rawlins AB, Clark RG, et al. Ser(P)-1292 LRRK2 in urinary exosomes is elevated in idiopathic Parkinson’s disease. Mov Disord. Epub 2016 Jun 14. | ||
Winner B, Melrose HL, Zhao C, et al. Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol Dis. 2011;41(3):706–716. | ||
Marder K, Wang Y, Alcalay RN, et al. Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology. 2015;85(1):89–95. | ||
Sierra M, Gonzalez-Aramburu I, Sanchez-Juan P, et al. High frequency and reduced penetrance of LRRK2 G2019S mutation among Parkinson’s disease patients in Cantabria (Spain). Mov Disord. 2011;26(13):2343–2346. | ||
Latourelle JC, Sun M, Lew MF, et al. The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson’s disease: the GenePD study. BMC Med. 2008;6:32. | ||
Dzamko N, Rowe DB, Halliday GM. Increased peripheral inflammation in asymptomatic leucine-rich repeat kinase 2 mutation carriers. Mov Disord. 2016;31(6):889–897. | ||
Mirelman A, Bernad-Elazari H, Thaler A, et al. Arm swing as a potential new prodromal marker of Parkinson’s disease. Mov Disord. Epub 2016 Jul 19. | ||
Mirelman A, Gurevich T, Giladi N, Bar-Shira A, Orr-Urtreger A, Hausdorff JM. Gait alterations in healthy carriers of the LRRK2 G2019S mutation. Ann Neurol. 2011;69(1):193–197. | ||
Bergareche A, Rodriguez-Oroz MC, Estanga A, et al. DAT imaging and clinical biomarkers in relatives at genetic risk for LRRK2 R1441G Parkinson’s disease. Mov Disord. 2016;31(3):335–343. | ||
Mirelman A, Alcalay RN, Saunders-Pullman R, et al. Nonmotor symptoms in healthy Ashkenazi Jewish carriers of the G2019S mutation in the LRRK2 gene. Mov Disord. 2015;30(7):981–986. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.