")
Back to Journals » International Journal of General Medicine » Volume 7
Lowering low-density lipoprotein cholesterol levels in patients with type 2 diabetes mellitus
Authors Bays H
Received 31 March 2014
Accepted for publication 13 May 2014
Published 5 July 2014 Volume 2014:7 Pages 355—364
DOI https://doi.org/10.2147/IJGM.S65148
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Harold E Bays
Louisville Metabolic and Atherosclerosis Research Center, Louisville, KY, USA
Abstract: Type 2 diabetes mellitus (T2DM) is characterized by hyperglycemia, insulin resistance, and/or progressive loss of β-cell function. T2DM patients are at increased risk of micro- and macrovascular disease, and are often considered as representing an atherosclerotic coronary heart disease (CHD) risk equivalent. Interventions directed at glucose and lipid level control in T2DM patients may reduce micro- and macrovascular disease. The optimal T2DM agent is one that lowers glucose levels with limited risk for hypoglycemia, and with no clinical trial evidence of worsening CHD risk. Lipid-altering drugs should preferably reduce low-density lipoprotein cholesterol and apolipoprotein B (apo B) and have evidence that the mechanism of action reduces CHD risk. Statins reduce low-density lipoprotein cholesterol and apo B and have evidence of improving CHD outcomes, and are thus first-line therapy for the treatment of hypercholesterolemia. In patients who do not achieve optimal lipid levels with statin therapy, or who are intolerant to statin therapy, add-on therapy or alternative therapies may be indicated. Additional available agents to treat hypercholesterolemic patients with T2DM include bile acid sequestrants, fibrates, niacin, and ezetimibe. This review discusses the use of these alternative agents to treat hypercholesterolemia in patients with T2DM, either as monotherapy or in combination with statin therapy.
Keywords: dyslipidemia, statin, colesevelam
Introduction
Type 2 diabetes mellitus (T2DM) is a disease characterized by hyperglycemia, insulin resistance, and/or progressive loss of β-cell function. T2DM is associated with high cardiovascular disease (CVD) risk,1 and hyperglycemia induces vascular changes that contribute to atherosclerosis and vasculopathy (Table 1).2–4
 | Table 1 Effects of hyperglycemia on atherosclerotic processes |
Intensive glucose control in patients with T2DM may reduce CVD, depending upon how early and the speed at which such intervention is implemented, the types of agents used for glucose control, and the medical status of the patient.4,5 Overall, the best approach for reducing CVD risk is a comprehensive one that not only includes glucose and lipid control, but also the introduction of therapeutic lifestyle changes such as smoking cessation, optimal nutrition, increased physical activity, appropriate body weight management, blood pressure management, and possible aspirin therapy for patients with high CVD risk.6
In some patients, T2DM may be considered a coronary heart disease (CHD) risk equivalent,7 which may necessitate more stringent lipid control for primary prevention than in individuals without diabetes mellitus. While the recent American College of Cardiology/American Heart Association guidelines emphasize reducing risk in patient groups at high risk for CVD rather than focusing on specific low-density lipoprotein (LDL) cholesterol (LDL-C) lipid treatment goals, other guidelines recommend a LDL-C treatment goal of <100 mg/dL for high risk patients with diabetes mellitus, and <70 mg/dL for those at very high CVD risk (eg, diabetes mellitus patients with existing CVD or multiple other risk factors).6,8,9 Unfortunately, a substantial proportion of T2DM patients do not achieve those goals. In a study of 17,306 patients with diabetes that aimed to determine levels of therapeutic goal achievement, only 42% of patients achieved an LDL-C goal of <100 mg/dL over the 7-year period from 1999 to 2006.10
Hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins) are first-line lipid-lowering therapy for patients with T2DM. In patients with T2DM, statins generally reduce LDL-C levels by about 24%–52%, depending upon the statin and dose (eg, atorvastatin, fluvastatin, lovastatin, and rosuvastatin).11–13 The reduction in LDL-C levels achieved by statins is associated with reductions in CVD events. A meta-analysis by the Cholesterol Treatment Trialists’ Collaborators showed that, among 18,686 patients with diabetes mellitus (92% type 2/8% type 1) receiving statin therapy, for each mmol/L (39 mg/dL) reduction in LDL-C, there was a 21% proportional reduction in major vascular events (P<0.0001).14
Although the efficacy of statins is well established, a considerable proportion of patients do not achieve lipid goals with statin monotherapy and may require add-on or alternative therapies to statins to better achieve LDL-C treatment goals. In the National Cholesterol Education Program Evaluation ProjecT Utilizing Novel E-Technology (NEPTUNE) II study, the percentage of patients who achieved LDL-C treatment goals decreased as the number of risk factors increased; 89% and 76% of patients with zero to one and two or more risk factors, respectively, achieved LDL-C goal, while only 57% of patients with CHD or CHD risk equivalents achieved goal.15 Moreover, in the subset of patients with CHD or CHD risk equivalents, 55% of patients with diabetes mellitus but without CHD achieved LDL-C goal compared with 62% of patients with CHD and only 40% of patients with other CHD risk equivalents (without CHD).15 Of the patients in the CHD or CHD risk equivalents subgroup who had triglycerides ≥200 mg/dL (≥2.25 mmol/L), 50% of patients with diabetes mellitus (without CHD) and 57% of patients with CHD achieved LDL-C goal, whereas 44% of patients with other CHD risk equivalents (without CHD) achieved LDL-C goal.15
One strategy for improving LDL-C goal attainment is to increase statin therapy, often to the maximal approved dose; however, doubling the statin dose does not double the LDL-C lowering efficacy. In a pooled analysis of 37 studies of 32,258 patients receiving rosuvastatin, atorvastatin, or simvastatin, doubling the statin dose reduced LDL-C levels by only an additional 5%–7%.16 High-dose statin therapy is generally well tolerated in many patients, at least as determined by clinical trial data.11 However, increasing statin dose to the highest doses may not be the best strategy for all patients. A meta-analysis of studies investigating intensive- and moderate-dose statin regimens showed that patients receiving higher-dose statin were more likely to experience an adverse event, discontinue therapy because of an adverse event, and demonstrate liver abnormalities and increased creatine kinase levels compared with patients receiving moderate-dose statins.17 Also, while the clinical significance is unclear, statins (particularly at intensive doses) may be associated with increased risk of developing new-onset diabetes and/or unfavorable glycemic effects.18–21
In patients who are unable to achieve desired LDL-C treatment goals with statin therapy, a number of other agents, including bile acid (BA) sequestrants (BASs), fibrates, niacin, and cholesterol absorption inhibitors (eg, ezetimibe), may be combined with statins to facilitate goal achievement or be used in place of statins for patients who cannot tolerate statins.6,22 This review describes these add-on or alternative therapy options for the lowering of LDL-C levels in patients with T2DM. Because of their dual effects on lowering glucose and LDL-C, the role of BASs will be discussed in greater detail.
BASs
Before statins were approved as agents to reduce elevated cholesterol levels, BASs were recommended as first-line therapy for reducing LDL-C levels.23 In the Lipid Research Clinics Coronary Primary Prevention Trial, the BAS cholestyramine was shown to improve cardiovascular (CV) outcomes in a population of asymptomatic middle-aged men with primary hypercholesterolemia (diabetes mellitus was an exclusion criteria). This was the first study to demonstrate that a reduction in LDL-C levels (mean reduction of 12.6% compared with placebo) significantly reduced CV risk (primary endpoint of CHD death and/or nonfatal myocardial infarction reduced by 19%, P<0.05), which was associated with a 24% reduction in CHD death and a 19% reduction in nonfatal myocardial infarction.24 BASs are nonsystemic agents; however, BASs may bind to certain drugs in the gastrointestinal tract and it is therefore recommended that agents such as warfarin, digoxin, thyroid hormones, and fat-soluble vitamins be taken either 1 hour before or 4–6 hours after BAS administration. The primary adverse events reported for the older BASs cholestyramine and colestipol are constipation and flatulence, and these agents are associated with high discontinuation rates within clinical trials of 40%–60%.25,26 In comparison, the primary adverse events reported for the specifically engineered BAS colesevelam are constipation and dyspepsia, with an observed compliance rate within clinical trials of 88%–93%.27,28
The synthesis of BAs occurs exclusively in the liver, and the BA pool is tightly regulated within the liver and intestine. BAs are known ligands for the nuclear receptor farnesoid X receptor (FXR) and self-regulate their own synthesis. Published literature suggests the following proposed model for the regulation of BAs. In the intestine, BAs secreted in response to an ingested meal activate FXR, which induces expression of fibroblast growth factor (FGF)-19.29,30 FGF-19 binds to surface hepatocyte FGF receptor 4 (FGFR4), which subsequently results in a c-Jun N-terminal kinase-mediated repression of cytochrome P450 enzyme cholesterol 7 α-hydroxylase (CYP7A1), thus inhibiting the rate-limiting step in the conversion of cholesterol to BAs, and subsequently resulting in the downregulation of HMG-CoA reductase (the rate-limiting step of cholesterol synthesis) (Figure 1A). Metabolic pathways in the liver also play a major role in the regulation of BAs. More specifically, increasing BA levels in the liver upregulate the small heterodimer partner (SHP) via increased FXR activation, which results in both inhibition of the liver X receptor (LXR) and liver receptor homolog-1 (LRH-1), and ultimately further repression of CYP7A1 to reduce BA synthesis.30,31
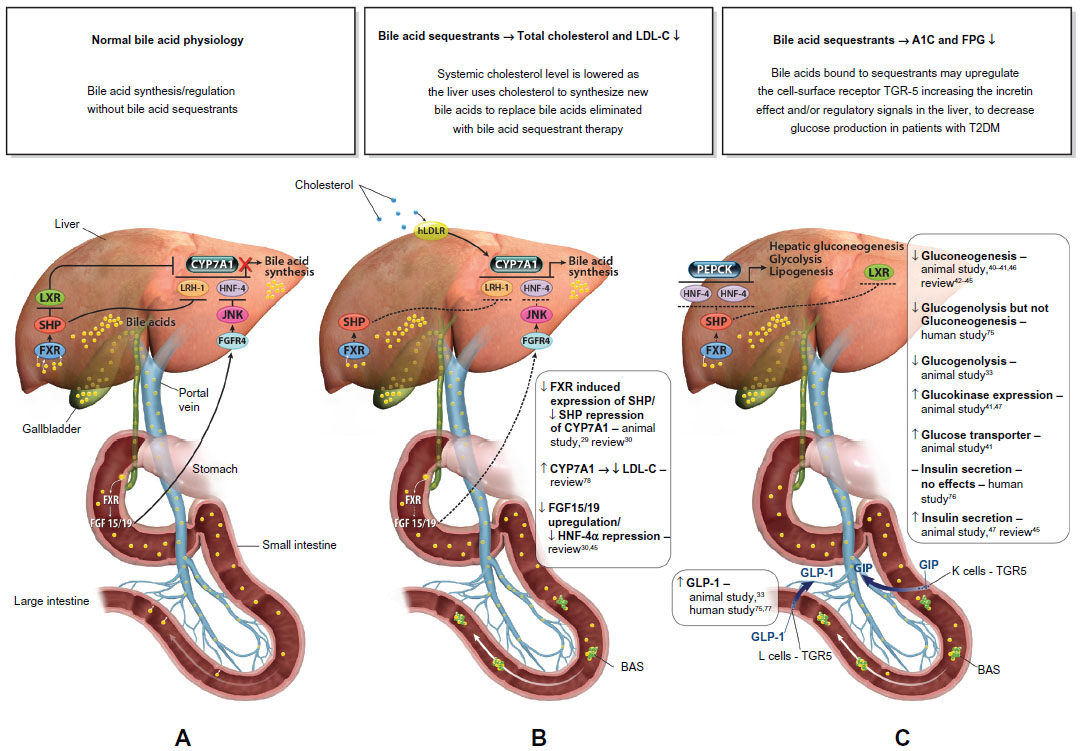 | Figure 1 Proposed mechanism of action for the lipid-lowering and glycemic effects of a BAS. |
BASs bind BAs in the intestine, thus increasing BA excretion in the feces. Consequently, fewer BAs are returned to the liver. Binding BAs also “deactivate” FXR activity. Thus, the alteration of the BA pool reduces nuclear receptor FXR-mediated repression of key regulatory elements (eg, FGF15/19, FGFR4, SHP) responsible for BA synthesis, in particular, CYP7A, which ultimately results in increased BA synthesis.29,30 The upregulation of CYP7A in the BA synthesis pathway increases HMG-CoA transcriptional activity. Subsequent increased conversion of cholesterol to synthesize BAs results in a compensatory upregulation in hepatic LDL receptors (hLDLR), increased hepatic LDL-C uptake, and decreased circulating LDL-C.32,33 The proposed mechanism by which BA sequestration leads to LDL-C lowering is shown in Figure 1B.
In studies in patients with T2DM, the BASs colesevelam and cholestyramine reduced LDL-C levels,34–37 which may be accompanied by a modest increase in high-density lipoprotein (HDL) cholesterol (HDL-C) and triglyceride levels. In a double-blind, randomized, crossover study of 21 patients with well-controlled T2DM but fasting LDL-C levels of >130 mg/dL receiving cholestyramine or placebo for 6 weeks, cholestyramine produced a 28% reduction in LDL-C (P<0.001 versus placebo), a 13.5% increase in triglycerides (P=0.02 versus placebo), and a non-statistically significant increase in HDL-C (1 mg/dL; P>0.2 versus placebo).34 In three pivotal randomized, double-blind, placebo-controlled studies in patients with T2DM (n>280), colesevelam reduced LDL-C by 13%–17% compared with placebo (P<0.001 for all); the placebo-adjusted mean change from baseline in triglyceride levels in colesevelam recipients ranged from +5% to +22% (Table 2) and HDL-C changed by −0.9% to +0.9% (P=not significant for all).35–37 Among patients from these studies who were taking concomitant statins, the addition of colesevelam reduced LDL-C by 16% compared with an increase of 1% with placebo, and had no significant effect on HDL-C levels (+0.02% versus placebo; P=not significant).38
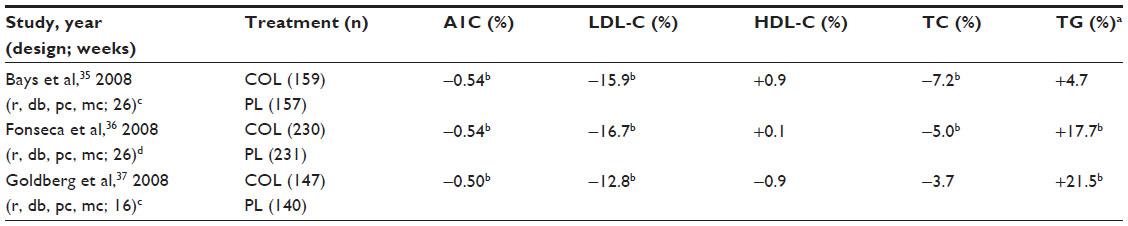 | Table 2 Least squares mean percent treatment difference in glycemic and lipid parameters in patients with type 2 diabetes mellitus receiving COL or PL |
BASs may also lower glucose levels in patients with T2DM.34–37,39 The BAS colesevelam was approved in 2008 by the US Food and Drug Administration (FDA) to improve glycemic control in adults with T2DM. The precise glucose-lowering mechanisms of BASs are unknown. Possible mechanisms involved with the glucose-lowering effects of BASs are summarized in Figure 1C. In brief, decreased activity of both FXR and SHP resulting from the reduction of the enterohepatic BA pool after BAS administration promotes phosphoenolpyruvate carboxykinase (PEPCK) production, which increases hepatic gluconeogenesis and glycolysis.30,31 However, increased LXR activity suppresses expression of PEPCK and results in a reduction in gluconeogenesis,40–46 as well as increased insulin secretion45,47 and increased expression of glucokinase32,41,47 and glucose transporter.41 Furthermore, BA bound to a BAS may activate the G-protein-coupled receptor TGR5 in the intestine leading to the increased secretion of glucagon-like peptide-1 ([GLP-1] [L cells]) resulting in reduced hepatic glucose production via the suppression of hepatic glycogenolysis.33,48
In three randomized, double-blind, placebo-controlled studies, colesevelam significantly lowered hemoglobin A1C by 0.5% or more compared with placebo (P<0.001 for all) in adults with T2DM when added to stable metformin-, insulin-, or sulfonylurea-based therapy (Table 2).35–37 Subgroup analysis of the metformin-based therapy study by pre-study use or nonuse of statins (which continued during the study) indicated that, regardless of any potential effect of statins on glycemia, concomitant statins did not attenuate the effects of colesevelam.49 In both statin users and nonusers, colesevelam produced significantly greater reductions than placebo in hemoglobin A1C (mean treatment differences −0.63% [P=0.0003] and −0.49% [P=0.001], respectively) and LDL-C (−16.4% [P=0.0024] and −15.8% [P<0.0001], respectively).
Fibrates
Fibrates are synthetic ligands for peroxisome proliferator-activated α-receptors. It is through binding to these nuclear receptors that they act to alter lipid levels.50 Fibrates primarily reduce triglycerides (which are often elevated in patients with T2DM),51 have a modest effect on HDL-C levels, and, depending upon the baseline triglyceride levels, may decrease LDL-C levels (in patients without baseline elevation in triglyceride levels) or may substantially increase LDL-C levels (in patients with very high baseline triglyceride levels). In the Diabetes Atherosclerosis Intervention Study,52 the improvements in lipid levels with fenofibrate were associated with reductions in the angiographic progression of coronary artery disease. However, while fenofibrate significantly improved LDL-C, HDL-C, triglyceride, and total cholesterol levels compared with placebo in the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study (relative treatment difference: −5.8%, 1.2%, −21.9%, and −6.9%, respectively; all P<0.05), it did not significantly reduce the risk of CHD death or nonfatal myocardial infarction in patients with T2DM, although there was a significant reduction in the rate of total CVD events, a composite of CVD death, myocardial infarction, stroke, and coronary and carotid revascularization (hazard ratio, 0.89; 95% confidence interval: 0.80–0.99; P=0.035).53 It is noteworthy that, in the FIELD study, fenofibrate did significantly reduce the need for retinal laser treatment in patients with retinopathy (5.2% versus 3.6%; P=0.0003), and resulted in significantly less albuminuria progression (P=0.002) in patients with T2DM.53 Thus, fenofibrate may have a beneficial effect in reducing microvascular complications in this population.
The Action to Control Cardiovascular Risk in Diabetes (ACCORD) lipid trial investigated the efficacy of fenofibrate versus placebo in 5,518 patients with T2DM at high risk of CVD who were receiving simvastatin therapy; the changes from baseline to the end of the study in lipid parameters are presented in Figure 2. While fenofibrate treatment resulted in significant improvements in total cholesterol, triglycerides and HDL-C compared with placebo, it did not significantly reduce the rate of fatal CV events, nonfatal myocardial infarction, or nonfatal stroke.54 However, an analysis by lipid subgroup suggested a possible benefit among patients with both a high baseline triglyceride level and a low baseline HDL-C level (P=0.057).54
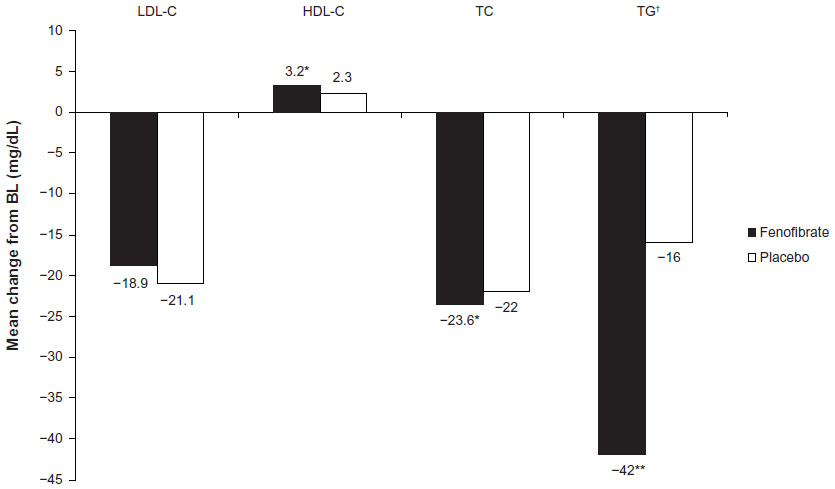 | Figure 2 Mean change from BL in lipid parameters in fenofibrate and placebo recipients from the ACCORD study. |
Some reports suggest fibrates may mildly reduce glucose levels, which, in addition to the triglyceride lowering, likely helps to account for the reduction in metabolic syndrome in patients treated with fibrates. When combined with statins, fibrates may mitigate the increase in glucose levels sometimes found associated with statins.55 The risk of rhabdomyolysis associated with combination therapy with statins and fibrates appears to differ among the fibrates, with a higher incidence observed with gemfibrozil, at least partially due to a higher risk of drug–drug interactions with statins.56
Niacin
Niacin is believed to exert its effects via a number of potential mechanisms including: 1) directly and noncompetitively inhibiting hepatocyte diacylglycerol acyltransferase 2, thereby reducing hepatic triglyceride synthesis and subsequent very low-density lipoprotein/LDL secretion; 2) inhibiting the surface expression of β-chain adenosine triphosphate synthase by hepatocytes, which inhibits HDL-apolipoprotein (apo) A-I removal, thus increasing apo A-I containing HDL particles; and 3) potentially stabilizing the circulation of secreted apo A-I via increased HDL biogenesis resulting from increased hepatic adenosine triphosphate-binding cassette transporter A-I-mediated apo A-I lipidation.57 At therapeutic doses, niacin significantly reduces LDL-C, non-HDL-C, apo B, and triglyceride levels and increases HDL-C levels. Niacin may also influence lipoprotein particle size and the distribution of lipid subparticles and improve lipid ratios.58 The Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) trial investigated the addition of extended-release niacin to simvastatin in 3,414 patients with established CVD (~34% of whom had diabetes), aiming to determine its effect on lipid levels and the composite endpoint of death from CHD, nonfatal myocardial infarction, ischemic stroke, hospitalization for an acute coronary syndrome, or symptom-driven coronary or cerebral revascularization.59 After 2 years of treatment, HDL-C levels had increased 25% with niacin treatment (versus 10% with placebo; P<0.001), while triglyceride and LDL-C levels had decreased by 29% and 12% (versus decreases of 8% and 6% with placebo), respectively; however, improvement in the lipid profile did not translate into a reduction of adverse CV events, with the primary endpoint occurring in 16.4% of patients receiving add-on niacin and 16.2% receiving placebo.59
Niacin causes flushing, which can be intolerable to some patients. Niacin-induced flushing is caused primarily by the promotion of prostaglandin D2 release from skin cells, which stimulates the action of prostaglandin D2 receptors in smooth muscle cells in the dermal arteriole vasculature. Stimulated dermal arterioles then dilate, increasing blood flow and causing flushing.58 Flushing is reduced with extended-release formulations and the fixed combination of extended-release niacin and laropiprant, a selective inhibitor of the prostaglandin D2 receptor subtype.58 As was observed in previous studies, preliminary results from the large Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) trial demonstrated no reduction in risk of CV events with extended-release niacin/laropiprant combination therapy. As a result of these disappointing findings, the extended-release niacin/laropiprant development program was discontinued.60
Examination of the literature regarding the safety profile of niacin shows that, in patients without diabetes, niacin therapy may result in insulin resistance and hyperglycemia; in patients with diabetes, niacin treatment may worsen glucose and hemoglobin A1C control. Although the changes are generally small, in clinical trials this translates into a need for intensification of antidiabetes medications.58,61–64 During a 9-month study in 796 patients with T2DM, a significantly greater proportion of those receiving niacin/laropiprant, compared with placebo, required intensification of their antihyperglycemic regimen (17.6% versus 8.2%; P<0.001).64
Ezetimibe
Ezetimibe acts to block intestinal cholesterol absorption, which leads to a reduction in cholesterol delivery to the liver and an enhanced clearance of LDL-C, which reduces plasma LDL-C levels.65 In patients with T2DM, the addition of ezetimibe to statin therapy provides a significantly greater reduction in LDL-C,66–68 even more so than doubling the statin dose.69–71
While ezetimibe has no known effect on glycemic parameters in patients with T2DM, ezetimibe was studied in diabetes mellitus patients. In two studies (>500 patients) investigating the addition of ezetimibe to existing statin therapy in patients with and without T2DM, patients receiving the combination therapy had significantly greater reductions in LDL-C levels compared with statin therapy alone, irrespective of diabetes status, and similarly, improvements were observed in total cholesterol, triglycerides, and HDL-C levels (Table 3).66,68 A study comparing the efficacy of simvastatin/ezetimibe combination therapy (10/20 or 40 mg/day) with atorvastatin monotherapy (10, 20, or 40 mg/day) in 1,229 patients with T2DM showed that simvastatin + ezetimibe recipients had significantly (P≤0.001) greater improvements in LDL-C, total cholesterol, and HDL-C levels than patients receiving any dose of atorvastatin alone.67 Generally, patients receiving simvastatin/ezetimibe combination therapy in this trial achieved LDL-C goals (<100 mg/dL or <70 mg/dL) more frequently than patients receiving atorvastatin.67
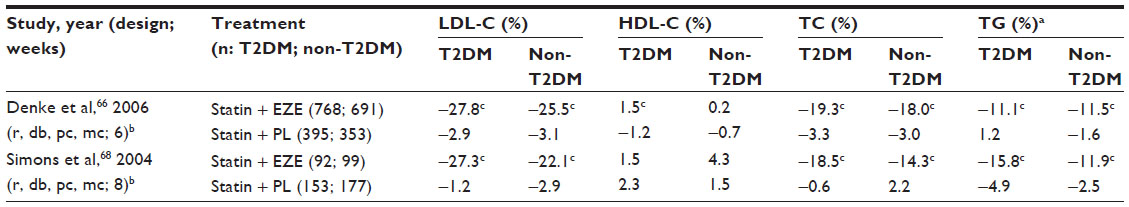 | Table 3 Least squares mean percent change from baseline in lipid parameters in patients with or without T2DM receiving EZE or PL on a background of statin therapy |
Simvastatin/ezetimibe (Vytorin®; Merck & Co., Inc., Whitehouse Station, NJ, USA) combination therapy is generally well tolerated. But, as with simvastatin monotherapy, the simvastatin/ezetimibe combination agent may increase the risk for myopathy and rhabdomyolysis, which increases among patients taking higher simvastatin doses, as is often true with other statins at higher doses.72 In addition, the prescribing information lists other very rare adverse effects, including anaphylaxis, angioedema, rash, and urticaria.72
The IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) is an ongoing trial that aims to determine if simvastatin/ezetimibe combination therapy improves CV outcomes in patients with acute coronary syndromes to a greater degree than simvastatin alone.73 The study has enrolled >18,000 patients, and follow-up will continue until >5,250 patients experience the primary endpoint (a composite of CV-related death, nonfatal coronary events, and nonfatal stroke) and each patient is followed for >2.5 years; at present, the trial is expected to report results in September 2014.74
Conclusion
For patients with T2DM, the therapeutic goal is to lower LDL-C and favorably affect other CV risk factors. Statins are LDL-C-lowering agents with the best clinical trial evidence of CVD outcome benefits, and are first-line therapy for hypercholesterolemia. However, statins may not be tolerated by all patients in doses large enough to attain LDL-C goal. Moreover, evidence suggests that statins may be associated with risk (particularly at high doses) for increasing new-onset diabetes and unfavorable glycemic effects. Other lipid-lowering agents, eg, fibrates and ezetimibe, have little to no impact on glucose parameters. BASs are the only class of agents with dual benefits in the management of glucose and lipids in patients with T2DM. Colesevelam is currently the only BAS with an approved indication for use in combination with other classes of lipid- (and glucose-) lowering drugs in patients with T2DM to both lower LDL-C and improve glycemic control.
Acknowledgments
Sheridan Henness, PhD, Alan J Klopp, PhD, CMPP, and Sushma Soni of inScience Communications, Springer Healthcare, provided medical writing support funded by Daiichi Sankyo, Inc.
Disclosure
In the past year, Dr Harold Bays has served as a clinical investigator for (and has received research grants from) pharmaceutical companies such as Abbott, Amarin, Arena, Cargill, California Raisin Board, Daiichi Sankyo, Inc., Esperion, Essentialis, Forest, Gilead, GlaxoSmithKline, Johnson & Johnson, Merck, Novo Nordisk, Omthera, Orexigen, Pfizer, Pozen, Schering Plough, Shionogi, Stratum Nutrition, Takeda, Trygg, and TWI Bio. Dr Bays has received consultant, advisory, or speaking fees from Amarin, AstraZeneca, Boston Scientific, Essentialis, Daiichi Sankyo, Inc., Merck, Novartis, Regeneron, Sanofi, Valeant, Vivus, and Zeomedex. The development of this manuscript was supported by Daiichi Sankyo, Inc. Medical writing support was funded by Daiichi Sankyo, Inc.
References
Kannel WB, McGee DL. Diabetes and cardiovascular risk factors: the Framingham study. Circulation. 1979;59(1):8–13. | |
Aronson D, Rayfield EJ. How hyperglycemia promotes atherosclerosis: molecular mechanisms. Cardiovasc Diabetol. 2002;1:1. | |
Bays HE. “Sick fat,” metabolic disease, and atherosclerosis. Am J Med. 2009;122(Suppl 1):S26–S37. | |
Bays HE. Adiposopathy, diabetes mellitus, and primary prevention of atherosclerotic coronary artery disease: treating “sick fat” through improving fat function with antidiabetes therapies. Am J Cardiol. 2012;110(Suppl 9):4B–12B. | |
Ray KK, Seshasai SR, Wijesuriya S, et al. Effect of intensive control of glucose on cardiovascular outcomes and death in patients with diabetes mellitus: a meta-analysis of randomised controlled trials. Lancet. 2009;373(9677):1765–1772. | |
American Diabetes Association. Standards of medical care in diabetes – 2013. Diabetes Care. 2013;36 Suppl 1:S11–S66. | |
Grundy SM, Cleeman JI, Merz CN, et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110(2):227–239. | |
Jellinger PS, Smith DA, Mehta AE, et al; AACE Task Force for Management of Dyslipidemia and Prevention of Atherosclerosis. American Association of Clinical Endocrinologists’ Guidelines for Management of Dyslipidemia and Prevention of Atherosclerosis. Endocr Pract. 2012;18 Suppl 1:1–78. | |
Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. Epub November 12, 2013. | |
Cheung BM, Ong KL, Cherny SS, Sham PC, Tso AW, Lam KS. Diabetes prevalence and therapeutic target achievement in the United States, 1999 to 2006. Am J Med. 2009;122(5):443–453. | |
Berne C, Siewert-Delle A; URANUS study investigators. Comparison of rosuvastatin and atorvastatin for lipid lowering in patients with type 2 diabetes mellitus: results from the URANUS study. Cardiovasc Diabetol. 2005;4:7. | |
Garg A, Grundy SM. Lovastatin for lowering cholesterol levels in non-insulin-dependent diabetes mellitus. N Engl J Med. 1988; 318(2):81–86. | |
Knopp RH, Frohlich J, Jokubaitis LA, Dawson K, Broyles FE, Gomez-Coronado D. Efficacy and safety of fluvastatin in patients with non-insulin-dependent diabetes mellitus and hyperlipidemia. Am J Med. 1994;96(6A):69S–78S. | |
Cholesterol Treatment Trialists’ (CTT) Collaborators, Kearney PM, Blackwell L, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet. 2008;371(9607):117–125. | |
Davidson MH, Maki KC, Pearson TA, et al. Results of the National Cholesterol Education (NCEP) Program Evaluation ProjecT Utilizing Novel E-Technology (NEPTUNE) II survey and implications for treatment under the recent NCEP Writing Group recommendations. Am J Cardiol. 2005;96(4):556–563. | |
Nicholls SJ, Brandrup-Wognsen G, Palmer M, Barter PJ. Meta-analysis of comparative efficacy of increasing dose of atorvastatin versus rosuvastatin versus simvastatin on lowering levels of atherogenic lipids (from VOYAGER). Am J Cardiol. 2010;105(1):69–76. | |
Silva M, Matthews ML, Jarvis C, et al. Meta-analysis of drug-induced adverse events associated with intensive-dose statin therapy. Clin Ther. 2007;29(2):253–260. | |
Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA. 2011;305(24):2556–2564. | |
Rautio N, Jokelainen J, Oksa H, et al. Do statins interfere with lifestyle intervention in the prevention of diabetes in primary healthcare? One-year follow-up of the FIN-D2D project. BMJ Open. 2012;2(5). | |
Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375(9716):735–742. | |
Zaharan NL, Williams D, Bennett K. Statins and risk of treated incident diabetes in a primary care population. Br J Clin Pharmacol. 2013;75(4):1118–1124. | |
Handelsman Y, Mechanick JI, Blonde L, et al; AACE Task Force for Developing Diabetes Comprehensive Care Plan. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for developing a diabetes mellitus comprehensive care plan. Endocr Pract. 2011;17 Suppl 2:1–53. | |
[No authors listed]. Report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. The Expert Panel. Arch Intern Med. 1988;148(1):36–69. | |
[No authors listed]. The Lipid Research Clinics Coronary Primary Prevention Trial results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. JAMA. 1984;251(3):365–374. | |
Andrade SE, Walker AM, Gottlieb LK, et al. Discontinuation of antihyperlipidemic drugs – do rates reported in clinical trials reflect rates in primary care settings? N Engl J Med. 1995;332(17):1125–1131. | |
Avorn J, Monette J, Lacour A, et al. Persistence of use of lipid-lowering medications: a cross-national study. JAMA. 1998;279(18):1458–1462. | |
Florentin M, Liberopoulos EN, Mikhailidis DP, Elisaf MS. Colesevelam hydrochloride in clinical practice: a new approach in the treatment of hypercholesterolaemia. Curr Med Res Opin. 2008;24(4):995–1009. | |
WELCHOL® (colesevelam hydrochloride) [prescribing information]. Daiichi Sankyo Inc. Parsippany, NJ; 2014 [revised Jan 2014]. | |
Goodwin B, Jones SA, Price RR, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6(3):517–526. | |
Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89(1):147–191. | |
Staels B. A review of bile acid sequestrants: potential mechanism(s) for glucose-lowering effects in type 2 diabetes mellitus. Postgrad Med. 2009;121(3 Suppl 1):25–30. | |
Bays HE, Goldberg RB. The ‘forgotten’ bile acid sequestrants: is now a good time to remember? Am J Ther. 2007;14(6):567–580. | |
Potthoff MJ, Potts A, He T, et al. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am J Physiol Gastrointest Liver Physiol. 2013;304(4):G371–G380. | |
Garg A, Grundy SM. Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Ann Intern Med. 1994;121(6):416–422. | |
Bays HE, Goldberg RB, Truitt KE, Jones MR. Colesevelam hydrochloride therapy in patients with type 2 diabetes mellitus treated with metformin: glucose and lipid effects. Arch Intern Med. 2008;168(18):1975–1983. | |
Fonseca VA, Rosenstock J, Wang AC, Truitt KE, Jones MR. Colesevelam HCl improves glycemic control and reduces LDL cholesterol in patients with inadequately controlled type 2 diabetes on sulfonylurea-based therapy. Diabetes Care. 2008;31(8):1479–1484. | |
Goldberg RB, Fonseca VA, Truitt KE, Jones MR. Efficacy and safety of colesevelam in patients with type 2 diabetes mellitus and inadequate glycemic control receiving insulin-based therapy. Arch Intern Med. 2008;168(14):1531–1540. | |
Jialal I, Abby SL, Misir S, Nagendran S. Concomitant reduction in low-density lipoprotein cholesterol and glycated hemoglobin with colesevelam hydrochloride in patients with type 2 diabetes: a pooled analysis. Metab Syndr Relat Disord. 2009;7(3):255–258. | |
Yamakawa T, Takano T, Utsunomiya H, Kadonosono K, Okamura A. Effect of colestimide therapy for glycemic control in type 2 diabetes mellitus with hypercholesterolemia. Endocr J. 2007;54(1):53–58. | |
Cao G, Liang Y, Broderick CL, et al. Antidiabetic action of a liver X receptor agonist mediated by inhibition of hepatic gluconeogenesis. J Biol Chem. 2003;278(2):1131–1136. | |
Laffitte BA, Chao LC, Li J, et al. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc Natl Acad Sci U S A. 2003;100(9):5419–5424. | |
Baranowski M. Biological role of liver X receptors. J Physiol Pharmacol. 2008;59 Suppl 7:31–55. | |
Goldfine AB. Modulating LDL cholesterol and glucose in patients with type 2 diabetes mellitus: targeting the bile acid pathway. Curr Opin Cardiol. 2008;23(5):502–511. | |
Reasner CA. Reducing cardiovascular complications of type 2 diabetes by targeting multiple risk factors. J Cardiovasc Pharmacol. 2008;52(2):136–144. | |
Staels B, Handelsman Y, Fonseca V. Bile acid sequestrants for lipid and glucose control. Curr Diab Rep. 2010;10(1):70–77. | |
Potthoff MJ, Boney-Montoya J, Choi M, et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1α pathway. Cell Metab. 2011;13(6):729–738. | |
Efanov AM, Sewing S, Bokvist K, Gromada J. Liver X receptor activation stimulates insulin secretion via modulation of glucose and lipid metabolism in pancreatic beta-cells. Diabetes. 2004;53 Suppl 3:S75–S78. | |
Harach T, Pols TW, Nomura M, et al. TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Sci Rep. 2012;2:430. | |
Jones M, Bays H. Colesevelam HCl: glycemic and lipid parameter effects in patients with type 2 diabetes mellitus treated with metformin-based therapy and a statin. Paper presented at: AACE 21st Annual Scientific and Clinical Congress; May 23–27, 2012; Philadelphia, PA. | |
Goldenberg I, Benderly M, Goldbourt U. Update on the use of fibrates: focus on bezafibrate. Vasc Health Risk Manag. 2008;4(1):131–141. | |
Mooradian AD. Dyslipidemia in type 2 diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2009;5(3):150–159. | |
[No authors listed]. Effect of fenofibrate on progression of coronary-artery disease in type 2 diabetes: the Diabetes Atherosclerosis Intervention Study, a randomised study. Lancet. 2001;357(9260):905–910. | |
Keech A, Simes RJ, Barter P, et al; FIELD study investigators. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366(9500):1849–1861. | |
ACCORD Study Group, Ginsberg HN, Elam MB, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362(17):1563–1574. | |
Bays HE, Roth EM, McKenney JM, et al. The effects of fenofibric acid alone and with statins on the prevalence of metabolic syndrome and its diagnostic components in patients with mixed dyslipidemia. Diabetes Care. 2010;33(9):2113–2116. | |
Jones PH, Davidson MH. Reporting rate of rhabdomyolysis with fenofibrate + statin versus gemfibrozil + any statin. Am J Cardiol. 2005;95(1):120–122. | |
Kamanna VS, Ganji SH, Kashyap ML. Recent advances in niacin and lipid metabolism. Curr Opin Lipidol. 2013;24(3):239–245. | |
Bays HE, Ballantyne C. What’s the deal with niacin development: is laropiprant add-on therapy a winning strategy to beat a straight flush? Curr Opin Lipidol. 2009;20(6):467–476. | |
AIM-HIGH Investigators, Boden WE, Probstfield JL, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–2267. | |
Merck Sharp & Dohme Corp. Merck Announces HPS2-THRIVE Study of TREDAPTIVE™ (Extended-Release Niacin/Laropiprant) Did Not Achieve Primary Endpoint [press release]. Whitehouse Station, NJ: Merck Sharp & Dohme Corp; 2012 [December 20]. Available from: http://www.mercknewsroom.com/press-release/prescription-medicine-news/merck-announces-hps2-thrive-study-tredaptive-extended-relea#sthash.8dgN5z3w.dpuf. Accessed June 6, 2014. | |
Elam MB, Hunninghake DB, Davis KB, et al. Effect of niacin on lipid and lipoprotein levels and glycemic control in patients with diabetes and peripheral arterial disease: the ADMIT study: a randomized trial. Arterial Disease Multiple Intervention Trial. JAMA. 2000;284(10):1263–1270. | |
Grundy SM, Vega GL, McGovern ME, et al; Diabetes Multicenter Research Group. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the assessment of diabetes control and evaluation of the efficacy of niaspan trial. Arch Intern Med. 2002;162(14):1568–1576. | |
Guyton JR, Bays HE. Safety considerations with niacin therapy. Am J Cardiol. 2007;99(6A):22C–31C. | |
MacLean A, McKenney J, Scott R, et al. Efficacy and safety of extended-release niacin/laropiprant in patients with type 2 diabetes mellitus. Br J Cardiol. 2011;18:37–45. | |
Bays HE, Neff D, Tomassini JE, Tershakovec AM. Ezetimibe: cholesterol lowering and beyond. Expert Rev Cardiovasc Ther. 2008;6(4):447–470. | |
Denke M, Pearson T, McBride P, Gazzara RA, Brady WE, Tershakovec AM. Ezetimibe added to ongoing statin therapy improves LDL-C goal attainment and lipid profile in patients with diabetes or metabolic syndrome. Diabetes Vasc Dis Res. 2006;3(2):93–102. | |
Goldberg RB, Guyton JR, Mazzone T, et al. Ezetimibe/simvastatin vs atorvastatin in patients with type 2 diabetes mellitus and hypercholesterolemia: the VYTAL study. Mayo Clin Proc. 2006;81(12):1579–1588. | |
Simons L, Tonkon M, Masana L, et al. Effects of ezetimibe added to on-going statin therapy on the lipid profile of hypercholesterolemic patients with diabetes mellitus or metabolic syndrome. Curr Med Res Opin. 2004;20(9):1437–1445. | |
Bardini G, Giorda CB, Pontiroli AE, Le Grazie C, Rotella CM. Ezetimibe + simvastatin versus doubling the dose of simvastatin in high cardiovascular risk diabetics: a multicenter, randomized trial (the LEAD study). Cardiovasc Diabetol. 2010;9:20. | |
Gaudiani LM, Lewin A, Meneghini L, et al. Efficacy and safety of ezetimibe co-administered with simvastatin in thiazolidinedione-treated type 2 diabetic patients. Diabetes Obes Metab. 2005;7(1):88–97. | |
Conard S, Bays H, Leiter LA, et al. Ezetimibe added to atorvastatin compared with doubling the atorvastatin dose in patients at high risk for coronary heart disease with diabetes mellitus, metabolic syndrome or neither. Diabetes Obes Metab. 2010;12(3):210–218. | |
Vytorin® [prescribing information]. Whitehouse Station, NJ: Merck Sharp and Dohme Corp, a subsidiary of Merck & Co, Inc.; 2014. | |
Cannon CP, Giugliano RP, Blazing MA, et al; IMPROVE-IT Investigators. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimbe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J. 2008;156(5):826–832. | |
Merck Sharp & Dohme Corp. IMPROVE-IT: Examining Outcomes in Subjects With Acute Coronary Syndrome: Vytorin (Ezetimibe/Simvastatin) vs Simvastatin (P04103 AM5). Available from: http://clinicaltrials.gov/show/NCT00202878. Accessed January 12, 2014. NLM identifier: NCT00202878. | |
Beysen C, Murphy EJ, Deines K, et al. Effect of bile acid sequestrants on glucose metabolism, hepatic de novo lipogenesis, and cholesterol and bile acid kinetics in type 2 diabetes: a randomised controlled study. Diabetologia. 2012;55(2):432–442. | |
Smushkin G, Sathananthan M, Piccinini F, et al. The effect of a bile acid sequestrant on glucose metabolism in subjects with type 2 diabetes. Diabetes. 2013;62(4):1094–1101. | |
Suzuki T, Oba K, Igari Y, et al. Colestimide lowers plasma glucose levels and increases plasma glucagon-like PEPTIDE-1 (7–36) levels in patients with type 2 diabetes mellitus complicated by hypercholesterolemia. J Nippon Med Sch. 2007;74(5):338–343. | |
Bays H, Dujovne C. Colesevelam HCl: a non-systemic lipid-altering drug. Expert Opin Pharmacother. 2003;4(5):779–790. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.