")
Back to Journals » OncoTargets and Therapy » Volume 10
Low platelet count is potentially the most important contributor to severe bleeding in patients newly diagnosed with acute promyelocytic leukemia
Authors Song Y , Peng P, Qiao C, Zhang R, Li J, Lu H
Received 20 June 2017
Accepted for publication 31 August 2017
Published 9 October 2017 Volume 2017:10 Pages 4917—4924
DOI https://doi.org/10.2147/OTT.S144438
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Samir Farghaly
Yu-hua Song,1,2 Peng Peng,3 Chun Qiao,1 Run Zhang,1 Jian-yong Li,1 Hua Lu1
1Department of Hematology, The First Affiliated Hospital of Nanjing Medical University, Jiangsu Province Hospital, 2Department of Hematology, 3Department of Oncology, The Second Hospital of Nanjing, Nanjing, People’s Republic of China
Abstract: The objective of the current study was to provide more appropriate therapeutic strategies for reducing severe hemorrhaging by assessing the recovery of abnormal coagulation indexes in patients with acute promyelocytic leukemia (APL) during induction therapy. Retrospective analyses of 112 patients newly diagnosed with APL were performed during initial treatment. In our study, the early death rate was 5.36%. Hemorrhage was the leading cause of death during the induction period (4/6). The values of white blood cell count, lactate dehydrogenase, prothrombin time (PT), fibrinogen (Fbg), hemoglobin, and bone marrow leukemic promyelocytes were significantly different in the high-risk group compared to the low/intermediate-risk groups. There were significant differences in the white blood cell count, bone marrow leukemic promyelocytes, platelet (PLT) count, and the levels of lactate dehydrogenase, d-dimer, PT, and Fbg, as well as in FLT3-ITD mutations between patients with major bleeding and those with minor bleeding. Hemostatic variables significantly improved over time during induction therapy. The recovery times of the PLT, PT, and Fbg values were significantly slower in patients with major bleeding than in those with minor bleeding. Specifically, the PLT level in patients with major bleeding was not similar to that in the minor bleeding group until after 4 weeks of treatment. Hemorrhages were the most common cause of induction death in this study. High-risk patients were more prone to serious clinical bleeding symptoms. Patients with major bleeding had more rapid proliferation characteristics and an increased incidence of FLT3-ITD mutations compared to patients with minor bleeding. Hemostatic variables recovered significantly more slowly in patients with major bleeding than in those with minor bleeding. Active induction therapy and blood product infusion are effective in preventing severe bleeding. Our results suggested that low PLT count might be the leading cause of fatal bleeding in patients newly diagnosed with APL.
Keywords: acute promyelocytic leukemia, all-trans retinoic acid, arsenic trioxide, coagulation, hemorrhage
Introduction
Acute promyelocytic leukemia (APL) is identified by the predominance of abnormal promyelocytes in the bone marrow (BM) and a specific chromosomal translocation – t(15;17) – resulting in a fusion transcript between the promyelocytic (PML) gene on chromosome 15 and the retinoic acid receptor alpha (RARα) gene on chromosome 17; this transcript is known as PML–RARα.1 The t(15;17) chromosomal translocation can induce hyperexpression of tissue factor (TF) in cells of patients with APL and render the patient hypercoagulable.2 All-trans retinoic acid (ATRA) and arsenic trioxide (ATO) are the current cornerstones of APL therapy, and these treatments have dramatically improved patient outcomes. ATRA improves the terminal differentiation of leukemic promyelocytes in patients with APL, and ATRA-induced remission is associated with an improvement in the coagulopathy of these patients.3 ATO also promotes the molecular remission of APL and addresses the associated coagulopathy by reducing membrane procoagulant activity (PCA) and TF expression.4,5 Despite this tremendous progress, APL still remains associated with a high incidence of premature death due to the frequent occurrence of abrupt bleeding diathesis. The presence of coagulopathy is an important risk factor for early hemorrhagic death in APL patients.6 Changes in clinical and laboratory parameters before and during induction therapy were examined in this study to identify more appropriate therapeutic strategies in order to reduce hemorrhage-mediated mortality.
Patients and ethics statement
A total of 112 patients newly diagnosed with APL were treated at Jiangsu Province Hospital, The First Affiliated Hospital of Nanjing Medical University (Nanjing, People’s Republic of China), between May 2009 and April 2016. This cohort included 66 males and 46 females with a median age of 41 years (range: 12–75). The APL diagnostic criteria were based on the World Health Organization Classification of Tumors–Pathology and Genetics of Tumors of Hematopoietic and Lymphoid Tissues (2008) and French–American–British classification systems (1976) criteria.7 A molecular diagnosis was confirmed by identification of t(15;17) in a cytogenetic analysis and/or positive detection of PML–RARα using either fluorescence in situ hybridization or reverse transcription-polymerase chain reaction. The immune phenotype diagnosis of APL was established by positivity for CD33, CD9, CD13, and CD117, and low expression of HLA-DR and CD34. Early death (ED) was defined as death due to any cause within 30 days of diagnosis. Other inclusion criteria were as follows: no severe liver disease (abnormal liver function, alanine aminotransferase/aspartate aminotransferase greater than 2.5 times the normal value, or total bilirubin greater than 1.5 times the normal value), no cardiovascular disorders or other hemorrhagic diseases, and no usage of anticoagulants during induction therapy. We collected data from May 2009 to April 2016 and identified the diagnostic information during and after data collection. The study was approved by the ethics committee at Jiangsu Province Hospital, affiliated with Nanjing Medical University. Written informed consent was obtained from all the enrolled subjects, including legal guardians on behalf of the minors recruited in our study.
Treatment strategies
The treatment followed the Shanghai APL protocol with some minor adjustments during the introduction period.8 Daily administration of intravenous ATO (10 mg/day) was started immediately when APL was suspected, and oral ATRA (25 mg/m2/d) was then prescribed upon genetic confirmation until the patient achieved complete remission (CR) after induction therapy. Additional chemotherapy (idarubicin 8 mg/m2/d for 3–4 days or daunorubicin 45 mg/m2/d for 3–4 days) was administered to control hyperleukocytosis if the peripheral white blood cell (WBC) count was greater than 10×109/L or on the second day in high-risk patients (pretreatment WBC count >10×109/L).
In addition to systemic treatment, patients were given central nervous system prophylaxis via six intrathecal injections of 10 mg of methotrexate, 50 mg of cytosine arabinoside, and 5 mg of dexamethasone. Therapeutic platelets (PLTs), fresh frozen plasma (FFP), or cryoprecipitate was transfused when clinically relevant bleeding occurred. A prophylactic PLT transfusion strategy (when available) was administered when the PLT count was lower than 50×109/L. Patients received only random ABO identical (non-HLA-typed) apheresis PLTs for PLT transfusions. In China, one apheresis unit is standardized to contain at least 2.5×1011 PLTs with less than 5×108 leukocytes. Prophylactic transfusion of either FFP or cryoprecipitate was mainly based on fibrinogen (Fbg) levels lower than 1.5 g/L. One unit of cryoprecipitate was converted into 200 mL of plasma for the subsequent statistical analysis.
Laboratory studies and clinical outcomes
The obtained information included demographic (age, gender), clinical (initial bleeding events and early hemorrhagic death events), and laboratory variables (WBC count, hemoglobin [HB] levels, PLT count, prothrombin time [PT], activated partial thromboplastin time [APTT], Fbg, D-dimer, lactate dehydrogenase [LDH], FLT3-ITD transcript type, and bone marrow leukemic promyelocyte [BMP] percentage). Routine blood tests were performed using a Sysmex XT-4000i Hematology Analyzer (Sysmex, Kobe, Japan) on EDTA-anticoagulated blood samples. The Sysmex CS-2000i Automated Hemostasis Analyzer (Sysmex) was used to detect coagulation and fibrinolysis parameters such as the PT, APTT, and Fbg levels (Clauss method) and D-dimer (immuno-turbidimetric method). Biochemical blood tests were done on a Beckman Coulter AU5800 Biochemical Analyzer (Beckman Coulter, Brea, CA, USA) using heparin-anticoagulated blood samples. Blast counts and promyelocyte percentages were determined by microscopic examination of the BM by two experienced physicians separately. Fusion gene transcripts from chromosomal aberrations were analyzed by reverse transcription-polymerase chain reaction. FLT3-ITD mutations and the PML–RARα gene fusion were detected in BM samples collected at the time of APL diagnosis.
Statistical analysis
The prognostic risk stratification of APL is based on a widely recognized risk evaluation standard that originated from the Italian GIMEMA and the Spanish PETHEMA trials:26 low risk, WBC ≤10×109/L and PLT >40×109/L; intermediate risk, WBC ≤10×109/L and PLT ≤40×109/L; and high risk, WBC >10×109/L.1 The outline of bleeding scores in idiopathic immune thrombocytopenia in China is based on the immune thrombocytopenia-specific bleeding assessment tool.9 Bleeding manifestations were classified (with gradations of severity) based on age and three major domains: skin, visible mucosae, and deep organs. Severity was scored from 1 to either 3 or 5, with a score of 8 assigned for any fatal bleeding (Table 1). We classified ≥6 as major bleeding and <6 as minor bleeding. Statistical analysis was conducted using SPSS software 16.0 (SPSS Inc., Chicago, IL, USA). The results are presented as the mean ± standard deviation for normally distributed data and as the median for nonnormally distributed data. Comparisons were made using Student’s t-test, Fisher’s exact test, and analysis of variance. P-values less than 0.05 indicated statistical significance.
 | Table 1 Outline of bleeding scores in idopathic immune thrombocytopenia |
Results
In our study, 112 patients were newly diagnosed with APL based on morphology and detection of the PML–RARα mutation and/or t(15;17)(q22;q21). Sixty-six of the 112 evaluated patients (58.93%) were male and 46 (41.07%) were female (ratio: 1.43:1). The median age at diagnosis was 41 years (range: 12–75 years). Among the entire cohort, 22 patients (19.64%) were classified as low risk, 63 (56.25%) were intermediate risk, and 27 (24.11%) were high risk. The patient characteristics are summarized in Table 2. The ED rate in our study was 5.36% (6/112). Among these six deaths, five were extremely early (<7 days of induction). The causes of ED in these 5 cases were intracranial hemorrhage (3), pulmonary hemorrhage (1), and acute renal failure (1). One of the patients with intracranial bleeding also had concomitant pulmonary hemorrhage. The sixth patient died of severe infection, and he also suffered from type 2 diabetes with poor glycemic control. All the patients who survived past 1 week achieved remission. Thus, the CR rate was 95.53% (107/112). Hemorrhagic events account for the majority (66.67%) of the EDs.
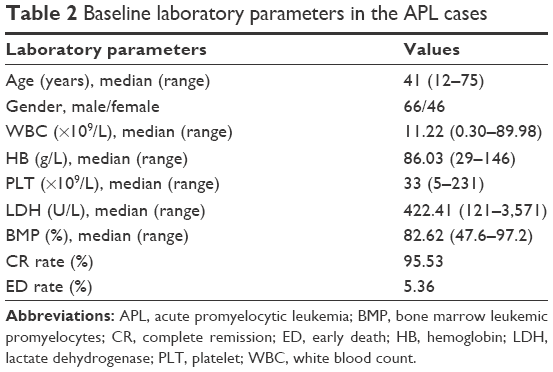 | Table 2 Baseline laboratory parameters in the APL cases |
We analyzed the clinical characteristics of all 112 patients. The median age of the cohort was 43 years (range: 12–75 years). Stratification of APL risk according to the Sanz stratification, which is based on WBC and PLT counts, remains the most reliable and valid method for rapidly identifying high-risk APL patients. We divided the patients into two groups: 85 (75.89%) were low/intermediate risk, and 27 (24.11%) were high risk. Among the 27 high-risk patients, 18 were male, and 9 were female; there was no statistically significant difference in the gender distribution of the two risk groups (low/intermediate group: 48 males and 37 females, P=0.455). The median age of the high-risk patients was 43.19 years versus 40.32 years for the low/intermediate-risk patients (P=0.383). The WBC count at presentation was higher in the high-risk group (median: 38.30×109/L) than in the low/intermediate-risk group (median: 2.62×109/L, P=0.000). The average Fbg concentration was lower in high-risk patients than in low/intermediate-risk patients (0.90±0.51 vs 1.48±0.85; P=0.000). The levels of HB, LDH, and BMP at presentation were higher in high-risk patients (median: 94.44 g/L, 847.26 U/L, and 85.81%, respectively) than in low/intermediate-risk patients (median: 81.61 g/L, 287.45 U/L, and 81.61%, respectively; P=0.044, P=0.000, and P=0.038, respectively). The PT value at presentation was higher in high-risk patients (median: 17.33 s) than in low/intermediate-risk patients (median: 13.77 s, P=0.003). However, the PLT, APTT, and D-dimer levels at presentation were not significantly different in high-risk patients compared to low/intermediate-risk patients (Table 3). The bleeding scores of high-risk patients were all ≥10, and the percentage of low/intermediate-risk patients with severe bleeding (≥6) was 48.24% (41/85).
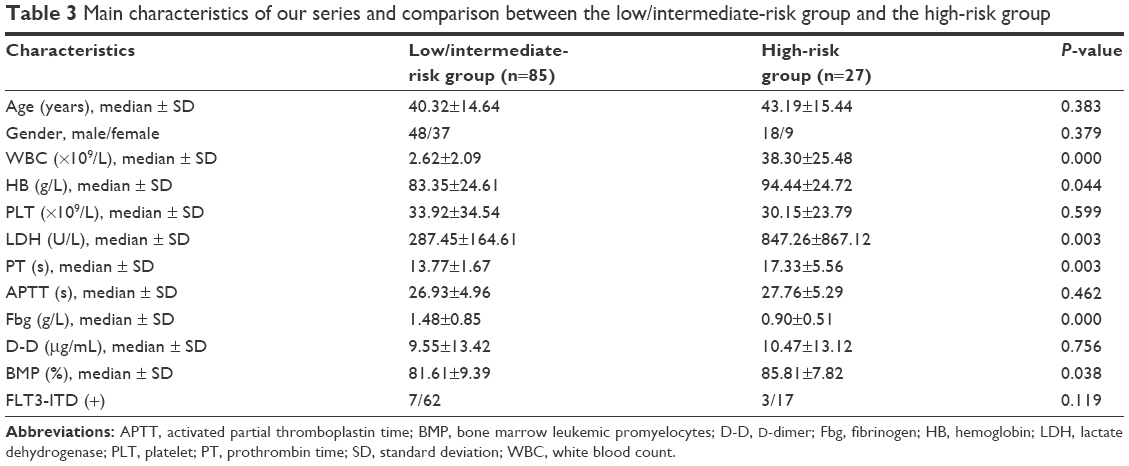 | Table 3 Main characteristics of our series and comparison between the low/intermediate-risk group and the high-risk group |
Then 6 cases of ED were excluded, and the remaining 106 cases (61 males and 45 females) were analyzed in the subsequent studies. We divided the 106 patients into the major bleeding and minor bleeding groups based on the bleeding rating scale. Among the 60 patients with major bleeding, 35 were male and 25 were female; there was no statistically significant difference in the gender distribution of the two bleeding groups (minor bleeding: total 46, 26 males and 20 females, P=0.852). The median age of the patients who suffered from major bleeding was 40.77 years versus 41.15 years for those with minor bleeding (P=0.893). There was a significant difference in the number of WBCs (17.90×109/L vs 1.16×109/L, P=0.000) and PLTs (25.72×109/L vs 43.65×109/L, P=0.011) between the major bleeding group and the minor bleeding group. However, there was no significant difference in the mean HB level (86.62 g/L vs 85.54 g/L, P=0.828) or the APTT (26.36 s vs 26.79 s, P=0.544). There were significant differences in LDH (519.39 U/L vs 249.35 U/L, P=0.002), D-dimer (7.09 μg/mL vs 13.23 μg/mL, P=0.026), PT (15.39 s vs 13.60 s, P=0.004), Fbg (1.09 g/L vs 1.66 g/L, P=0.001), and BMP (84.04% vs 80.18%, P=0.033) (Table 4).
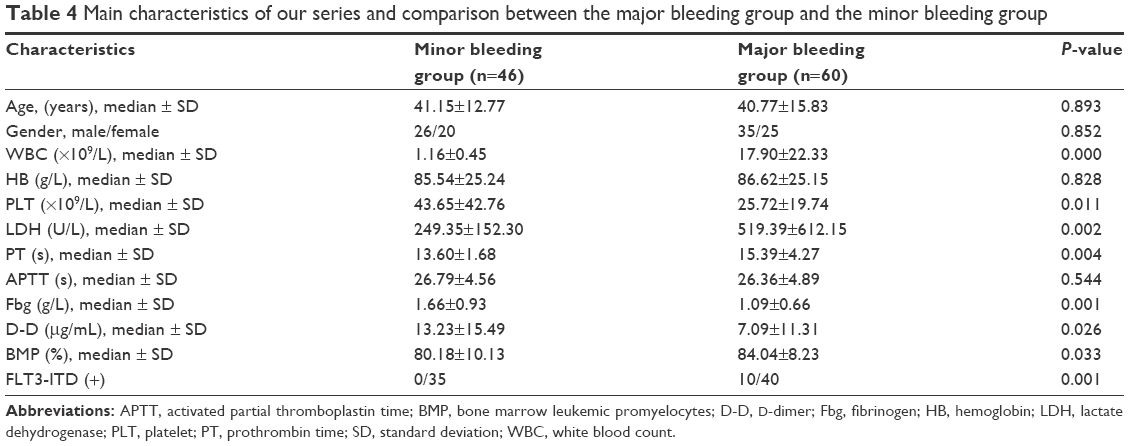 | Table 4 Main characteristics of our series and comparison between the major bleeding group and the minor bleeding group |
We also analyzed the changes in the hemostatic variables in 106 patients during induction therapy. The recorded time points were the time of diagnosis, before initial treatment, and at 0.5, 1, 1.5, 2, and 4 weeks after initiating treatment with ATO + ATRA and/or chemotherapy. All the observed variables showed statistically significant differences that quickly changed over time. The PLT, PT, APTT, and Fbg values appeared to significantly improve at 0.5 weeks after initiating induction therapy, and the D-dimers began to decrease after 1 week of therapy. There were significant differences in the changes in PLT, PT, and Fbg (P=0.005, P=0.018, and P=0.022) between the major and minor bleeding groups after 4 weeks of introduction therapy, but there were no significant differences in the changes in APTT and D-dimer (P=0.746, P=0.333). Further comparisons showed that differences in the Fbg levels were present only at diagnosis (P<0.001), with no significant difference between the two groups after initiating therapy (P0.5W =0.053, P1W =0.246, P1.5W =0.160, P2W =0.506, P4W =0.510). The PT differed between groups at 0 weeks (P0W =0.008) and 0.5 weeks (P0.5W =0.019), but there was no significant difference starting 1 week after initiating introduction therapy (P1W =0.563, P1.5W =0.615, P2W =0.857, P4W =0.070). However, differences in the PLT levels were present during the first 2 weeks (P0W =0.005, P0.5W =0.009, P1W =0.000, P1.5W =0.047, P2W =0.007) but disappeared by the end of the fourth week of therapy (P4W =0.740) (Figures 1–5).
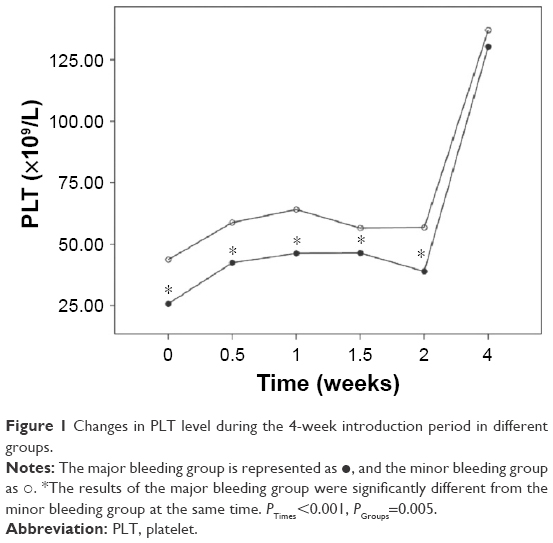 | Figure 1 Changes in PLT level during the 4-week introduction period in different groups. |
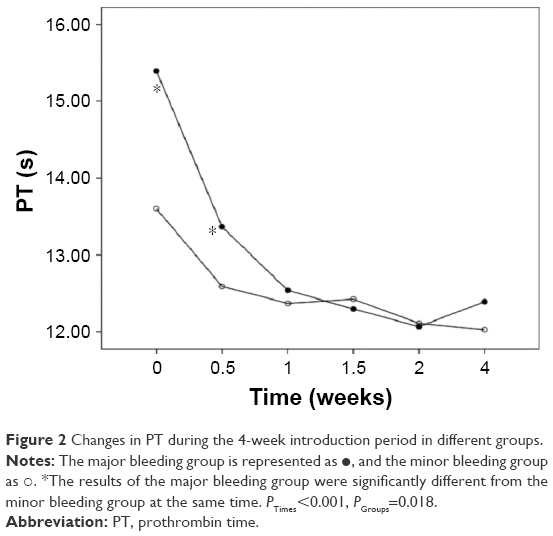 | Figure 2 Changes in PT during the 4-week introduction period in different groups. |
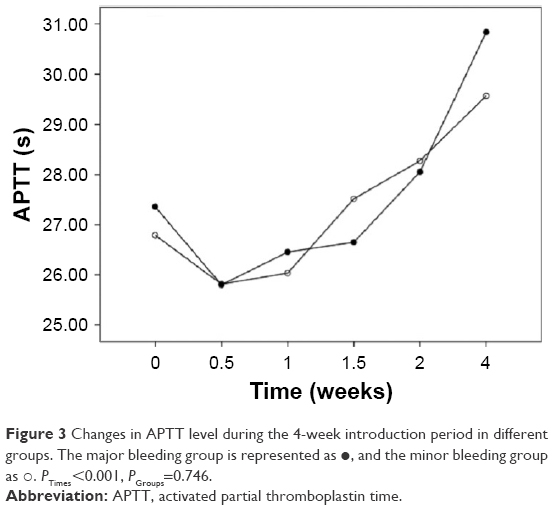 | Figure 3 Changes in APTT level during the 4-week introduction period in different groups. The major bleeding group is represented as •, and the minor bleeding group as ○. PTimes<0.001, PGroups=0.746. |
 | Figure 4 Changes in D-dimer level during the 4-week introduction period in different groups. |
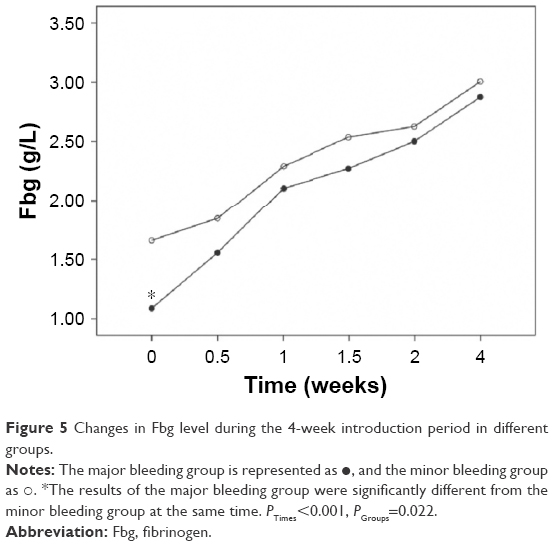 | Figure 5 Changes in Fbg level during the 4-week introduction period in different groups. |
Discussion
APL is a clinical and biological variant of acute myeloid leukemia that is characterized by a cytogenetic abnormality, t(15;17)(q22;q12); PML–RARα.1 APL has a good prognosis based on the high rates of CR and survival achieved with therapies containing ATRA and/or ATO. Unlike other subtypes of acute myeloid leukemia, the primary cause of treatment failure in patients with APL is ED, which is defined as death within the first 30 days of diagnosis. There are multiple causes of ED in patients with APL, although death during induction therapy is most frequently related to hemorrhagic diathesis due to hyperfibrinolysis, proteolysis, and disseminated intravascular coagulation, which is further complicated by thrombocytopenia.10 In this study, the overall ED rate was 5.36% (6/112). In comparison with developed countries, the mortality in the current study was within previously described ranges of 5%–10%.6,11–13 All the patients who succumbed to ED had a low PLT count, with a mean PLT count of 24.33×109/L at presentation. Hemorrhages have been reported to occur in 90% of APL patients.14 ED due to hemorrhage before and during induction therapy currently remains the main cause of treatment failure in APL.6,15 Hemorrhage is responsible for most cases of death during the induction period, and it accounted for 66.67% (4/6) of the EDs in this study. Furthermore, most hemorrhagic mortalities in this study were attributed to intracranial and pulmonary hemorrhages. All these fatal events occurred within the first week of hospitalization.
ED is particularly frequent among high-risk patients, and a high WBC count (>10×109/L) was suggested as an adverse prognostic factor for bleeding complications in APL.16,17 Almost all patients had laboratory findings of intravascular coagulation, increased fibrinolysis, and decreased PLT production due to marrow invasion upon initial presentation. It is common for patients newly diagnosed with APL to present elevated D-dimer levels, fibrin split products, prolonged PT, and partial thromboplastin time and hypofibrinogenemia.14 These unusual indicators are related to the clinical manifestations of a high risk of bleeding and thrombosis. In addition to an elevated WBC count at presentation, high-risk patients with APL in our study also had high LDH, HB, and PT levels; increased BMP percentage; and low Fbg levels. Despite the rapid proliferation characteristics, high-risk patients had more abnormal coagulation variables. Based on bleeding score, all patients in the high-risk group had severe bleeding (score ≥10). There was no doubt that high-risk patients were more prone to severe bleeding.
Reported risk factors for severe hemorrhage include low initial Fbg levels, poor performance status, and high WBC count.18 We identified patients with major bleeding based on decreased values of Fbg, D-dimer, and PLT; high values of WBC, LDH, PT, and BMP; and positivity for FLT3-ITD. The repeated finding that total WBC count is an independent predictor of major bleeding complications is not surprising. Elevated BMP and LDH levels were also a reflection of excessive proliferation. The presence of coagulopathy and low PLT levels were associated with the development of hemorrhages. Low PLT counts are usually due to impaired PLT production and consumption. Increases in D-dimer and decreases in Fbg levels are evidence of hyperfibrinolysis.19,20 Notably, patients with major bleeding had lower D-dimer levels compared to patients with minor bleeding; this finding might explain why none of the randomized data showed a benefit of the clinical application of antifibrinolytics, which could block primary hyperfibrinolysis. Some studies have suggested a possible correlation between FLT3-ITD and the occurrence of ED in APL patients.21,22 Patients with major bleeding had a significantly higher prevalence of the FLT3-ITD mutation in the current study. Our results suggested that the bleeding score, which is mainly based on the hemorrhage severity, was a good approach to distinguish APL patients with different clinical and laboratory characteristics. The establishment of a bleeding scale that is more consistent with the bleeding characteristics of APL has realistic clinical value.
Leukemic cells isolated from APL patients with the typical t(15;17) chromosomal balanced translocation exhibit high levels of PCA, including TF and cancer procoagulant, which are associated with the induction of hypercoagulability. ATRA induces cell differentiation of APL in vitro, which is associated with the loss of cancer procoagulant or TF expression. ATO, another effective agent for treating APL, also decreases TF expression and PCA in malignant APL cells both in vitro and in vivo.4 Therapy with ATRA and ATO leads to an improvement in the coagulopathy and a reduced risk of severe hemorrhage.23 After implementing the aggressive induction regimen, the hemostatic variables were significantly restored within 0.5–1 week compared to the initial levels. This confirmed that rapid initiation of therapy is important for treating APL. The ATO + ATRA regimen could quickly relieve the coagulopathy burden during the induction period. Thus, if an APL diagnosis is suspected based upon cytologic criteria, the recommendation is to immediately start introduction therapy without delay – even before obtaining definitive genetic confirmation of the diagnosis.
After aggressive treatment, the recovery rates of PLT, PT, and Fbg levels were significantly slower in patients with major bleeding than in those with minor bleeding. In the major bleeding group, Fbg and PT levels increased to similar levels as those in the minor bleeding group after 0.5–1 week of induction therapy. Interestingly, the PLT levels in the major bleeding group were significantly lower than those in the minor bleeding group during the 2 weeks of induction therapy; this index was not similar in the two groups until after 4 weeks of treatment. Patients with major bleeding require aggressive prophylactic transfusions based on frequent complete blood count and clotting tests. Blood products consisting of PLT transfusions, FFP, and Fbg concentrate (typically cryoprecipitate) are the cornerstone of prohemostatic treatments. Our findings suggested that sustained low PLT levels were the most important factor in major bleeding. Possible causes of sustained low PLT levels include malignant clonal proliferation (which reduces PLT production), PLT infusion resistance (which causes ineffective infusion), and chemotherapy-induced BM suppression. Clinicians should aim for higher PLT counts in patients with APL during induction therapy, which may help prevent death due to hemorrhage in these patients. Therefore, in addition to aggressive induction therapy and supportive care, a reduction in the use of chemotherapy should also be considered. Lo-Coco et al have shown the efficacy and safety of ATRA in combination with ATO in patients with low/intermediate-risk APL. However, the ATRA + ATO regimen alone may be inadequate for high-risk patients.24,25 New immunotherapies such as CD33 monoclonal antibodies may be a viable method for better reducing the incidence of severe bleeding in APL.
This study had some limitations, including small sample size, short follow-up period, and single-center, retrospective design. There is an urgent need for more randomized, prospective trials of different induction regimens and new drug applications.
In conclusion, fatal hemorrhage remains the main cause of ED during the induction period in patients newly diagnosed with APL. High-risk patients had severe coagulopathy and were more prone to serious bleeding. The establishment of a bleeding scale that is more consistent with the bleeding characteristics of APL may better distinguish APL patients with different clinical and laboratory characteristics. Rapid onset of introduction therapy with the ATO + ATRA regimen could relieve the coagulopathy burden of APL patients. Aggressive prophylactic transfusion is the cornerstone of prohemostatic treatments. Finally, this study suggested that sustained low PLT levels were the most important factor in predicting severe bleeding. A higher PLT count should be the goal for patients with APL during induction therapy, as it may help prevent hemorrhagic death in these patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Candoni A, Damiani D, Michelutti A, et al. Clinical characteristics, prognostic factors and multidrug-resistance related protein expression in 36 adult patients with acute promyelocytic leukemia. Eur J Haematol. 2003;71:1–8. | ||
Falanga A, Barbui T, Rickles FR. Hypercoagulability and tissue factor gene upregulation in hematologic malignancies. Semin Thromb Hemost. 2008;34(2):204–210. | ||
Falanga A, Rickles FR. Pathogenesis and management of the bleeding diathesis in acute promyelocytic leukemia. Best Pract Res Clin Haematol. 2003;16(3):463–482. | ||
Liu Y, Wang Z, Jiang M, et al. The expression of annexin II and its role in the fibrinolytic activity in acute promyelocytic leukemia. Leuk Res. 2011;35(7):879–884. | ||
Zhou J, Shi J, Hou J, et al. Phosphatidylserine exposure and procoagulant activity in acute promyelocytic leukemia. J Thromb Haemost. 2010;8(4):773–782. | ||
de la Serna J, Montesinos P, Vellenga E, et al. Causes and prognostic factors of remission induction failure in patients with acute promyelocytic leukemia treated with alltrans retinoic acid and idarubicin. Blood. 2008;111:3395–3402. | ||
Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. | ||
Hu J, Liu YF, Wu CF, et al. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 2009;106(9):3342–3347. | ||
Wang L, Hou M. Outline of bleeding scores in idopathic immune thrombocytopenia. Chin J Hematol. 2015;36(12):1053–1055. | ||
Iland HJ, Seymour JF, Wei A. Optimal approach for high-risk acute promyelocytic leukemia. Curr Opin Hematol. 2014;21:102–113. | ||
Lo-Coco F, Avvisati G, Vignetti M, et al. Front-line treatment of acute promyelocytic leukemia with AIDA induction followed by risk-adapted consolidation for adults younger than 61 years: results of the AIDA-2000 trial of the GIMEMA Group. Blood. 2010;116:3171–3179. | ||
Avisati G, Lo-Coco F, Paoloni FP, et al. AIDA 0493 protocol for newly diagnosed acute promyelocytic leukemia: very long-term results and role of maintenance. Blood. 2011;117:4716–4725. | ||
Ades L, Guerci A, Raffoux E, et al. Very long-term outcome of acute promyelocytic leukemia after treatment with all-trans retinoic acid and chemotherapy: the European APL Group experience. Blood. 2010;115:1690–1696. | ||
Kwaan HC, Wang J, Boggio LN. Abnormalities in hemostasis in acute promyelocytic leukemia. Hematol Oncol. 2002;20:33–41. | ||
Lehmann S, Ravn A, Carlsson L, et al. Continuing high early death rate in acute promyelocytic leukemia: a population-based report from the Swedish Adult Acute Leukemia Registry. Leukemia. 2011;25:1128–1134. | ||
Dally N, Hoffman R, Haddad N, Sarig G, Rowe JM, Brenner B. Predictive factors of bleeding and thrombosis during induction therapy in acute promyelocytic leukemia–a single center experience in 34 patients. Thromb Res. 2005;116(2):109–114. | ||
Kwaan HC, Cull EH. The coagulopathy in acute promyelocytic leukemia – what have we learned in the past twenty years. Best Pract Res Clin Haematol. 2014;27(1):11–18. | ||
Yanada M, Matsushita T, Asou N, et al. Severe hemorrhagic complications during remission induction therapy for acute promyelocytic leukemia: incidence, risk factors, and influence on outcome. Eur J Haematol. 2007;78:213–219. | ||
Stein E, McMahon B, Kwaan H, et al. The coagulopathy of acute promyelocytic leukemia revisited. Best Pract Res Clin Haematol. 2009;22(1):153–163. | ||
Kwaan HC. The unique hemostatic dysfunction in acute promyelocytic leukemia. Semin Thromb Hemost. 2014;40(3):332–336. | ||
Gale RE, Hills R, Pizzey A, Kottaridis PD, Swirsky D, Gilkes AF. Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood. 2005;106:3768–3778. | ||
Kutny MA, Moser BK, Laumann K, et al. FLT3 mutation status is a predictor of early death in pediatric acute promyelocytic leukemia: a report from Children’s Oncology Group. Pediatr Blood Cancer. 2012;59:662–667. | ||
Sanz MA, Grimwade D, Tallman MS, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113(9):1875–1891. | ||
Estey E, Garcia-Manero G, Ferrajoli A, et al. Use of all-trans retinoic acid plus arsenic trioxide as an alternative to chemotherapy in untreated acute promyelocytic leukemia. Blood. 2006;107:3469–3473. | ||
Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369(2):111–121. | ||
Sanz MA, Lo Coco F, Martin G, et al. Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: a joint study of the PETHEMA and GIMEMA cooperative groups. Blood. 2000:96(4):1247–1253. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.