")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 11
Low aquaporin-2 excretion in the nephrotic syndrome: an escape from the vasopressin regulating effect
Authors Brovko M , Kozlovskaya L , Pulin A, Moiseev S, Sholomova V, Shchekochikhin D, Gognieva D , Milovanova L , Fomin V
Received 17 June 2018
Accepted for publication 31 August 2018
Published 17 October 2018 Volume 2018:11 Pages 271—277
DOI https://doi.org/10.2147/IJNRD.S177469
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Mikhail Brovko,1 Lidia Kozlovskaya,1 Andrey Pulin,1,2 Sergey Moiseev,1 Victoria Sholomova,1 Dmitry Shchekochikhin,1 Daria Gognieva,1 Ludmila Milovanova,1 Victor Fomin1
1Sechenov First Moscow State Medical University, Moscow, Russia; 2Laboratory for Cell Technologies and Developmental Pathology, Federal State Budgetary Scientific Institution “Institute of General Pathology and Pathophysiology,” Moscow, Russia
Purpose: Experimental studies suggest that the nephrotic syndrome is associated with “vasopressin escape”, characterized by low aquaporin-2 (AQP2) expression in the collecting duct despite high vasopressin secretion. We investigated this phenomenon in patients with the nephrotic syndrome.
Patients and methods: We recruited 47 patients with proteinuric kidney disease who were distributed into the following four groups: 1) nephrotic syndrome with kidney dysfunction (n=10); 2) nephrotic syndrome with normal kidney function (n=16); 3) partial remission of nephrotic syndrome (n=10); and 4) minimal proteinuria (n=11). Nine healthy volunteers comprised a control group. Serum copeptin level (as a marker of vasopressin secretion) and urinary AQP2 were measured using ELISA.
Results: Nephrotic syndrome was associated with a significant increase in serum copeptin levels compared with those in the other groups (all P<0.05). In patients with nephrotic syndrome and a partial remission of nephrotic syndrome combined, there was more than a ten-fold decrease in the median urinary AQP2 excretion (0.03 ng/mL) compared with healthy volunteers (0.41 ng/mL; P<0.001) and more than a five-fold decrease compared with patients with minimal proteinuria (0.21 ng/mL; P<0.05). Unlike copeptin levels, the median urinary AQP2 excretion in patients with minimal proteinuria also decreased but less significantly than in those with nephrotic syndrome. There was a negative correlation between the urinary AQP2 excretion and daily proteinuria (R=−0.41; P=0.005).
Conclusion: Our clinical study was the first to demonstrate low AQP2 excretion in nephrotic syndrome that may indicate an escape from the vasopressin regulating effect.
Keywords: nephrotic syndrome, aquaporin-2, copeptin, vasopressin escape
Introduction
Aquaporins (AQPs) are a family of membrane proteins that mediate water transport across plasma membranes in many cells and tissues.1 The discovery of the first water channel (AQP1) led to the awarding of the Nobel Prize in Chemistry in 2003 to Peter Agre.2 At least eight AQPs have been found in the mammalian kidneys. AQP2 is the water-channel protein expressed in the connecting tubule and collecting duct, and is the chief target for the short-term and long-term regulation of water reabsorption by arginine–vasopressin (AVP), also known as the antidiuretic hormone.1 Short-term (acute) regulation occurs within a period of minutes and involves the trafficking of AQP2 from an intracellular reservoir to the apical membrane in response to activation of vasopressin V2 receptors by AVP, whereas long-term (chronic) regulation takes hours or days and is achieved through an increase in the AQP2 expression.3 These processes are essential for water balance homeostasis; moreover, understanding the pathophysiology of clinical syndromes characterized by the retention of water in the human body may provide the basis for their treatment.
Numerous studies have shown that dysregulation of AQP2 could be associated with hereditary and acquired nephrogenic diabetes insipidus, congestive heart failure, liver cirrhosis, and experimental nephrotic syndrome.4 In patients with congestive heart failure, increased secretion of AVP stimulates the expression and translocation of AQP2 in the collecting duct, and thereby facilitates water retention and hyponatremia. A higher urinary AQP2 level was a predictor of a response to tolvaptan – a novel V2 receptor antagonist that blocks the AVP–AQP2 pathway and increases diuresis in patients with heart failure who are refractory to conventional diuretics.5 However, rats with experimental nephrotic syndrome did not develop hyponatremia despite extensive extracellular fluid volume expansion and displayed downregulation of AQP2 expression in the kidneys that mediated relative resistance to AVP.6,7 This phenomenon of the AVP resistance of the collecting ducts was called a “vasopressin escape.”8 In fact, it may be a protective mechanism limiting AVP-induced antidiuretic action and water retention.
Decreased AQP2 expression in the nephrotic kidney, such that it mediated vasopressin escape, was shown in vivo in the experimental models; however, it is currently unknown whether this phenomenon exists in humans. Therefore, we investigated the serum levels of copeptin and the urinary excretion of AQP2 in patients with the nephrotic syndrome. Copeptin is a glycosylated peptide derived from the C-terminal part of the AVP precursor. Unlike AVP, copeptin remains stable for several days in serum or plasma and can serve as a biomarker of AVP release.9
Material and methods
Patients with proteinuric kidney disease (n=47), with or without the nephrotic syndrome, were recruited from March 2015 to October 2017. Exclusion criteria were age <18 years, pregnancy, active urinary infection, heart failure, diabetes mellitus, diabetes insipidus, severe arterial hypertension, obesity, liver disease, and diffuse connective tissue diseases. Nine healthy volunteers (three males and six females, 22–62 years of age; mean age 45.9±14.5 years) comprised a control group. All subjects provided written informed consent prior to study participation. The study had approval from the ethics committee of Sechenov University (Moscow, Russia).
Blood and urine samples were collected on the same day in a fasting state. The serum copeptin level (Copeptin Human EIA; Phoenix Pharmaceuticals, Inc., Burlingame, CA, USA) and urinary AQP2 (EIA Kit for aquaporin-2, Collecting duct; USCN Life Science Inc., Houston, TX, USA) were measured using ELISA. Serum sodium, albumin, and creatinine concentrations and 24-hour proteinuria were evaluated by standard methods. The glomerular filtration rate (GFR) was estimated by the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula. Serum cystatin C was measured using ELISA.
Data were summarized with descriptive statistics. Normality tests were conducted with the Shapiro–Wilk test. Patient and disease characteristics and laboratory parameters are reported as the number and percentage for categorical variables, mean±SD for continuous variables with a normal distribution, and median and IQR for continuous variables with a non-normal distribution. Differences in proportions and continuous data were tested using Pearson’s chi-squared test and the Mann–Whitney U-test, respectively. Correlations between laboratory variables were evaluated with the Spearmen correlation coefficient. A two-sided P-value <0.05 was considered to indicate statistical significance. Statistical analysis was conducted using IBM SPSS version 21 (IBM Corporation, Armonk, NY, USA).
Results
Characteristics of patients
Characteristics of the study participants are shown in Table 1. Causes of kidney disease were idiopathic membranous nephropathy (n=12), focal segmental glomerulosclerosis (n=10), membranoproliferative glomerulonephritis (n=2), mesangial proliferative glomerulonephritis (n=8), minimal change disease (n=4), and primary chronic glomerulonephritis, no histology (n=11). According to the proteinuria range and kidney function, patients were distributed into the following four groups: 1) nephrotic syndrome with kidney dysfunction; 2) nephrotic syndrome with normal kidney function; 3) partial remission of the nephrotic syndrome (proteinuria 1–3.5 g/day); and 4) minimal proteinuria (<1 g/day). Patients with minimal change disease were equally distributed within the first two groups. All patients with minimal proteinuria had no history of the nephrotic syndrome. In 30 patients, blood and urine samples were collected prior to the administration of immunosuppressive agents and diuretics. The remaining patients, at the time of laboratory testing, were treated with glucocorticoids as a monotherapy or in combination with immunosuppressive medications with or without loop diuretics (Table 1). The mean serum sodium concentration in all groups of patients was comparable to that in healthy volunteers.
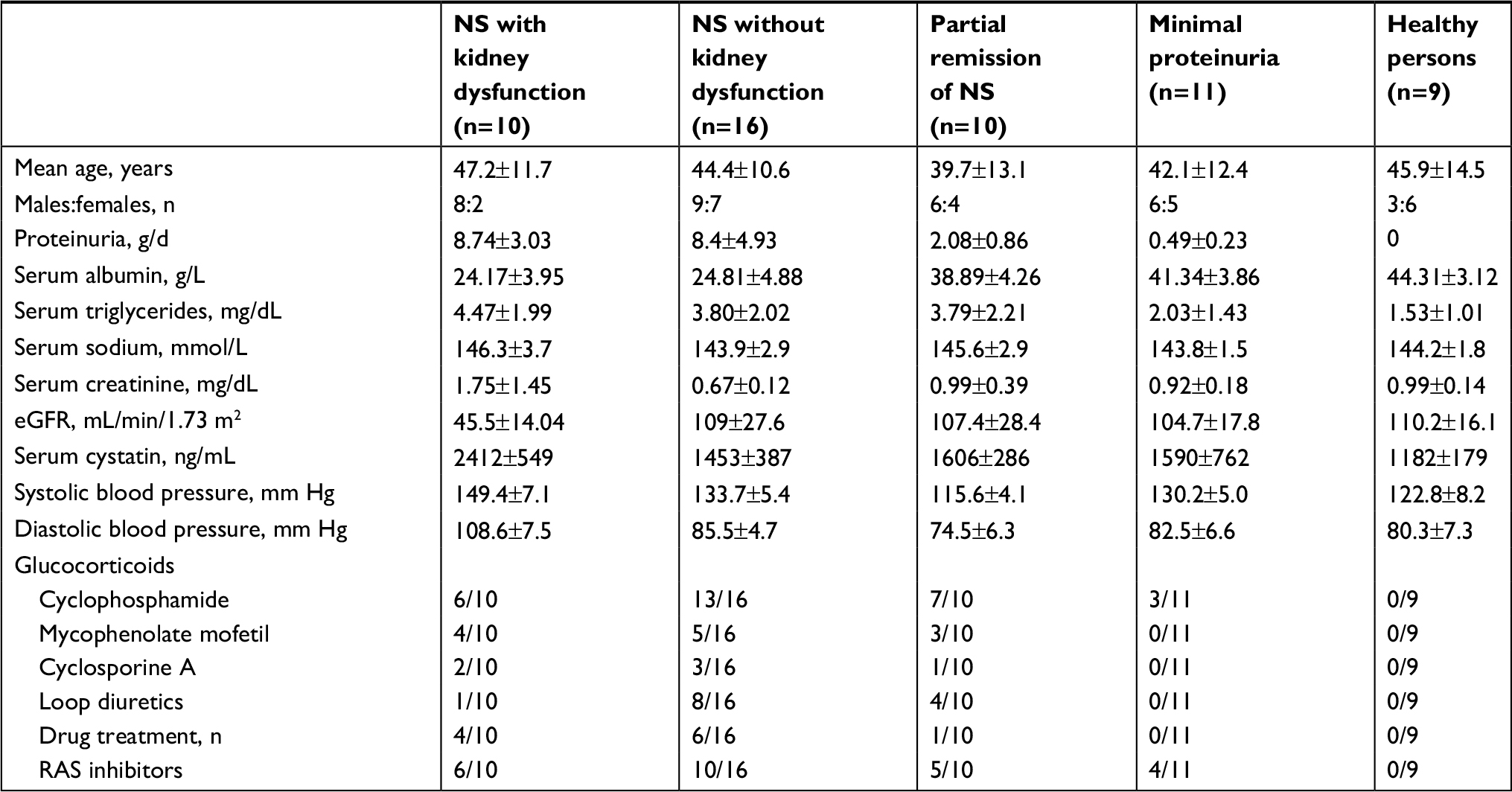 | Table 1 Demographic and clinical characteristics of the study participants Notes: Continuous data are shown as mean (SD). eGFR was calculated using the CKD-EPI formula. Abbreviations: CKD-EPI, Chronic Kidney Disease Epidemiology Collaboration; eGFR, estimated glomerular filtration rate; NS, nephrotic syndrome; RAS, renin–angiotensin system. |
Serum copeptin levels
The median serum copeptin level in patients with the nephrotic syndrome and a partial remission of the nephrotic syndrome combined (2.93 ng/mL; interquartile range [IQR] 1.66–4.57) was significantly higher than in patients with minimal proteinuria (0.99 ng/mL; IQR 0.92–2.36; P<0.05) and healthy volunteers (0.95 ng/mL; IQR 0.85–2.28; P<0.05).
Active nephrotic syndrome with or without kidney dysfunction was associated with a significant increase in serum copeptin levels compared with those in patients with a partial remission of the nephrotic syndrome and minimal proteinuria or healthy volunteers (all P<0.05; Figure 1). In patients with a partial remission of the nephrotic syndrome, the median serum copeptin level was numerically higher than in the control group, whereas it was close to normal in patients with minimal proteinuria (Table 2).
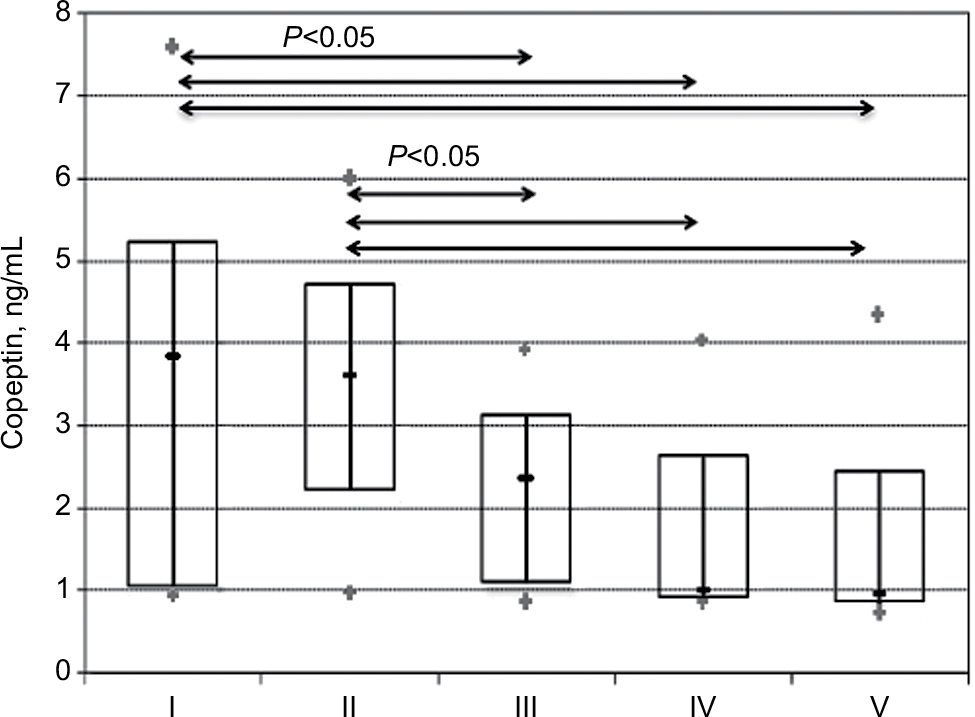 | Figure 1 Serum copeptin in patients with NS with kidney dysfunction (I), NS without kidney dysfunction (II), partial remission of NS (III), minimal proteinuria (IV), and healthy volunteers (V). Abbreviation: NS, nephrotic syndrome. |
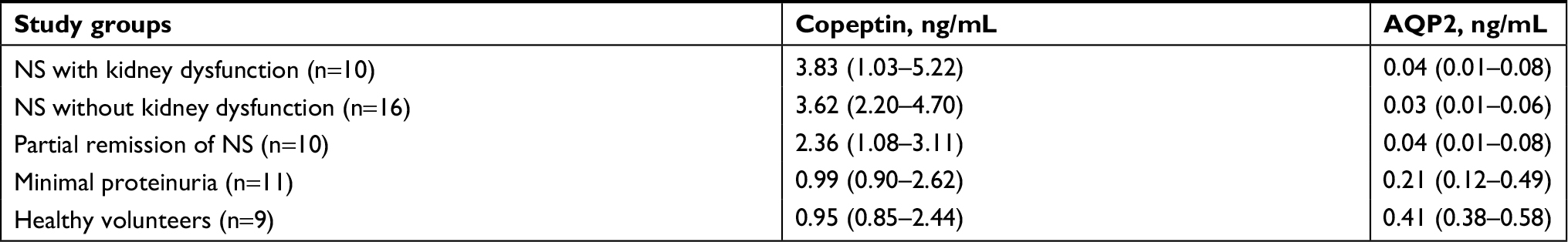 | Table 2 Serum copeptin and urinary AQP2 levels in study participants Note: Data are shown as median values (IQR). Abbreviations: AQP2, aquaporin-2; NS, nephrotic syndrome. |
There was a strong negative correlation between serum copeptin and albumin levels (R=−0.51; P=0.0003), and a strong positive correlation between serum copeptin concentration and daily proteinuria (R=0.46; P=0.0017). Copeptin levels did not correlate with serum sodium, creatinine, and cystatin concentrations.
Urinary AQP2 excretion
In patients with the nephrotic syndrome and a partial remission of the nephrotic syndrome combined, there was more than a ten-fold decrease in the median urinary AQP2 excretion (0.03 ng/mL; IQR 0.01–0.08) compared with the normal value in healthy volunteers (0.41 ng/mL; IQR 0.39–0.58; P<0.001) and more than a five-fold decrease compared with that in patients with minimal proteinuria (0.21 ng/mL; IQR 0.12–0.49; P<0.05; Table 2). Urinary AQP2 levels were similar in patients with nephrotic and sub-nephrotic range proteinuria. Moreover, the median urinary AQP2 excretion in patients with minimal proteinuria was decreased, albeit less significantly than in patients with the nephrotic syndrome (Figure 2).
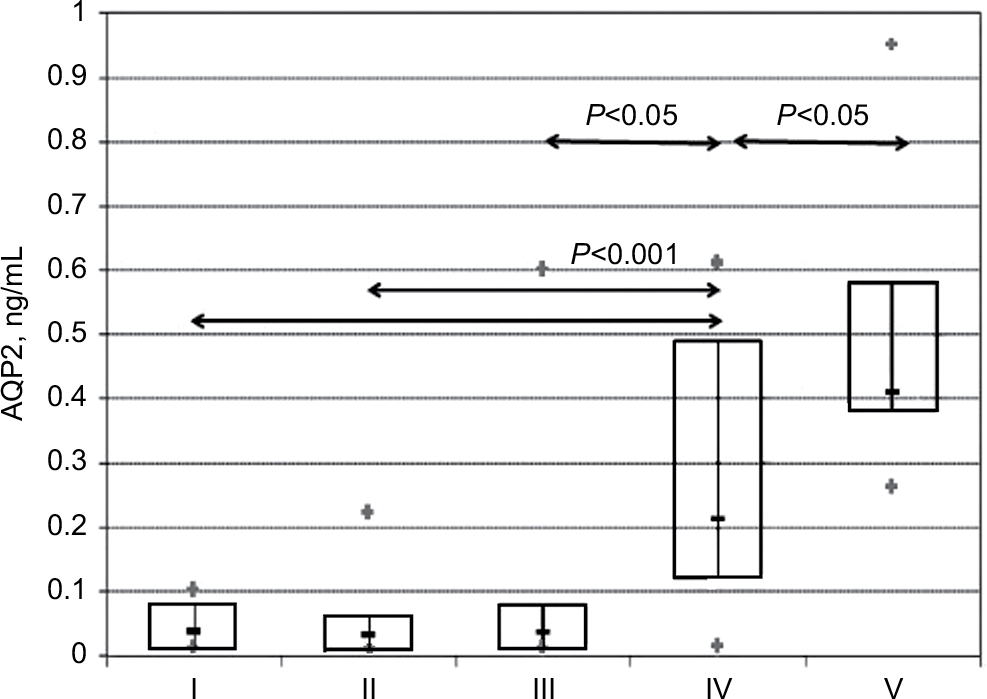 | Figure 2 Serum copeptin in patients with NS with kidney dysfunction (I), NS without kidney dysfunction (II), partial remission of NS (III), minimal proteinuria (IV), and healthy volunteers (V). Abbreviation: NS, nephrotic syndrome. |
There was a strong positive correlation between the urinary AQP2 level and serum albumin (R=0.41; P=0.005), and a strong negative correlation between the urinary AQP2 excretion and daily proteinuria (R=−0.41; P=0.005). Urinary AQP2 levels did not correlate with serum copeptin, sodium, creatinine, and cystatin C concentrations.
Discussion
In our study, we showed that the nephrotic syndrome was associated with a marked increase in serum copeptin levels, reflecting higher AVP secretion. These changes were more prominent in patients with active nephrotic syndrome, although a numerical rise in serum copeptin concentration was also found in patients with a partial remission of the nephrotic syndrome who presented with sub-nephrotic range proteinuria. Moreover, there was no upregulation of AQP2 excretion, expected consequent to increased AVP activity, as was demonstrated in patients with congestive heart failure.10 In contrast, urinary AQP2 levels were reduced significantly (more than ten-fold) in patients with active nephrotic syndrome and a partial remission of the nephrotic syndrome; further, a lower AQP2 excretion correlated with the severity of the nephrotic syndrome. Therefore, our data suggest that patients with the nephrotic syndrome develop the vasopressin escape that was previously shown in animal studies.6
The results of our study contradict the data of Rocchetti et al, who reported a rise in the renal excretion of AQP2 in untreated patients with immunoglobulin A (IgA) nephropathy.11 Furthermore, patients with a greater deterioration of kidney function, higher proteinuria, and arterial hypertension displayed even higher urinary AQP2 excretion; moreover, a decrease of daily proteinuria following treatment was associated with a fall of the urinary AQP2 level. The authors suggested that an increase in the urinary excretion of AQP2 may reflect enhanced intrarenal AVP and renin–angiotensin system (RAS) activity, as well as the severity of renal damage. Of note, patients with the nephrotic syndrome and edema were excluded from the study by Rocchetti et al, whereas we specifically aimed to investigate the AVP–AQP2 axis in the nephrotic syndrome.
In a histological study, Bedford et al have recently demonstrated, by means of immunohistochemistry and real-time RT-PCR, a reduction in AQP-2 expression in renal biopsy samples in patients with different types of glomerulonephritis and other chronic renal diseases, including IgA nephropathy.12 AQP-2 immunostaining was reduced in the lesions of interstitial fibrosis. In our study, interstitial fibrosis could account for a reduction in the urinary AQP2 excretion in patients with minimal proteinuria who had a relatively long-term history of kidney disease; however, it could not explain more than a five-fold further decrease in the urinary AQP2 level in patients with a first episode of the nephrotic syndrome or a short history of renal disease.
AVP, with a cAMP-dependent nature of its insertion into collecting ducts, or other ligands acting at V2 receptors are not the only factors regulating AQP2 expression, which can be altered in a manner that is independent of the activity of vasopressin although the signaling transduction pathways involved in the regulation of AQP2 during vasopressin escape are, at present, unknown.1 Acute ischemic kidney injury has been shown to decrease collecting-duct AQP2 protein expression in rats.13 Reduced AQP2 expression and a vasopressin-resistant downregulation of AQP2 have been found in rats with chronic renal failure induced by a 5/6 nephrectomy.14 In our study, transient kidney dysfunction in patients with active nephrotic syndrome was not associated with a further reduction in AQP2 excretion, and there was no correlation between urinary AQP2 levels and serum creatinine or cystatin C concentrations. Therefore, active inflammation in the kidney that accounted for a rise in serum creatinine levels in patients with transient kidney dysfunction had no significant impact on the urinary AQP2 excretion.
The results of experimental studies suggest a cross talk between AVP and angiotensin II in the kidneys. Urinary AQP2 levels are increased in patients with congestive heart failure as a result of inappropriate secretion of AVP accompanied by the activation of the RAS.5 Wang et al showed the upregulation of AQP2 expression in the mouse cortical collecting-duct cell line under angiotensin II exposure.15 The combination of AVP and angiotensin II demonstrated the most profound increase in AQP2 expression. These effects were reversible with specific AT1-receptor antagonists. In a recent study, hypertensive rats treated with the ACE inhibitor imidapril exhibited a reduced expression of AQP2 mRNA and AQP2 protein in renal tissue.16 RAS inhibitors are frequently used in patients with kidney disorders as nephroprotective agents. More than half of our patients were treated with AT1-receptor antagonists or ACE inhibitors at the time of testing. Therefore, RAS inhibition might have contributed to reduced AQP2 excretion, both in patients with nephrotic or sub-nephrotic range minimal proteinuria. However, it seems unlikely that it can account for a marked reduction in the urinary AQP2 levels in patients with the nephrotic syndrome; furthermore, it cannot explain a difference between urinary AQP2 excretion in the groups of patients with and without nephrotic/sub-nephrotic range proteinuria. Further investigations seem to be warranted.
In our study, there was a negative correlation between the urinary AQP2 levels and daily protein loss with urine. Proteinuria is a well-known mediator of tubulointerstitial injury.17 Urinary proteins themselves may elicit proinflammatory and profibrotic effects, including induction of tubular chemokine expression, cytokines, monocyte chemotactic proteins, different growth factors, and complement activation.18,19 It is tempting to speculate that these factors may be directly or indirectly involved in the modulation of AQP2 expression in the collecting ducts. Recently, it was shown that protein-rich urine from nephrotic rats and from patients with nephrotic syndrome activates the epithelial sodium channel (ENaC) in cultured M-1 mouse collecting-duct cells and in Xenopus laevis oocytes heterologously expressing ENaC.20 The activation of ENaC involves the conversion of aberrantly filtered plasminogen to plasmin as well as the cleavage and release of an inhibitory peptide from the ENaC gamma subunit ectodomain. These data suggest that a defective glomerular filtration barrier allows the passage of proteolytic enzymes and, probably, other substances that may modulate expression of ion and water channels in the kidney tissue.
Our study has several limitations. First, the total number of patients was relatively small. Nevertheless, our data (unlike those of Rocchetti et al) were consistent with the previous studies in rodents with experimental nephrotic syndrome. Second, renal biopsy was not available in a proportion of patients, and we were not able to correlate urinary AQP2 excretion with a degree of interstitial fibrosis. Third, we did not measure AQP2 expression in the kidney tissue. Although urine AQP2 measurement was shown as a marker of vasopressin action in the collective ducts by Elliot et al in 1996,21 it is not clear whether reduced AQP2 loss with urine reflects the increased tissue degradation of AQP2 in patients with the nephrotic syndrome. Fourth, besides RAS inhibitors, approximately 30% of patients were treated with glucocorticoids with or without immunosuppressive agents and loop diuretics at the time of testing. Medications can have different impacts on AQP2 expression in the kidney tissue, but it seems unlikely that immunosuppressive or diuretic treatment accounted for the results of our study. In the rat kidney, cyclophosphamide induced a vasopressin-independent activation of AQP222 whereas, in healthy subjects, furosemide treatment upregulated the urinary AQP2 excretion and plasma AVP concentrations – probably, as a compensatory mechanism to prevent an excessive loss of sodium and water.23 Rocchetti et al suggested that steroids can decrease AQP2 expression through both AVP-dependent and AVP-independent mechanisms.11 In contrast, experimental studies showed that glucocorticoids promote AQP2 expression, mainly via inhibition of AQP2 protein degradation.24 Finally, we did not conduct water-loading tests to document the vasopressin escape phenomenon because it is contraindicated in patients with severe nephrotic syndrome.
Conclusion
Our clinical study was the first to demonstrate low AQP2 excretion in the nephrotic syndrome that may indicate an escape from the vasopressin regulating effect. This compensatory phenomenon that limits AVP-induced water retention was described in animals with experimental nephrotic syndrome, but was never studied in humans with the nephrotic syndrome. What is the clinical significance of our data? Reduced urinary AQP2 excretion may predict a poor response to novel diuretics that block V2 receptors in patients with severe nephrotic syndrome who are refractory to conventional therapy, although empiric treatment is probably an easier way to test their activity. A persistent reduction in the loss of AQP2 with urine may be a marker of interstitial fibrosis, but it seems unlikely that this biomarker can replace renal biopsy or the more readily available laboratory signs of irreversible kidney damage, such as a sustained increase in serum creatinine. Our findings shed more light on the pathophysiology of the nephrotic syndrome and the water-retention states in general.
Disclosure
The authors report no conflicts of interest in this work. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
Nielsen S, Frøkiaer J, Marples D, Kwon TH, Agre P, Knepper MA. Aquaporins in the kidney: from molecules to medicine. Physiol Rev. 2002;82(1):205–244. | ||
Agre P, Preston GM, Smith BL, et al. Aquaporin CHIP: the archetypal molecular water channel. Am J Physiol. 1993;265(4 Pt 2):463–476. | ||
Kwon TH, Frøkiær J, Nielsen S. Regulation of aquaporin-2 in the kidney: A molecular mechanism of body-water homeostasis. Kidney Res Clin Pract. 2013;32(3):96–102. | ||
Schrier RW. Vasopressin and aquaporin 2 in clinical disorders of water homeostasis. Semin Nephrol. 2008;28(3):289–296. | ||
Imamura T, Kinugawa K, Fujino T, et al. Increased urine aquaporin-2 relative to plasma arginine vasopressin is a novel marker of response to tolvaptan in patients with decompensated heart failure. Circ J. 2014;78(9):2240–2249. | ||
Apostol E, Ecelbarger CA, Terris J, Bradford AD, Andrews P, Knepper MA. Reduced renal medullary water channel expression in puromycin aminonucleoside – induced nephrotic syndrome. J Am Soc Nephrol. 1997;8(1):15–24. | ||
Fernández-Llama P, Andrews P, Ecelbarger CA, Nielsen S, Knepper M. Concentrating defect in experimental nephrotic syndrone: altered expression of aquaporins and thick ascending limb Na+ transporters. Kidney Int. 1998;54(1):170–179. | ||
Ecelbarger CA, Chou CL, Lee AJ, Digiovanni SR, Verbalis JG, Knepper MA. Escape from vasopressin-induced antidiuresis: role of vasopressin resistance of the collecting duct. Am J Physiol. 1998;274(6 Pt 2):1161–1166. | ||
Lewandowski KC, Brabant G. Potential Clinical Utility of Copeptin (C-terminal provasopressin) measurements in clinical medicine. Exp Clin Endocrinol Diabetes. 2016;124(3):173–177. | ||
Funayama H, Nakamura T, Saito T, et al. Urinary excretion of aquaporin-2 water channel exaggerated dependent upon vasopressin in congestive heart failure. Kidney Int. 2004;66(4):1387–1392. | ||
Rocchetti MT, Tamma G, Lasorsa D, et al. Altered urinary excretion of aquaporin 2 in IgA nephropathy. Eur J Endocrinol. 2011;165(4):657–664. | ||
Bedford JJ, Leader JP, Walker RJ. Aquaporin expression in normal human kidney and in renal disease. J Am Soc Nephrol. 2003;14(10):2581–2587. | ||
Kwon TH, Frøkiaer J, Fernández-Llama P, Knepper MA, Nielsen S. Reduced abundance of aquaporins in rats with bilateral ischemia-induced acute renal failure: prevention by alpha-MSH. Am J Physiol. 1999;277(3 Pt 2):413–427. | ||
Kwon TH, Frøkiaer J, Knepper MA, Nielsen S. Reduced AQP1, -2, and -3 levels in kidneys of rats with CRF induced by surgical reduction in renal mass. Am J Physiol. 1998;275(5 Pt 2):724–741. | ||
Wang W, Li C, Summer S, Falk S, Schrier RW. Interaction between vasopressin and angiotensin II in vivo and in vitro: effect on aquaporins and urine concentration. Am J Physiol Renal Physiol. 2010;299(3):F577–F584. | ||
Zhao W, Xu AG, Wu J, et al. Effects of imidapril treatment on aquaporin-2 expression in the kidneys and excretion in the urine of hypertensive rats. Exp Ther Med. 2013;5(5):1327–1331. | ||
Abbate M, Remuzzi G. Proteinuria as a mediator of tubulointerstitial injury. Kidney Blood Press Res. 1999;22(1–2):37–46. | ||
Gorriz JL, Martinez-Castelao A. Proteinuria: detection and role in native renal disease progression. Transplant Rev. 2012;26(1):3–13. | ||
Bobkova IN, Chebotareva NV, Kozlovskaia LV, Varshavskiĭ VA, Golitsyna EP. Urine excretion of a monocytic chemotaxic protein-1 and a transforming growth factor beta1 as an indicator of chronic glomerulonephritis progression. Ter Arkh. 2006;78(5):9–14. | ||
Svenningsen P, Bistrup C, Friis UG, et al. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol. 2009;20(2):299–310. | ||
Elliot S, Goldsmith P, Knepper M, Haughey M, Olson B. Urinary excretion of aquaporin-2 in humans: a potential marker of collecting duct responsiveness to vasopressin. J Am Soc Nephrol. 1996;7(3):403–409. | ||
Kim S, Choi HJ, Jo CH, Park JS, Kwon TH, Kim GH. Cyclophosphamide-induced vasopressin-independent activation of aquaporin-2 in the rat kidney. Am J Physiol Renal Physiol. 2015;309(5):F474–F483. | ||
Starklint J, Bech JN, Pedersen EB. Urinary excretion of aquaporin-2 after furosemide and felodipine in healthy humans. Scand J Clin Lab Invest. 2005;65(3):249–261. | ||
Chen M, Cai H, Klein JD, Laur O, Chen G. Dexamethasone increases aquaporin-2 protein expression in ex vivo inner medullary collecting duct suspensions. Front Physiol. 2015;6:310. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.