")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Long-term safety and efficacy of tobramycin in the management of cystic fibrosis
Authors Vázquez-Espinosa E, Giron RM, Gómez-Punter RM, García-Castillo E , Valenzuela C, Cisneros C, Zamora E, García-Pérez FJ, Ancochea J
Received 1 October 2014
Accepted for publication 8 January 2015
Published 12 March 2015 Volume 2015:11 Pages 407—415
DOI https://doi.org/10.2147/TCRM.S75208
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Emma Vázquez-Espinosa, Rosa María Girón, Rosa Mar Gómez-Punter, Elena García-Castillo, Claudia Valenzuela, Carolina Cisneros, Enrique Zamora, F Javier García-Pérez, Julio Ancochea
Pulmonology Department, La Princesa Institute for Health Research, Hospital Universitario de La Princesa, Madrid, Spain
Abstract: Cystic fibrosis (CF) is a fatal inherited disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene whose mortality is conditioned by a progressive decline in lung function. Bacterial infections play a key role in this decline. Chronic bacterial infection in CF patients varies over time and the presence of Pseudomonas aeruginosa in sputum is a marker of poor prognosis. P. aeruginosa is eradicated from the airways using inhaled antibiotics administered in various formulations and devices. Antipseudomonal antibiotics have extended the survival of CF patients to 40 years. Tobramycin is a bactericidal aminoglycoside antibiotic with demonstrated activity against gram-negative microorganisms. Initially, the drug was administered as an inhaled parenteral solution. Subsequently, a specific tobramycin inhalation solution was developed. PulmoSphere™ technology enables dry tobramycin powder to be formulated for inhalation (tobramycin inhalation powder) using a small and portable capsule-based breath-activated device (T-326). Chronic colonization by P. aeruginosa is the main indication for aerosol antibiotic therapy. The American Cystic Fibrosis Foundation, European guidelines, and Spanish consensus guidelines provide different recommendations for eradication.
Keywords: cystic fibrosis, Pseudomonas aeruginosa, tobramycin, chronic infection, inhaled antibiotic
Introduction
Cystic fibrosis (CF) is a fatal hereditary disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene, resulting in alterations of electrolyte transport across the airway epithelium. The mortality of this multisystemic illness is conditioned by a progressive decline in lung function, in which bacterial infections play a key role.1 The life expectancy for children born and diagnosed with CF in 2010 was 37 years for females and 40 years for males.1 Factors such as improved treatments and prevention or control of pulmonary exacerbations have contributed to the extended survival of CF patients.1
Chronic bacterial infection in CF patients varies over time. Patients are initially infected with Staphylococcus aureus and Haemophilus influenza, and then by Pseudomonas aeruginosa. The most frequent antibiotic-resistant pathogens include P. aeruginosa, Burkholderia cepacia, methicillin-resistant S. aureus, Stenotrophomonas maltophilia, and Achromobacter xylosoxidans.2 Chronic P. aeruginosa infection usually occurs during adolescence, when most patients (60%–80%) are colonized.3,4
Currently, the role of microorganisms in CF is more complex due to new nonculture methods, and the preferred sample is expectorated or hypertonic saline-induced sputum; other samples are bronchoalveolar lavage fluid, as well as cough and oropharyngeal swabs.5 Environmental factors substantially influence the development of the CF pediatric microbiome.6
The morbidity and mortality of CF ultimately depend on the degree of deterioration in lung function. CF patients produce abnormally thick lung secretions that cause obstruction of the airway, thus facilitating infection, inflammation, and bronchiectasis. Lung function is impaired, and the patient dies of progressive respiratory failure.7–11 The presence of P. aeruginosa in sputum is a marker of poor prognosis.7 Antipseudomonal antibiotics have extended survival until 40 years.8,11 Aerosolized antibiotics reach higher concentrations in the lower airways, and the associated minimal systemic exposure means that toxicity is low. P. aeruginosa is eradicated from the airways using inhaled antibiotics in many formulations and devices. Currently commercialized inhaled antibiotics are shown in Table 1.
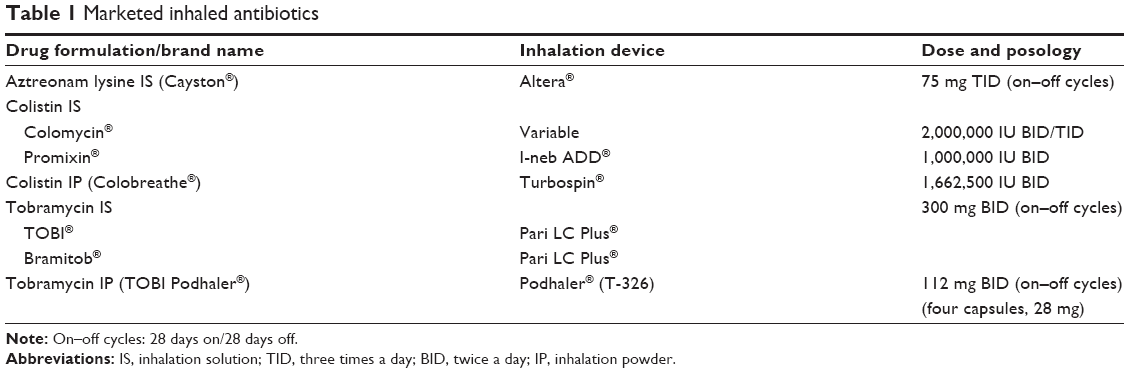 | Table 1 Marketed inhaled antibiotics |
The antipseudomonal efficacy of tobramycin is well established.4 Tobramycin is a bactericidal aminoglycoside antibiotic with demonstrated activity against gram-negative microorganisms. It works by disrupting protein synthesis and irreversibly binding the 30S bacterial ribosome, thus leading to alterations in cell membrane permeability and, eventually, cell death.
Parenteral tobramycin preparations administered via aerosol were first described in 1983.12 Tobramycin was the first commercial aminoglycoside designed for aerosol delivery. Aerosolized tobramycin is effective and improves delivery to the site of infection, leading to sputum drug levels that are >1,000-fold higher than in serum.4,12,13
Sputum can inhibit the biological activity of inhaled aminoglycosides. Constituents such as mucin, glycoprotein, and free eukaryotic and bacterial DNA bind aminoglycosides, owing to their highly cationic state and chemical properties such as pH and tonicity.14 It is therefore necessary to achieve a sputum concentration that is tenfold higher than the minimal inhibitory concentration (MIC) after administration of inhaled tobramycin.4 A controlled study reported that after the third cycle of active treatment with tobramycin at levels ≥15-fold higher than the MIC for P. aeruginosa, the microorganism was suppressed in ≥89% of patients.15
P. aeruginosa is able to develop several aminoglycoside resistance mechanisms, as follows: enzyme modification; low outer membrane permeability, which is often the most common mechanism in CF patients; active efflux MexXY proteins operating simultaneously with OprM and with other outer membrane proteins (OmpB, OmpG, OmpI); target modification (the first 16S rRNA methylase, RmtA, was reported in an aminoglycoside-resistant P. aeruginosa clinical isolate from Japan in 2003); nonenzyme-based mechanisms; and the accumulation of mutants, which leads to a gradual increase in resistance to aminoglycosides, as seen in CF patients.16–19
Bacteria growing on biofilm are much more resistant to antibiotics than planktonic cells; the MIC and minimal bactericidal concentration can be 100–1,000-fold greater for old biofilm, whereas young biofilm is less resistant.
Chronic P. aeruginosa lung infection in CF is characterized by growth of the mucoid phenotype on biofilm; this phenotype produces abundant alginate in vitro and in vivo. Microcolonies surrounded by alginate are seen in both sputum and postmortem samples. The bacteria remain inside the mucus and grow under anaerobic conditions using nitrate as an electron acceptor. The bacteria are not located on the epithelial cells, although they do induce bronchiolitis without spreading to the blood or to other organs. Patients develop high levels of antibodies against alginate and other P. aeruginosa antigens. P. aeruginosa has been shown to develop resistance to patients’ defense mechanisms and to antibiotic treatment.20
Oxygen radicals produced by polymorphonuclear leukocytes as part of the inflammatory response induce mutations leading to alginate production, which is characteristic of P. aeruginosa biofilm infection in CF.20 Quorum sensing is also involved in mature biofilm formation in vitro and in vivo. The biofilm of P. aeruginosa and alginate protects against antibiotics and the immune response. Therefore, it is necessary to administer prophylaxis and early aggressive therapy with oral ciprofloxacin, inhaled colistin, and inhaled tobramycin, colistin, aztreonam, and gentamicin before the biofilm is fully established. The addition of macrolides or N-acetylcysteine slows alginate production.
Administration of inhaled tobramycin
The first inhaled tobramycin solutions were parenteral tobramycin. Subsequently, a specific tobramycin inhalation solution (TIS) was developed, although many clinical trials were needed to assess doses, dosing intervals, and drug delivery devices.14,21,22
In a study by Ramsey et al23 (CF patients chronically infected with P. aeruginosa receiving TIS), tolerance was good, lung function improved, the density of P. aeruginosa in sputum decreased, and the risk of hospitalization was lower.2,24 Each cycle included a 4-week “on” period followed by a 4-week “off” period. Murphy et al25 did not find a significant improvement in 181 chronically infected patients after seven cycles of TIS 300 mg/5 mL (Table 2).
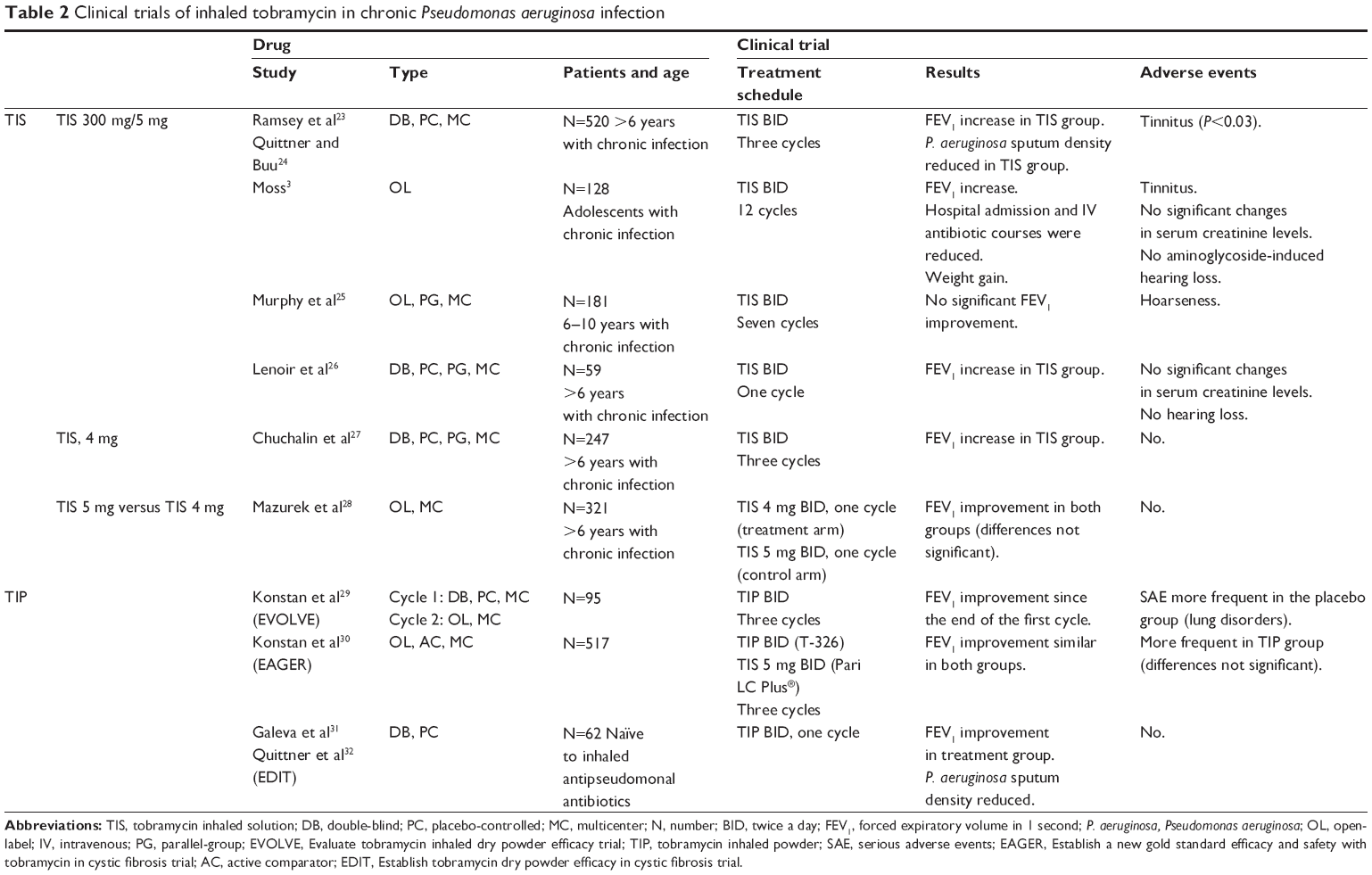 | Table 2 Clinical trials of inhaled tobramycin in chronic Pseudomonas aeruginosa infection |
The long-term effect of suppression of P. aeruginosa with inhaled TIS 300 mg/5 mL was assessed by Moss3 in 128 adolescents. After 96 weeks of treatment, lung function, weight gain, and body mass index had improved, and the number of hospital admissions and intravenous antibiotic courses per patient/year had fallen. The density of P. aeruginosa colony-forming units had also decreased, and the reduction was significantly correlated with improved lung function. No statistically significant changes in serum creatinine levels were found, and aminoglycoside-induced hearing loss was not recorded.
Two clinical trials performed with TIS 300 mg/4 mL in chronically infected CF patients also revealed that treatment produced a significant improvement in forced expiratory volume in 1 second (FEV1).26,27 Mazurek et al28,33 compared TIS 300 mg/5 mL with TIS 300 mg/4 mL in 321 patients and found improved FEV1 in both arms.
The recently developed PulmoSphere™ particles enable tobramycin powder to be formulated for inhalation (tobramycin inhalation powder [TIP]) using a small portable capsule-based breath-activated device (T-326). Compared with TIS, TIP improves the efficiency of drug deposition in the airway. The dry powder form of PulmoSphere™ is composed of small particles with a uniform size.4,34 The spheroid and porous structure of the particles diminishes the contact area between them, thus enabling efficient drug delivery.
PulmoSphere™ is advantageous for younger patients and those with compromised lung function. It also obviates the need for lactose carrier particles,34 thus facilitating a higher dose of active drug per capsule. The particle size (diameter <4 μm) helps to ensure that the aerosol powder is not deposited in the oropharynx and can reach the small airways. Furthermore, this system requires little inspiratory flow and a shorter administration time.30,35 The relative distribution between the large and small airways is similar with TIP and TIS. The results of pharmacokinetic studies show that intersubject variability was lower with TIP than with TIS.36
After a single dose of TIP or TIS, the drug is absorbed quickly through the lungs. Systemic exposure is similar in both formulations,35 and peak concentrations are reached quickly. After two daily doses of TIP 112 mg on a 4-week cycle, the maximum serum concentration was 1.99 mg/L 1 hour after administration. Evaluation of repeated doses of TIS revealed no evidence of accumulated tobramycin in respiratory secretions.4
No marked pharmacokinetic variations have been observed with respect to body mass index, lung function, sex, and age.15 The drug is cleared in urine after a single dose, and the half-life is about 2 hours in sputum and 3 hours in serum for both TIP and TIS. Although the efficacy of inhaled tobramycin (TIS and TIP) has been demonstrated, TIP was developed to improve some characteristics of TIS.
Three studies have been developed to assess the efficacy of TIP in chronically infected CF patients. EVOLVE29 (Evaluate tobramycin inhaled dry powder efficacy) and EDIT31,32 (Establish tobramycin dry powder efficacy in cystic fibrosis) compared TIP with placebo. EAGER30 (Establish a new gold standard efficacy and safety with tobramycin in cystic fibrosis) compared TIP with TIS.
The TIP dose used in all studies was 112 mg twice daily (4×28 mg capsules) in cycles of 28 days “on” followed by 28 days “off”. In the EDIT study,7 patients were naïve to inhaled antipseudomonal antibiotics.
In all three trials, efficacy was assessed based on relative changes in FEV1 from baseline in each group. In the EAGER study,14 patient satisfaction with each of the inhalation devices was evaluated using a version of the Treatment Satisfaction Questionnaire for Medication; patients treated with TIP were significantly more satisfied.
The EVOLVE study reported that the density of Pseudomonas in sputum was reduced more with TIP than placebo for both mucoid and nonmucoid forms.29 Clearance of P. aeruginosa cultures was significantly more frequent with TIP than placebo. In the EAGER study, the sputum density of P. aeruginosa followed through three cycles of use was similar to that recorded in the original TIS trial.30 Data on increased resistance to tobramycin were reported in both EVOLVE and EAGER.30,31 In EVOLVE, after three consecutive treatment cycles, an increasing trend of resistance to tobramycin compared with baseline was observed in the group of patients treated with TIP. However, this trend was also found in the placebo group. In EAGER, no differences were found in the MIC for P. aeruginosa when patients under treatment with TIS were compared with patients under treatment with TIP. Most of the adverse events observed are transient and mild to moderate. Cough is the most common adverse event in patients treated with TIP, although it seems to disappear with successive cycles.
The authors of the EAGER study30 reported cough in 48.4% of TIP-treated patients compared with 31.3% in the TIS arm. Dysphonia and dysgeusia were also more frequent in patients treated with TIP. The most frequent serious adverse events were pulmonary exacerbations and bronchospasm, although differences were not statistically significant. The differences between the TIS and TIP studies are shown in Table 2.
The EPIC (Early Pseudomonas Infection Control) and ELITE (EarLy Inhaled Tobramycin for Eradication) trials assessed patients with new-onset P. aeruginosa infection.37 The EPIC study37 assessed the efficacy and safety of inhaled and oral antipseudomonal antibiotics following the initial isolation of P. aeruginosa and compared tobramycin plus placebo with tobramycin plus ciprofloxacin. No differences were found between the treatment arms. The ELITE study38 is a multicenter European study that assessed the duration of P. aeruginosa eradication following a 28-day or 56-day course of inhaled tobramycin. TIS was administered at 300 mg/5 mL twice a day. Preliminary results from this study showed a high rate of successful and durable eradication with no differences in time to recurrence between the treatment arms. The rate of eradication was more than 90% at the end of the clinical trial and after 4 weeks of therapy2,39 (Table 3). In Figure 1, we show a flowchart with the progress and outcomes of all these studies.
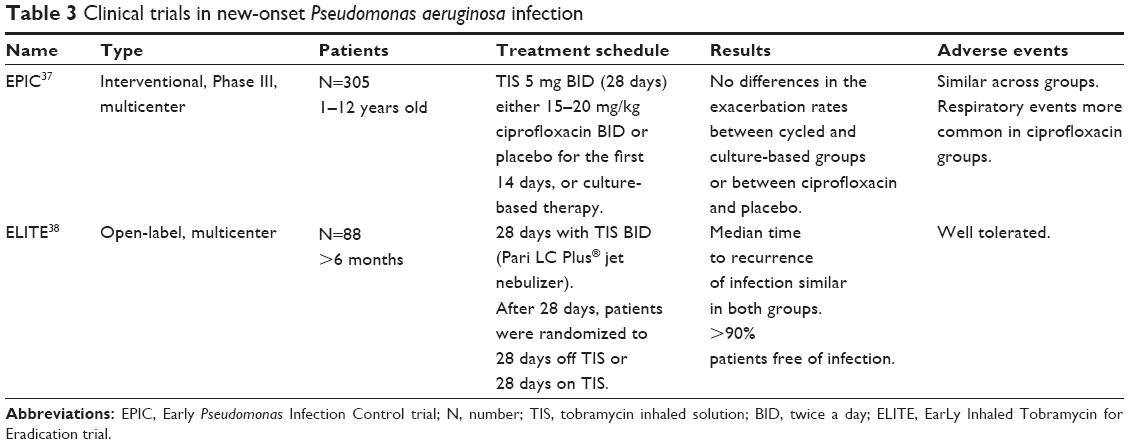 | Table 3 Clinical trials in new-onset Pseudomonas aeruginosa infection |
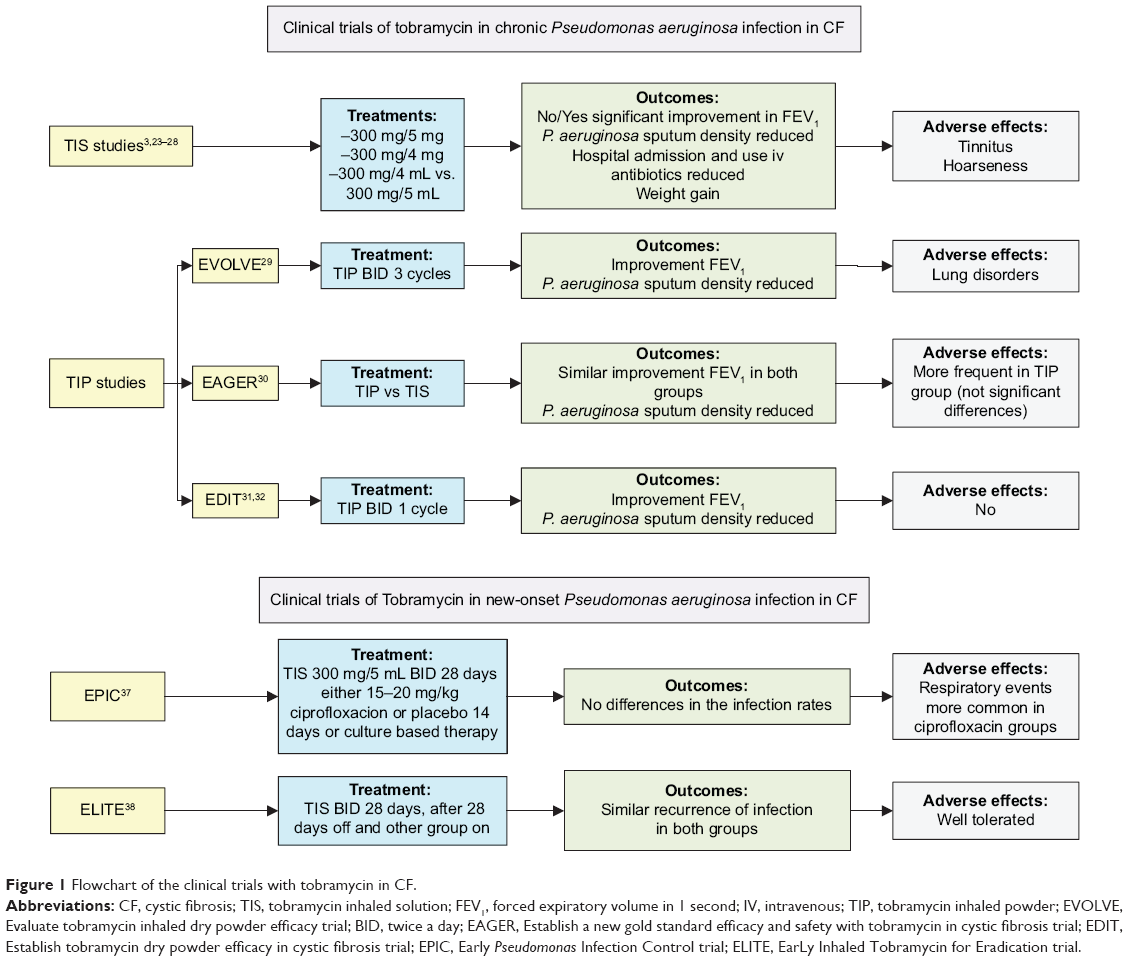 | Figure 1 Flowchart of the clinical trials with tobramycin in CF. |
The most widely tested inhaled antibiotic is tobramycin. Since it has demonstrated efficacy, it would be unethical to compare it with placebo. Studies comparing tobramycin with aztreonam showed that improvement was more frequent in patients treated with a new antibiotic than those receiving tobramycin during the first treatment cycles. Similarly, aztreonam was shown to be effective and safe in placebo-controlled studies for the treatment of chronic Pseudomonas infection. Aztreonam was superior to TIS in the comparative study of Assael et al.40
Colistin has been widely used in Europe, although it is not approved in the United States. A comparative study between inhaled colistin and tobramycin showed that colistin dry powder for inhalation was not inferior to tobramycin and that the safety and tolerability profile were similar to that of tobramycin.7
Very recent Phase I and II studies show that levofloxacin achieves fast and good local concentrations with little systemic distribution.41 They also showed an improvement in lung function that reduces the need for further cycles. Phase III studies have shown a good efficacy and safety profile for levofloxacin.41
Liposomal formulations of amikacin for inhalation have also proven useful against chronic P. aeruginosa infection. The drug penetrates lung secretions and biofilms. Phase I and II studies have shown a significant improvement in lung function after 12–14 days of treatment, and a Phase III study comparing amikacin with tobramycin showed a significant increase in FEV1 after three cycles.42 Amikacin is also a good option for lung infections by other microorganisms such as nontuberculous mycobacteria.42
Indication for aerosolized antibiotics
Life expectancy in patients with CF has increased in recent decades as a result of advances in clinical management. Early detection and aggressive treatment of respiratory tract infections are essential.4 Antibiotic prophylaxis is used to prevent infection and prevents decline in lung function, which remains the main cause of death in patients with CF.
Since P. aeruginosa infection is associated with increased morbidity and mortality, prophylaxis against P. aeruginosa can be planned. However, some studies report administration of tobramycin to patients with negative cultures.2,43 Given the lack of data on prophylaxis against Pseudomonas and the potential risk of drug toxicity and development of resistance, prophylaxis is not recommended.
Before treating P. aeruginosa, it is important to detect early colonization of the airway. The first isolate is often a nonmucoid phenotype that is sensitive to most antipseudomonal treatments. Furthermore, the patient does not usually have a large colony count. Consequently, eradication should be attempted before the immune response is activated and infection becomes chronic. Although several approaches for eradication can be taken, inhaled colistin and tobramycin (TIS or TIP) are now the most common.44
Current guidelines recommend combining different administration modalities (inhaled, oral, and intravenous). The 2009 Spanish consensus recommends oral ciprofloxacin (15–20 mg/kg/12 hours ×21 days) and inhaled tobramycin or colistin, even when no signs of infection are present.45 If positive cultures persist after 1 month, inhalation therapy can be maintained and a new cycle of oral ciprofloxacin can be started (21 days or 30 days). If the cultures do not become negative after 1 month or 2 months, a new 14-day cycle of intravenous antibiotic should be started. At this point, if positive cultures persist, the patient should be considered chronically infected. Therapy must be discontinued in patients whose cultures are negative after 6–12 months of inhaled treatment.45
The latest recommendations in the new version of the Spanish consensus advise early treatment of P. aeruginosa infection with inhaled antibiotics (eg, colistimethate sodium [3–6 months], or TIS [28 days], or inhaled aztreonam [1–3 cycles] with[out] oral ciprofloxacin [2–3 weeks]). If positive cultures persist, the same regimen should be administered, or treatment should be switched to a new regimen (ciprofloxacin combined with an inhaled antibiotic not used during the first cycle).46
Chronic infection is defined as continuously positive cultures or ≥3 positive cultures 1 month apart over 6 months in a clinically stable patient.47 The prevalence of chronic colonization by P. aeruginosa seems to have decreased in the last decade, perhaps as a result of early and aggressive eradication strategies. In 2007, over 50% of patients with CF had positive cultures for this organism, although the percentage rises to approximately 80% in patients aged >25 years.47,48
The 28-day on/28-day off cycles were established to avoid increased bacterial resistance, although lung function declines in many cases at the end of the off cycle.20 Therefore, it may be beneficial to use continuous alternative therapy with two antibiotics.7
Chronic colonization by P. aeruginosa is the main indication for aerosolized antibiotic therapy. In chronic colonization, P. aeruginosa is usually the mucoid phenotype associated with alginate production and biofilm growth, which lead to further decline in lung function.20 Therefore, antibiotic therapy is administered to reduce bacterial load by improving lung function, thus reducing morbidity and mortality.
The American Cystic Fibrosis Foundation recommends long-term aerosolized tobramycin in patients aged ≥6 years with moderate to severe disease (grade A recommendation) to improve lung function and quality of life and reduce exacerbations. For patients with mild disease, inhaled tobramycin is recommended to reduce exacerbations (grade B recommendation). These guidelines do not recommend this approach in patients younger than 6 years. Inhaled aztreonam is recommended for patients ≥6 years with moderate to severe disease (grade A recommendation) or mild disease.49
In contrast, European guidelines indicate inhaled antipseudomonal antibiotics for all patients chronically colonized with mucoid P. aeruginosa, regardless of age or lung function.2 Inhaled colistin is widely used in Europe.
Conclusion
- Inhaled tobramycin treatment seems to be safe and effective for the long-term management of primary and chronic Pseudomonas infection in patients chronically infected with CF. The clinical significance of the slight increase in MIC has not been established.
- If inhaled tobramycin is administered as soon as the primary infection is detected, lung function improves and morbidity and mortality decrease.
- Both formulations of inhaled tobramycin were compared with each other and placebo. Trial results show that the antipseudomonal efficacy of intermittent twice-daily TIP 112 mg was greater than that of placebo and noninferior to intermittent twice-daily nebulized TIS 300 mg/5 mL with regard to lung function and sputum density of P. aeruginosa.
- Patients using TIP were more satisfied with their treatment, and most adverse events were transient and mild to moderate in intensity. Cough was the most common adverse event and more frequent in patients receiving TIP.
Disclosure
The authors report no conflicts of interest in this work.
References
MacKenzie T, Gifford AH, Sabadosa KA, et al. Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the cystic fibrosis foundation patient registry. Ann Intern Med. 2014;161(4):233–241. | ||
Girón Moreno RM, Salcedo Posadas A, Mar Gómez-Punter R. [Inhaled antibiotic therapy in cystic fibrosis]. Arch Bronchoneumol. 2011;47 Suppl 6:14–18. Spanish. | ||
Moss RB. Long-term benefits of inhaled tobramycin in adolescent patients with cystic fibrosis. Chest. 2002;121(1):55–63. | ||
McKeage K. Tobramycin inhalation powder: a review of its use in the treatment of chronic Pseudomonas aeruginosa infection in patients with cystic fibrosis. Drugs. 2013;73(16):1815–1827. | ||
Burns JL, Rolain JM. Culture-based diagnostic microbiology in cystic fibrosis: can we simplify the complexity? J Cyst Fibros. 2014;13(1):1–9. | ||
Hampton TH, Green DM, Cutting GR, et al. The microbiome in pediatric cystic fibrosis patients: the role of shared environment suggests a window of intervention. Microbiome. 2014;2:14. | ||
Máiz L, Girón RM, Olveira C, et al. Inhaled antibiotics for the treatment of chronic bronchopulmonary Pseudomonas aeruginosa infection in cystic fibrosis: systematic review of randomised controlled trials. Expert Opin Pharmacother. 2013;14(9):1135–1149. | ||
Flume PA, O’Sullivan BP, Robinson KA, et al; Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176(10):957–969. | ||
Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34(2):91–100. | ||
Simmonds NJ, Macneill SJ, Cullinan P, Hodson ME. Cystic fibrosis and survival to 40 years: a case-control study. Eur Respir J. 2010;36(6):1277–1283. | ||
UK Cystic Fibrosis Trust Antibiotic Working Group. Antibiotic Treatment for Cystic Fibrosis. 3rd ed. Kent, UK: UK Cystic Fibrosis Trust; 2009. | ||
Stephens D, Garey N, Isles A, Levison H, Gold R. Efficacy of inhaled tobramycin in the treatment of pulmonary exacerbations in children with cystic fibrosis. Pediatr Infect Dis. 1983;2(3):209–211. | ||
Ratjen F, Brockhaus F, Angyalosi G. Aminoglycoside therapy against Pseudomonas aeruginosa in cystic fibrosis: a review. J Cyst Fibros. 2009;8(6):361–369. | ||
Lam J, Vaughan S, Parkins MD. Tobramycin inhalation powder (TIP): an efficient treatment strategy for the management of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Clin Med Insights Circ Respir Pulm Med. 2013;7:61–77. | ||
European Medicines Agency. Annex I: Summary of Product Characteristics. London, UK: European Medicines Agency; 2011. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002155/WC500110921.pdf. Accessed July 20, 2014. | ||
Poole K. Aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2005;49(2):479–487. | ||
Poole K. Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms. J Mol Microbiol Biotechnol. 2001;3(2):255–264. | ||
Wolter DJ, Smith-Moland E, Goering RV, Hanson ND, Lister PD. Multidrug resistance associated with mexXY expression in clinical isolates of Pseudomonas aeruginosa from a Texas hospital. Diagn Microbiol Infect Dis. 2004;50(1):43–50. | ||
Strateva T, Yordanov D. Pseudomonas aeruginosa – a phenomenon of bacterial resistance. J Med Microbiol. 2009;58(Pt 9):1133–1148. | ||
Høiby N. Understanding bacterial biofilms in patients with cystic fibrosis: current and innovative approaches to potential therapies. J Cyst Fibros. 2002;1(4):249–254. | ||
Smith AL, Ramsey BW, Hedges DL, et al. Safety of aerosol tobramycin administration for 3 months to patients with cystic fibrosis. Pediatr Pulmonol. 1989;7(4):265–271. | ||
Le Brun PP, de Boer AH, Gjaltema D, Hagedoorn P, Heijerman HG, Frijlink HW. Inhalation of tobramycin in cystic fibrosis. Part 1: the choice of a nebulizer. Int J Pharm. 1999;189(2):205–214. | ||
Ramsey BW, Pepe MS, Quan JM, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med. 1999;340(1):23–30. | ||
Quittner AL, Buu A. Effects of tobramycin solution for inhalation on global ratings of quality of life in patients with cystic fibrosis and Pseudomonas aeruginosa infection. Pediatr Pulmonol. 2002;33(4):269–276. | ||
Murphy TD, Anbar RD, Lester LA, et al. Treatment with tobramycin solution for inhalation reduces hospitalizations in young CF subjects with mild lung disease. Pediatr Pulmonol. 2004;38(4):314–320. | ||
Lenoir G, Antypkin YG, Miano A, et al. Efficacy, safety, and local pharmacokinetics of highly concentrated nebulized tobramycin in patients with cystic fibrosis colonized with Pseudomonas aeruginosa. Paediatr Drugs. 2007;9 Suppl 1:11–20. | ||
Chuchalin A, Csiszér E, Gyurkovics K, et al. A formulation of aerosolized tobramycin (Bramitob) in the treatment of patients with cystic fibrosis and Pseudomonas aeruginosa infection: a double-blind, placebo-controlled, multicenter study. Paediatr Drugs. 2007;9 Suppl 1:21–31. | ||
Mazurek H, Lenoir G, Pelikan L, et al. Head-to-head comparison of two inhaled tobramycin solutions in cystic fibrosis (CF) patients with chronic Pseudomonas aeruginosa (Pa) infection [abstract]. J Cyst Fibros. 2011;10(Suppl 1):S28. | ||
Konstan MW, Geller DE, Minić P, Brockhaus F, Zhang J, Angyalosi G. Tobramycin inhalation powder for P. aeruginosa infection in cystic fibrosis: the EVOLVE trial. Pediatr Pulmonol. 2011;46(3):230–238. | ||
Konstan MW, Flume PA, Kappler M, et al. Safety, efficacy and the convenience of tobramycin inhalation powder in cystic fibrosis patients: the EAGER trial. J Cyst Fibros. 2011;10(1):54–61. | ||
Galeva I, Konstan MW, Higgins M, et al. A challenging double-blind placebo-controlled study of tobramycin inhalation powder in cystic fibrosis: results of the EDIT trial [abstract]. J Cyst Fibros. 2012;11(Suppl 1):S12. | ||
Quittner AL, Higgins M, Angyalosi G, Brockhaus F, Thomas K, Zhang J. Trends in health-related quality of life (HRQoL) in cystic fibrosis with tobramycin inhalation powder: the EDIT trial [abstract]. J Cyst Fibros. 2012;11(Suppl 1):S73. | ||
Mazurek H, Chiron R, Pelikan L, et al. Comparison of two inhaled tobramycin solutions in cystic fibrosis patients with Pseudomonas aeruginosa infection: results in different age subgroups [abstract]. J Cyst Fibros. 2011;10(Suppl 1):S28. | ||
Geller DE, Weers J, Heuerding S. Development of an inhaled dry-powder formulation of tobramycin using PulmoSphere™ technology. J Aerosol Med Pulm Drug Deliv. 2011;24(4):175–182. | ||
Geller DE, Konstan MW, Smith J, Noonberg SB, Conrad C. Novel tobramycin inhalation powder in cystic fibrosis subjects: pharmacokinetics and safety. Pediatr Pulmonol. 2007;42(4):307–313. | ||
Newhouse MT, Hirst PH, Duddu SP, et al. Inhalation of a dry powder tobramycin PulmoSphere formulation in healthy volunteers. Chest. 2003;124(1):360–366. | ||
Treggiari MM, Retsch-Bogart G, Mayer-Hamblett N, et al; Early Pseudomonas Infection Control (EPIC) Investigators. Comparative efficacy and safety of 4 randomized regimens to treat early Pseudomonas aeruginosa infection in children with cystic fibrosis. Arch Pediatr Adolesc Med. 2011;165(9):847–856. | ||
Ratjen F, Munck A, Kho P. Short and long-term efficacy of inhaled tobramycin in early P. aeruginosa infection: the ELITE study. Pediatric Pulmonology. 2008;43 Suppl 31:319–320. | ||
Ratjen F, Munck A, Kho P, Angyalosi G; ELITE Study Group. Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis: the ELITE trial. Thorax. 2010;65(4):286–291. | ||
Assael BM, Pressler T, Bilton D, et al; AZLI Active Comparator Study Group. Inhaled aztreonam lysine vs inhaled tobramycin in cystic fibrosis: a comparative efficacy trial. J Cyst Fibros. 2013;12(2):130–140. | ||
Stockmann C, Sherwin CM, Ampofo K, Spigarelli MG. Development of levofloxacin inhalation solution to treat Pseudomonas aeruginosa in patients with cystic fibrosis. Ther Adv Respir Dis. 2014;8(1):13–21. | ||
Waters V, Ratjen F. Inhaled liposomal amikacin. Expert Rev Respir Med. 2014;8(4):401–409. | ||
Heinzl B, Eber E, Oberwaldner B, Haas G, Zach MS. Effects of inhaled gentamicin prophylaxis on acquisition of Pseudomonas aeruginosa in children with cystic fibrosis: a pilot study. Pediatr Pulmonol. 2002;33(1):32–37. | ||
Ratjen F, Döring G, Nikolaizik WH. Effect of inhaled tobramycin on early Pseudomonas aeruginosa conolisation in patients with cystic fibrosis. Lancet. 2001;358(9286):983–984. | ||
Barrio Gómez de Agüero MI, García Hernández G, Gartner S; Grupo de Trabajo de Fibrosis Quística. [Protocol for the diagnosis and follow up of patients with cystic fibrosis]. An Pediatr (Barc). 2009;71(3):250–264. Spanish. | ||
Cantón R, Maiz L, Escribano A, et al. Consenso español para la prevención y el tratamiento de la infección bronquial por Pseudomonas aeruginosa en el paciente con fibrosis quística. Arch Bonconeumol. In press 2015. Spanish. | ||
Razvi S, Quittell L, Sewall A, Quinton H, Marshall B, Saiman L. Respiratory microbiology of patients with cystic fibrosis in the United States, 1995 to 2005. Chest. 2009;136(6):1554–1560. | ||
Cystic Fibrosis Foundation. Patient Registry: Annual Data Report 2009. Bethesda, MD: Cystic Fibrosis Foundation; 2007. | ||
Mogayzel PJ, Naureckas ET, Robinson KA, et al; Pulmonary Clinical Practice Guidelines Committee. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2013;187(7):680–689. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.