")
Back to Journals » Cancer Management and Research » Volume 12
Long Noncoding RNA SNHG22 Induces Cell Migration, Invasion, and Angiogenesis of Gastric Cancer Cells via microRNA-361-3p/HMGA1/Wnt/β-Catenin Axis
Authors Cui X, Zhang H, Chen T, Yu W, Shen K
Received 15 September 2020
Accepted for publication 19 November 2020
Published 15 December 2020 Volume 2020:12 Pages 12867—12883
DOI https://doi.org/10.2147/CMAR.S281578
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eileen O'Reilly
Xiaofeng Cui, Huaiyu Zhang, Tong Chen, Wei Yu, Kexin Shen
Department of Gastrointestinal Colorectal and Anal Surgery, China-Japan Union Hospital of Jilin University, Changchun, Jilin 130033, People’s Republic of China
Correspondence: Wei Yu; Kexin Shen
Department of Gastrointestinal Colorectal and Anal Surgery, China-Japan Union Hospital of Jilin University, No. 126, Xiantai Street, Changchun, Jilin 130033, People’s Republic of China
Tel/Fax +86-0431-84996792
Email [email protected]; [email protected]
Background: The correlation between long non-coding RNAs (lncRNAs) and gastric cancer (GC) has been indicated. As a newly found lncRNA, small nucleolar RNA host gene 22 (SNHG22) functions as an oncogene in ovarian carcinoma and breast cancer. However, its action has not been explored in GC. Herein, the purpose of the current research was to examine the influence of SNHG22 on GC development.
Methods: RT-qPCR was used to identify SNHG22 and microRNA-361-3p (miR-361-3p) in GC tissues and cells. Functional assays were implemented to measure changes on biological activities of GC cells under different transfections. Besides, after human umbilical vein endothelial cells (HUVECs) were co-cultured with supernatant of transfected GC cells, angiogenesis was assessed by tube formation assay in vitro. HMGA1 and β-catenin expression were determined. Finally, mechanistic assays, including RNA pull-down assay and dual-luciferase reporter assay, were employed to assess relationships among SNHG22, miR-361-3p, and HMGA1.
Results: SNHG22 and HMGA1 were highly expressed but miR-361-3p was poorly expressed in GC tissues. Mechanistically, SNHG22 bound to miR-361-3p, and miR-361-3p targeted HMGA1 to disrupt the Wnt/β-catenin pathway. Following SNHG22 or HMGA1 silencing or miR-361-3p upregulation, we observed a decline of proliferation, migration, and invasion of GC cells and HUVEC angiogenesis but acceleration of GC cell apoptosis and cell cycle arrest.
Conclusion: Collectively, SNHG22 silencing possessed tumor-suppressing potentials in GC development via Wnt/β-catenin pathway by binding to miR-361-3p and downregulating HMGA1, highlighting a new promising road for GC treatment development.
Keywords: gastric cancer, long noncoding RNA SNHG22, microRNA-361-3p, HMGA1, Wnt/β-catenin pathway
Introduction
Gastric cancer (GC) ranks the fifth most prevalent cancer on a global scale with geographically varying incidence and prevalence, and 952 thousand new cases (7% of total cancer incidence rate) and 723 thousand deaths were estimated in 2012.1 Although the survival rate of GC patients has improved significantly in the past few decades, GC is often diagnosed at the late stage, and the prognosis is still unsatisfactory owing to the high recurrence rate.2 There are several risk factors for GC, like Helicobacter pylori infection, genetic susceptibility of the host, and other environmental factors.3 As a heterogeneous disease, GC is the result of the stepwise accumulation of numerous (epi)genetic alterations, triggering the imbalance of carcinogenic and anti-cancer pathways.4 These changes are essential to accelerate and maintain a series of pathways participating in tumor development, such as immune surveillance, cell-to-cell and cell-to-matrix interactions, cell cycle, metabolism, apoptosis, DNA repair, and angiogenesis.5 Interestingly, the crucial implication of various noncoding RNAs (ncRNAs), like microRNAs (miRs) and long noncoding RNAs (lncRNAs) has been documented in GC development.6
There are multiple reports about the oncogenic effect of various lncRNAs, including small nucleolar RNA host genes (SNHGs), like SHNG1, SHNG3, SHNG4, and SHNG15, on tumorigenesis and cancer progression.7–10 As a newly discovered lncRNA located on chromosome 18q21.1, SNHG22 has been detected to be overexpressed and to act as an oncogene in epithelial ovarian carcinoma through interacting with miR-2467.11 LncRNAs assume a sponge in the mediation of multiple miRNAs, and subsequently orchestrate miRNAs and mRNA function in various cancers.12 Moreover, the anti-oncogenic effects of miR-361-3p on cervical cancer have been documented by a prior study.13 Similarly, miR-361-3p was also reported to possess tumor-suppressing potentials in GC by targeting PDPK1.14 Furthermore, our bioinformatics prediction using Starbase showed that miR-361-3p targeted high mobility group A1 (HMGA1). It is well-known that HMGA1 is an oncofoetal gene to correlate to various malignant cancers.15 More importantly, HMGA1 could accelerate epithelial–mesenchymal transition (EMT) to facilitate the malignant progression of GC.16 HMGA1 was capable of activating Wnt/β-catenin pathway in circulating tumor cells from patients with gastrointestinal stromal tumor.17 A prior research provided evidence highlighting the tumor-promoting capacity of Wnt/β-catenin pathway in GC.18
These findings triggered a network of SNHG22/miR-361-3p/HMGA1/Wnt/β-catenin pathway in GC. Since the growth of newly formed blood vessels by the tumor (termed as angiogenesis) has been established as a vital mechanism for tumor cell survival, antiangiogenic treatment might be a novel target for the current cancer treatment regimens.19 Therefore, tissue and cell experiments were conducted in this study to investigate the influence of this network on cell proliferation, invasion, migration, apoptosis, cell cycle progression and angiogenesis in GC.
Materials and Methods
Clinical Sample Collection
Tumor and adjacent normal tissues were harvested from 60 GC patients who underwent surgery in China-Japan Union Hospital of Jilin University from 2012 to 2014 and preserved at −80ºC. These patients with complete medical history did not receive preoperative chemoradiotherapy, nor had other malignancies. Patients were followed-up every 6 months for 5 years after surgery. The experimental protocol was ratified by the Ethics Committee of China-Japan Union Hospital of Jilin University, all included patients provided signed informed consent and all procedures were aligned with the Declaration of Helsinki.
Cell Incubation and Transfection
Two human GC cell lines, SNU-1 (CRL-5971) and AGS (CRL-1739) were procured from American Type Culture Collection (Manassas, VA, USA), and another two human GC cell lines, KATO III (CL-0372) and MKN-45 (CL-0292) from Procell Life Science & Technology Co., Ltd. (Wuhan, China). Human normal gastric epithelial cell line, GES-1 (SCSP-308), was procured from CCTCC (Shanghai, China). Roswell Park Memorial Institute-1640 medium encompassing 10% fetal bovine serum (FBS, Wisent, Montreal, Canada) and 1% antibiotics (100 U/mL penicillin G and 100 mg/mL streptomycin) was adopted for incubation of all aforesaid cell lines at 37ºC with 5% CO2.
GenePharma (Shanghai, China) provided short hairpin RNA (sh)-SNHG22 1, 2, 3#, miR-361-3p mimic/inhibitor and their respective controls. The overexpression plasmid pcDNA-HMGA1 was designed and synthesized by GenePharma, and the empty PDNA3.1 vector served as the control. Then, cell transfection was conducted based on protocols of Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA).
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Through TRIzol reagent (Invitrogen), we conducted total RNA isolation from GC and adjacent normal tissues and cultured cells before synthesis of cDNA using PrimeScript RT kit (Takara, Shiga Prefecture, Japan). cDNA was mixed with TB Green Premix Ex Taq II (Takara) for RT-qPCR reaction. The relative expression was calculated by means of relative quantification (2−ΔΔCt method) and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH; for lncRNAs and mRNAs) and U6 (for miRs). The primers used are depicted in Table 1.
 |
Table 1 Primer Sequences for RT-qPCR |
Cell Counting Kit (CCK)-8 Assay
Cells were incubated in 96-well plates at 1 × 103 cells/well with 5% CO2. Cells were treated with 10 μL CCK-8 solution (Beyotime, Shanghai, China) at 0, 24, 48, 72 h, respectively, followed by another 2-h incubation. The growth rate of cells was evaluated by detecting the optical density at 450 nm using a SpectraMax M5 multimode microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA).
Colony Formation Assay
Cells were cultured in 6-well plates (1 × 103 cells/well) for 3 weeks, followed by a 15-min methanol fixing and a 1-h 2% crystal violet staining. Images of colonies were captured with ChemiDoc Imaging Systems (Bio-Rad, Hercules, CA, USA), followed by counting of colonies using Image J software.
5-Ethynyl-2ʹ-Deoxyuridine (EdU) Assay
The EdU Apollo DNA in vitro kit (RiboBio Co., Ltd., Guangzhou, China) was employed in this experiment. One day before transfection, cells were seeded in 96-well plates at 5 × 103 cells/well. Forty-eight hours post-transfection, 100 μL EdU solution (50 μM) was supplemented to each well for a 2-h incubation at 37ºC. After a 30-min fixing at room temperature using 100 μL phosphate buffer saline (PBS) containing 4% polyformaldehyde, the cells were incubated with 50 μL glycine (2 mg/mL) for 5 min. After permeating with 0.5% Triton X, the cells were reacted with 1 × Apollo solution in dark for 30 min at room temperature. Following 10-min nuclei staining with 4ʹ,6-Diamidino-2-Phenylindole (DAPI; Sigma-Aldrich, St. Louis, MO, USA), the cells were then observed and photographed under an inverted fluorescence microscope, and the positive cell rate, determined as EdU positive cells/DAPI positive cells, was counted using Image-J software.
TdT-Mediated dUTP-Biotin Nick End-Labeling (TUNEL) Assay
Apoptotic cells were assessed based on the manuals of a TUNEL Cell Apoptosis Detection Kit (Beyotime). Briefly, 30-min fixing of the transfected GC cells (1 × 105) was carried out with 4% paraformaldehyde (Solarbio, p1110, Beijing, China) at 4ºC, followed by a 5-min incubation with 0.3% TritonX-100 (Sigma-Aldrich, 9002–93-1). Afterwards, TUNEL solution (Beyotime, c1088) was supplemented to each well for a 60-min incubation in dark at 37ºC. After that, DAPI (Sigma-Aldrich) was used for nuclei staining in dark at room temperature for 10 min, followed by observation and photographing under the inverted fluorescence microscope. Finally, the positive cell rate, defined as TUNEL positive cells/DAPI positive cells, was counted using Image-J software.
Flow Cytometry
Flow cytometry was used to analyze the cell cycle. Briefly, GC cells were seeded into 6-well plates at a density of 1 × 106 cells/well for culture. Cells were detached by trypsin and then centrifuged at 500 g for 12 min. The harvested cells were fixed overnight at 4°C with 70% ethanol and resuspended in cold propidium iodide (PI) solution (Sigma-Aldrich) containing RNase for 40 min in the dark. A flow cytometer (Accuri C6 FACS, MA, USA) was applied to determine cell cycle progression, and data analysis was performed using FlowJo software (BD FACSCalibur, USA).
Wound Healing Test
The transfected 5 × 104 cells were cultured in 64-well plates for confluence. Linear wounds were formed with 10 μL sterile pipette tips, followed by cell culture in serum-free medium. Images were captured at 0 and 24 h, and 24-h migration rate was measured using Image J software.
Transwell Assay
GC cells (1 × 105) were positioned in Matrigel (40 μL, BD Biosciences, San Jose, CA, USA)-coated Transwell apical chamber, followed by dilution to 4 μg/μL with serum-free medium. Then, the medium encompassing 10% FBS (600 μL) was supplemented to the basolateral chamber, followed by a 24-h incubation at 37ºC. The upper surface of the membrane was wiped with a cotton swab before paraformaldehyde fixing and 15-min crystal violet staining of the cells attached to the lower surface. Cells in five random fields of view were counted under a microscope, followed by calculation of the average number of cells.
Tube Formation in vitro Assay
A 96-well plate was coated with Matrigel (BD Biosciences; 100 μL/well), followed by a 30-min standing at 37ºC. Afterwards, human umbilical vein endothelial cells (HUVECs) were seeded into the plates at a density of 1 × 104 cells/well. After the cells fully adherent to the wells, the medium was replaced with a conditioned medium of AGS and MKN-45 cells. After 24-h incubation, cells were monitored and imaged using an Olympus DP71 immunofluorescence microscope (Olympus, Tokyo, Japan), and the number of microvessels in the focus area was measured and analyzed using Chemi Imager 5500 V2.03 software (Alpha Innotech, San Leandro, CA, USA).
Nuclear-Cytoplasmic Separation Experiment
Nuclear and cytoplasmic RNAs were isolated from AGS and MKN-45 cells in the light of the protocols of a PARIS kit (Invitrogen), followed by RT-qPCR with GAPDH (cytoplasmic control) and U6 (nuclear control) for standardization.
Dual-Luciferase Reporter Assay
The putative miR-361-3p binding sites on SNHG22 or HMGA1 3ʹ-untranslated region (3ʹ-UTR) were obtained through Starbase (http://starbase.sysu.edu.cn/), followed by designing of the mutants of their binding sites. Luciferase reporter plasmids for SNHG22-wild type (WT)/SNHG22-mutant type (MT) and HMGA1-WT/HMGA1-MT were generated by cloning sequences into downstream of the luciferase gene in the pmirGLO luciferase vector (Promega, Madison, WI, USA). After the miR-361-3p mimic or mimic negative control (NC) were, respectively, co-transfected with pmirGLO reporter plasmids, a dual-luciferase reporter assay system (Promega) was adopted to assess relative luciferase activity.
RNA Pull-Down
After synthesis of biotin-labeled probes for miR-361-3p-WT/MT or NC (GenePharma Shanghai, China), probe-coated beads were prepared by culturing probes with streptomycin antibiotic magnetic beads (Thermo Fisher Scientific, Waltham, MA, USA) at 25ºC for 2 h. Beads coated with probes were incubated with GC cell lysates overnight at 4ºC, followed by RNA extraction for RT-qPCR to measure the enrichment capacity of SNHG22.
Western Blot Analysis
Following total protein extraction with cell lysis buffer (Beyotime) containing a mixture of protease and phosphatase inhibitors, protein concentration was estimated in the light of the manuals of the bicinchoninic acid protein assay kit (Pierce, San Jose, CA, USA). Following sodium dodecyl sulfate polyacrylamide gel electrophoresis, samples (30 μg protein/lane) were electroblotted to polyvinylidene fluoride membranes (0.22 μm pore, Roche Diagnostics GmbH, Mannheim, Germany). After blockage with Tris-buffered saline with Tween-20 buffer containing 5% skimmed milk, overnight membrane probing was conducted with primary antibodies against HMGA1 [1:1000, #7777, Cell Signaling Technologies (CST), Beverly, MA, USA], β-catenin (1:1000, #8480, CST), and the internal reference GAPDH (1:10,000, ab181602, Abcam, Cambridge, UK) at 4ºC. Then, the membrane was re-probed with the goat anti-rabbit Immunoglobulin G H&L [horseradish peroxidase (HRP)] secondary antibody (1:10,000, ab205718, Abcam) for 1 h at room temperature. Protein bands were visualized using Immobilon Western chemiluminescent HRP substrates (Millipore, Burlington, MA, USA) and quantified using Quantity One software (Bio-Rad).
Statistical Analysis
All data were summarized as mean ± standard deviation and analyzed by GraphPad Prism 8 software (GraphPad Software Inc., La Jolla, California, USA), with p < 0.05 as a level of statistically significance. Data between two groups were compared by paired or unpaired t test. Comparisons among multiple groups were performed using two-way analysis of variance (ANOVA), followed by Tukey’s post hoc test. Log-rank test was adopted for survival analysis, and Pearson correlation coefficient for analyzing correlation.
Results
SNHG22 Overexpression Was Observed in GC Tissues and Correlated to Disease Progression and Poor Prognosis
SNHG22 has shown cancer-promoting effects in numerous cancers.11,20,21 However, little is acknowledged about whether SNHG22 orchestrated GC. Therefore, we tested whether SNHG22 mediated GC development. GEPIA (http://gepia.cancer-pku.cn/) showed that SNHG22 was highly expressed in GC patients (Figure 1A). Then, RT-qPCR detection of SNHG22 expression was conducted in GC tissues and adjacent tissues. Its expression in GC tissues was significantly elevated versus adjacent tissues (Figure 1B). After the tumor tissues were grouped according to the tumor node metastasis (TNM) stage of the patients, we found that SNHG22 expression was significantly higher in the tissues of patients with high TNM stage than in the tissues of patients with low TNM stage (Figure 1C). Meanwhile, patients were assigned into high SNHG22 expression group and low SNHG22 expression group according to the mean value of SNHG22 expression in tumor tissues (3.13). The five-year survival rate of GC patients in low SNHG22 expression group was obviously augmented in contrast to high SNHG22 expression group (Figure 1D). Therefore, SNHG22 high expression correlated to disease progression and poor prognosis of GC patients.
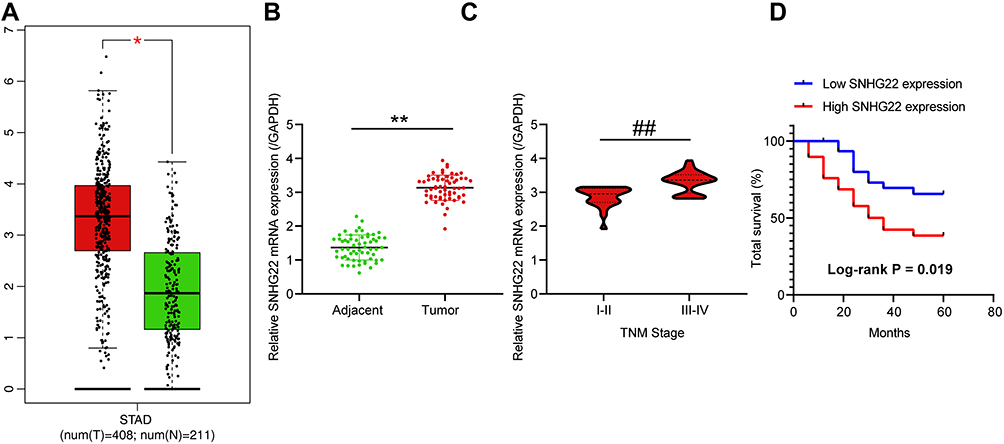 |
Figure 1 SNHG22 is overexpressed in GC tissues and is related to disease progression and poor prognosis of GC patients. (A) SNHG22 high expression in GC patients predicted by GEPIA (unpaired t test, * p < 0.05). (B) The expression of SNHG22 in tumor tissues and adjacent normal tissues of GC patients measured by RT-qPCR (paired t test, ** p < 0.01). (C) The relationship between SNHG22 expression in tumor tissues of GC patients and their TNM stage (unpaired t test, ## p < 0.01). (D) The effect of SNHG22 expression on the five-year survival rate of patients (Log-rank test, p = 0.019). All experiments were repeated three times independently, and the results were averaged. |
Inhibition of SNHG22 Repressed GC Cell Proliferation and Promoted Cell Apoptosis
To investigate whether SNHG22 had an effect on GC, we conducted the following experiments. RT-qPCR detection of SNHG22 expression was performed in GES-1 and GC cell lines. As depicted in Figure 2A, SNHG22 expression was significantly higher in GC cell lines than in GES-1 cells with the highest expression in MKN-45 and AGS cell lines, which were selected for subsequent experiments.
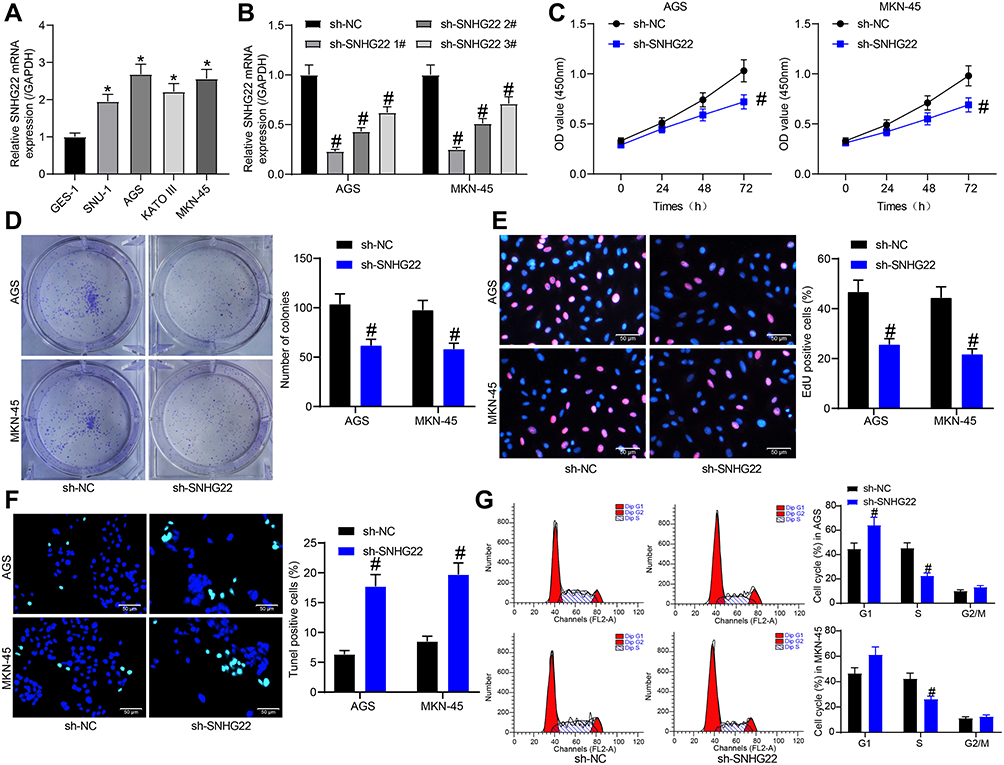 |
Figure 2 GC cell proliferation is repressed but cell apoptosis is accelerated by silencing of SNHG22. (A) RT-qPCR detection of SNHG22 expression in human normal gastric epithelial cells and GC cells. (B) RT-qPCR detection of the transfection efficiency of sh-SNHG22 1, 2, 3#. (C) CCK-8 assay of the effect of sh-SNHG22 on MKN-45 and AGS cell viability. (D) The effect of sh-SNHG22 on the ability of MKN-45 and AGS cell colony formation determined by colony formation assay. (E) EdU assay of the effect of sh-SNHG22 on the DNA synthesis ability of MKN-45 and AGS cells. (F) TUNEL assay of the effect of sh-SNHG22 on the apoptotic rate of MKN-45 and AGS cells. (G) Flow cytometry of the effect of sh-SNHG22 on MKN-45 and AGS cell cycle. In panel A, * p < 0.05 vs GES-1 cells according to one-way ANOVA; in panel B, # p < 0.05 vs MKN-45 and AGS cells transfected with sh-NC according to one-way ANOVA; in panel C–G, # p < 0.05 vs MKN-45 and AGS cells transfected with sh-NC according to two-way ANOVA. All experiments were repeated three times independently, and the results were averaged. |
MKN-45 and AGS cells were transfected with sh-SNHG22 1, 2, 3#, followed by RT-qPCR measurement of the transfection efficiency (Figure 2B). The sh-SNHG22 1# with best transfection efficiency was selected as sh-SNHG22 for following experiments. From results of CCK-8 (Figure 2C), colony formation (Figure 2D), and EdU (Figure 2E) assays, silencing of SNHG22 reduced the proliferation, colony formation, and DNA synthesis ability of MKN-45 and AGS cells. TUNEL staining revealed that the apoptotic rate of SNHG22-silenced MKN-45 and AGS cells was strikingly enhanced (Figure 2F). Consistently, flow cytometry showed that inhibition of SNHG22 in GC cells resulted in G1 phase block (Figure 2G) in GC cells.
Inhibition of SNHG22 Suppressed GC Cell Migration and Invasion and HUVEC Angiogenesis
To further investigate the effect of SNHG22 on GC cells, we detected the expression of EMT-related factors (E-cadherin and N-cadherin) in MKN-45 and AGS cells by RT-qPCR (Figure 3A). We found that sh-SNHG22 remarkably diminished N-cadherin expression and elevated E-cadherin expression. Further wound healing and Transwell assays described prominent decline of 24-h migration rate (Figure 3B) and invasion (Figure 3C) of MKN-45 and AGS cells after sh-SNHG22 treatment.
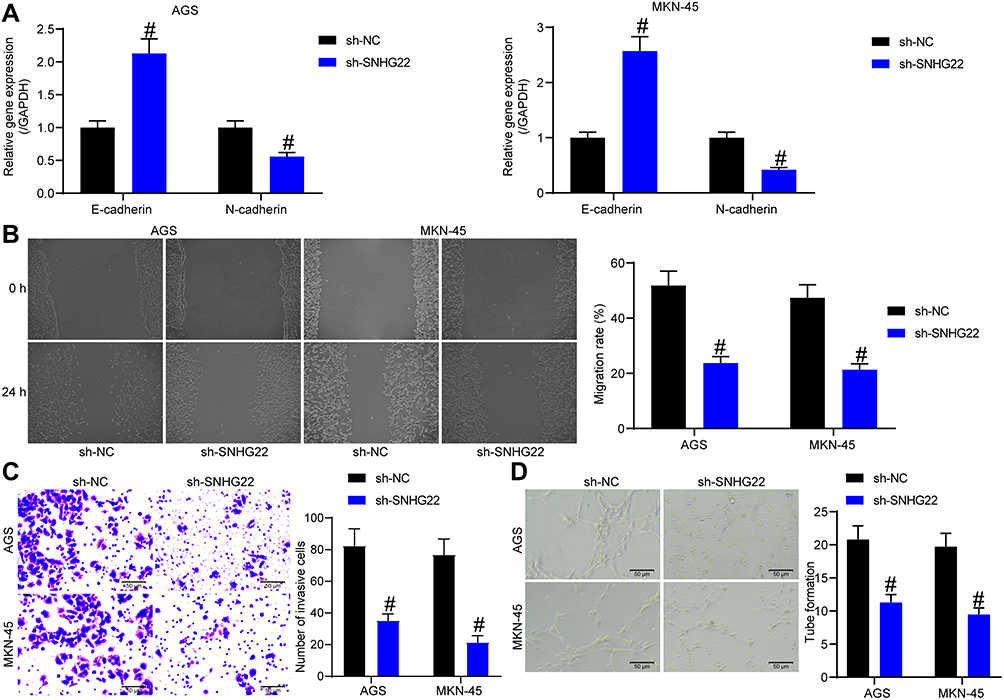 |
Figure 3 SNHG22 silencing causes declines in GC cell migration and invasion and HUVEC angiogenesis. MKN-45 and AGS cells were transfected with sh-NC or sh-SNHG22. (A) RT-qPCR to assess the expression of E-cadherin and N-cadherin in MKN-45 and AGS cells. (B) MKN-45 and AGS cell migration evaluated by wound healing assay. (C) Transwell assay to detect MKN-45 and AGS cell invasion. (D) HUVEC angiogenesis examined by tube formation assay in vitro. # p < 0.05 vs MKN-45 and AGS cells transfected with sh-NC according to two-way ANOVA. All experiments were repeated three times independently, and the results were averaged. |
Finally, the supernatant of MKN-45 and AGS cells stably transfected with sh-NC or sh-SNHG22 was harvested, and HUVECs were seeded into a conditioned medium containing the supernatant for co-culture. Then, tube formation assays in vitro (Figure 3D) displayed that after sh-SNHG22 treatment, the tube formation ability of MKN-45 and AGS cells was prominently reduced. In conclusion, GC cell migration and invasion and HUVEC angiogenesis were repressed by SNHG22 downregulation.
Overexpression of SNHG22 Promotes Malignant Biological Behavior in GC Cells
We have shown that silencing of SNHG22, which is highly expressed in GC cells, inhibited the malignant biological behavior of GC cells. We then selected the SNU-1 cells with the lowest SNHG22 expression relative to other GC cells to perform gain-of-function assay to study the effect of SNHG22 overexpression in GC cells. We transfected pcDNA-SNHG22 into SNU-1 cells and measured effective transfection by RT-qPCR (Figure 4A). We performed the following experiments to detect cell growth. By the CCK8, colony formation assay and EdU staining, we found that overexpression of SNHG22 led to increases in cell proliferation, colony formation ability and DNA synthesis ability of GC cells (Figure 4B-D). TUNEL staining showed that overexpression of SNHG22 inhibited apoptosis, while flow cytometry displayed that it significantly promoted the transition of GC cells from G1 to S phase (Figure 4E and F). We also observed that overexpression of SNHG22 significantly contributed to the elevated migratory and invasive abilities of GC cells (Figure 4G and H), as detected by the wound healing test and Transwell assay. Finally, by tube formation in vitro assay, we observed that overexpression of SNHG22 significantly promoted the tube formation of the cells (Figure 4I).
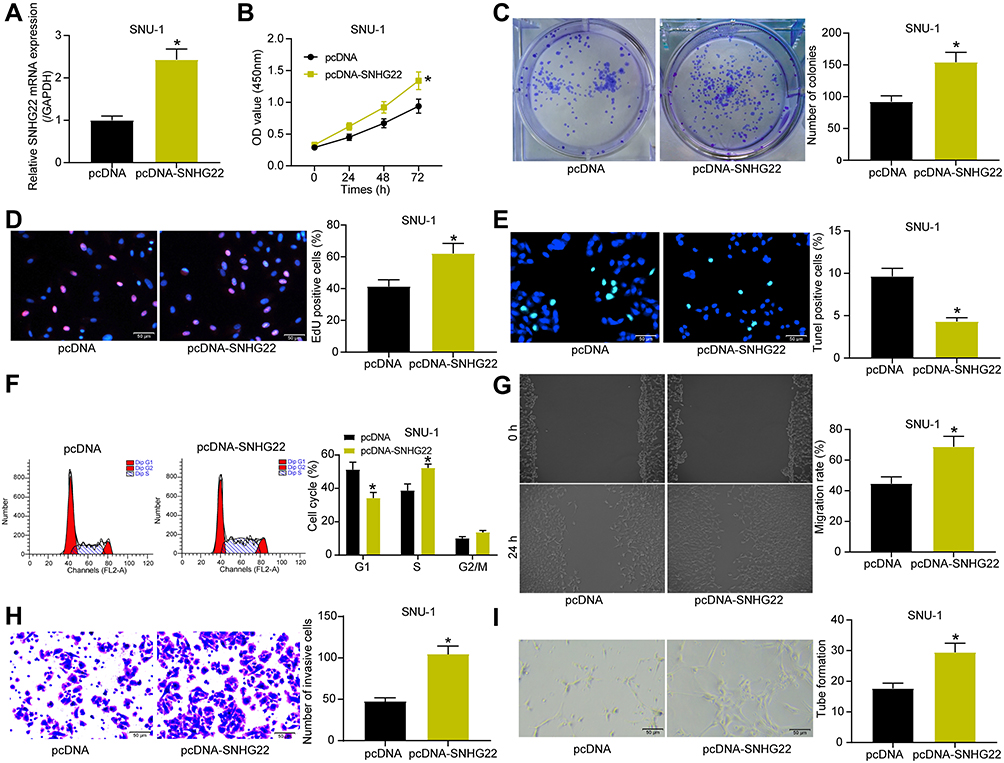 |
Figure 4 Overexpression of SNHG22 promotes the biological behavior of GC cells. SNU-1 cells were transfected with pcDNA-SNHG22 or pcDNA. (A) transfection efficiency of pcDNA-SNHG22 detected by RT-qPCR. (B) CCK-8 assay of the effect of pcDNA-SNHG22 on SNU-1 cell viability. (C) The effect of pcDNA-SNHG22 on the ability of SNU-1 cell colony formation determined by colony formation assay. (-D) EdU assay of the effect of pcDNA-SNHG22 on the DNA synthesis ability of SNU-1 cells. (E) TUNEL assay of the effect of pcDNA-SNHG22 on the apoptotic rate of SNU-1 cells. (F) Flow cytometry of the effect of pcDNA-SNHG22 on SNU-1 cell cycle. (G) SNU-1 cell migration evaluated by wound healing assay. (H) Transwell assay to detect SNU-1 cell invasion. (I) HUVEC angiogenesis examined by tube formation assay in vitro. In panel (A, C, D, E, G, H and I) * p < 0.05 vs pcDNA transfection according to unpaired t test; in panel B and F, * p < 0.05 vs pcDNA transfection according to unpaired t test. All experiments were repeated three times independently, and the results were averaged. |
SNHG22 Bound to miR-361-3p in GC Cells
To investigate the mechanism of SNHG22 in GC, SNHG22 localization was evaluated in GC cells. Through nuclear-cytoplasmic separation experiments, it was observed that SNHG22 was mainly localized in the cytoplasm (Figure 5A). miRs binding to SNHG22 were predicted in Starbase (http://starbase.sysu.edu.cn/) and Diana (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2%2Findex-experimental), with five intersected genes (Figure 5B). The expression of these miRNAs was measured by RT-qPCR in sh-NC or sh-SNHG22-transfected MKN-45 and AGS cells. As exhibited in Figure 5C, only miR-361-3p showed notable differences between the two GC cells at the same time. miR-361-3p expression in GC tissues was severely reduced in contrast to adjacent normal tissues (Figure 5D). Moreover, miR-361 expression was inversely correlated with SNHG22 expression in GC tissues (Figure 5E).
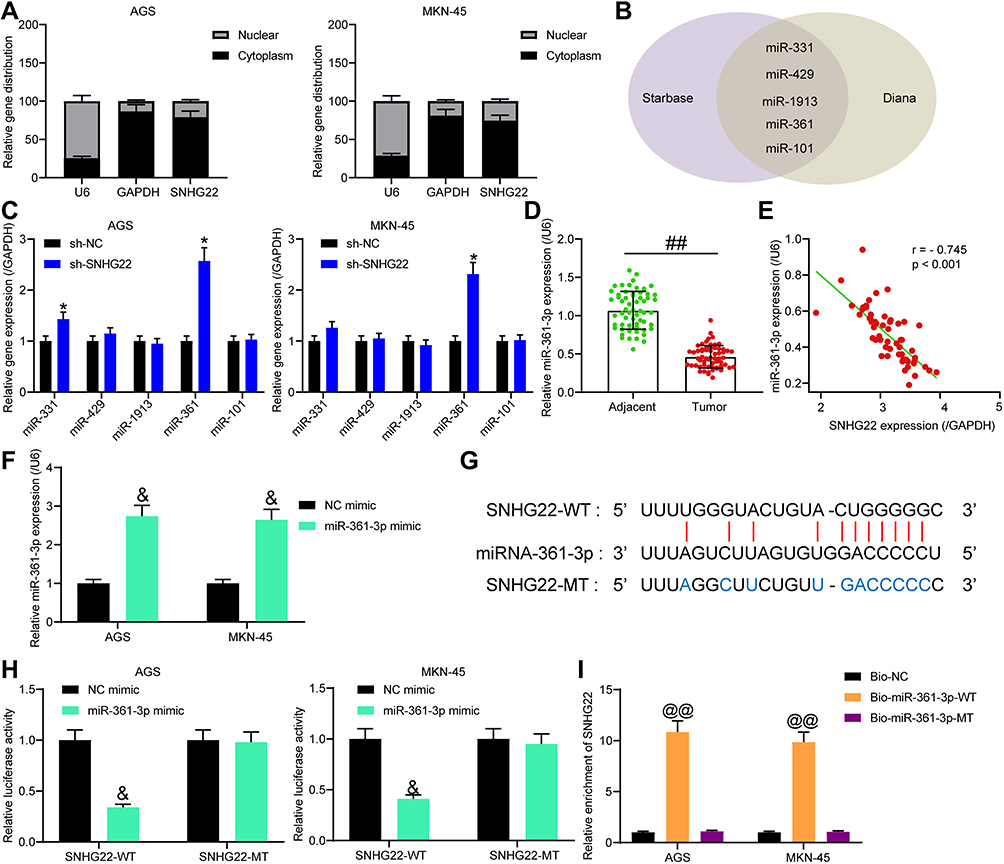 |
Figure 5 SNHG22 binds to miR-361-3p in GC cells. (A) The localization of SNHG22 in GC cells detected by nuclear-cytoplasmic separation experiment. (B) Predicted SNHG22-bound miRNAs in Starbase and Diana. (C) RT-qPCR to measure the effect of sh-SNHG22 on the expression of predicted binding miRs (* p < 0.05 according to two-way ANOVA). (D) RT-qPCR detection of miR-361-3p expression in GC and adjacent normal tissues (## p < 0.01 according to paired t test). (E) The correlation between miR-361-3p and SNHG22 expression in tumor tissues (Pearson correlation analysis, r = −0.745, p < 0.001). (F) RT-qPCR determination of the transfection efficiency of miR-361-3p mimic (&p < 0.05 according to two-way ANOVA). (G) The binding sites of miR-361-3p to SNHG22. (H) The effect of miR-361-3p mimic on the luciferase activity of SNHG22-WT/SNHG22-MT luciferase reporter plasmid detected by dual-luciferase reporter assay (&p < 0.05 according to two-way ANOVA). (I) RNA pull-down assay to detect the enrichment of SNHG22 by Bio-miR-361-3p-WT/Bio-miR-361-3p-MUT (@@ p < 0.01 vs Bio-NC according to two-way ANOVA). All experiments were repeated three times independently, and the results were averaged. |
Next, MKN-45 and AGS cells were transfected with miR-361-3p mimic and its control, followed by transfection efficiency measurement by RT-qPCR (Figure 5F). To verify the binding relationship between SNHG22 and miR-361-3p, we designed SNHG22-WT with the binding sites of miR-361-3p and SNHG22-MT with mutations in the binding site (Figure 5G). Then, dual-luciferase reporter assay (Figure 5H) manifested that miR-361-3p mimic markedly inhibited the luciferase activity of SNHG22-WT, but had no significant effect on that of SNHG22-MT. Finally, RNA pull-down (Figure 5I) showed that compared with Bio-NC, Bio-miR-361-3p-WT noticeably enriched SNHG22, while there was no significant difference in enrichment ability of Bio-miR-361-3p-MT for SNHG22. In summary, SNHG22 expression was inversely correlated with miR-361-3p expression in GC cells by binding to miR-361-3p.
Downregulation of miR-361-3p Attenuated the Effect of SNHG22 Silencing on GC Cell Biological Function
We validated that SNHG22 could bind to miR-361-3p in GC cells. Next, we investigated whether miR-361-3p would affect the role of SNHG22 in GC by rescue experiments.
MKN-45 and AGS cells were co-transfected with sh-SNHG22 + miR-361-3p inhibitor or NC inhibitor, and the transfection efficiency was assessed by RT-qPCR (Figure 6A). Moreover, in the presence of sh-SNHG22, MKN-45 and AGS cell viability (Figure 6B), colony formation (Figure 6C), and DNA synthesis (Figure 6D) were augmented by miR-361-3p inhibitor. While TUNEL staining showed that the apoptosis rate of GC cells after sh-SNHG22 treatment was significantly reduced after inhibition of miR-361-3p (Figure 6E). Flow cytometry results showed that the cell cycle blocking effect of SNHG22 was significantly attenuated after inhibition of miR-361-3p (Figure 6F).
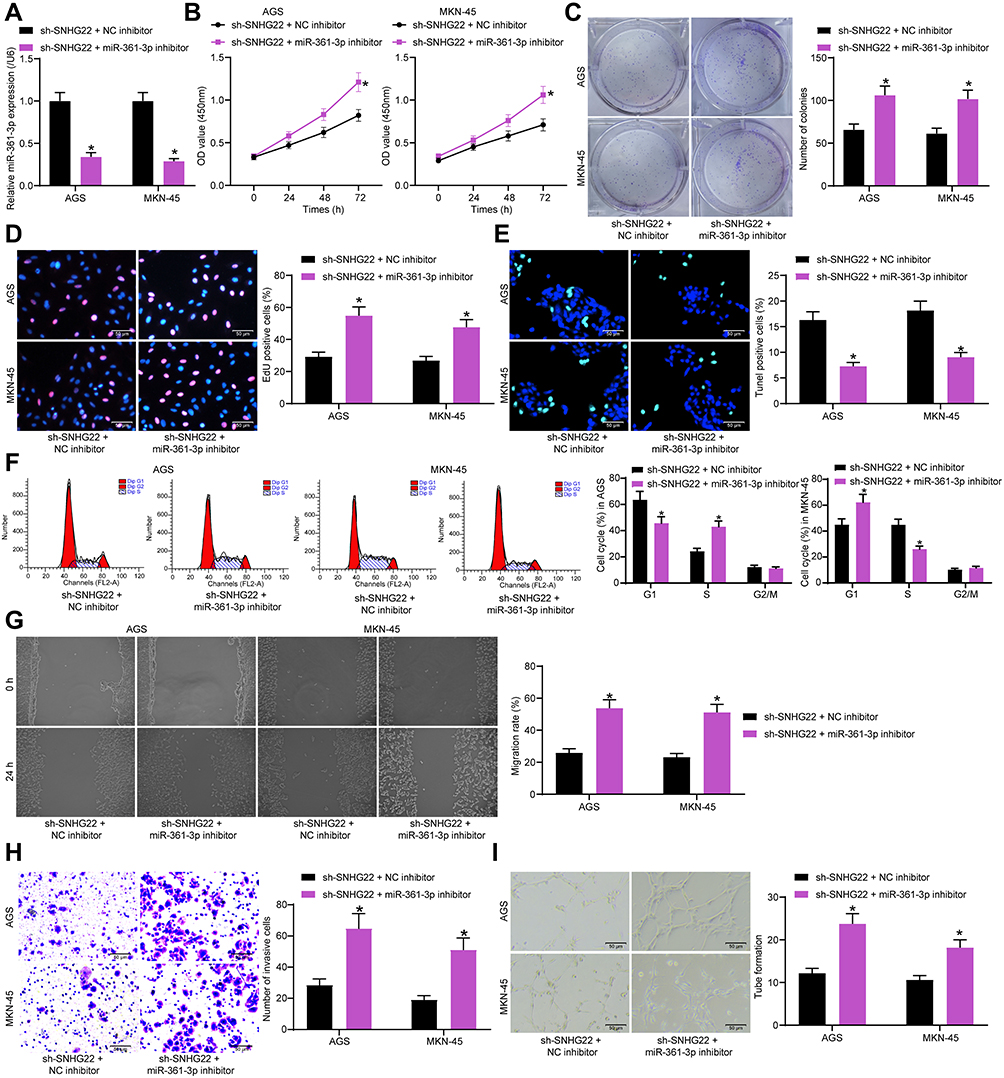 |
Figure 6 SNHG22 overexpression promotes cell proliferation, migration, invasion, and angiogenesis but inhibits apoptosis in GC by downregulating miR-361-3p. MKN-45 and AGS cells were co-transfected with sh-SNHG22 + NC inhibitor or sh-SNHG22 + miR-361-3p inhibitor. (A) RT-qPCR determination of the transfection efficiency of miR-361-3p inhibitor in SNHG22-silenced MKN-45 and AGS cells. (B) CCK-8 assay of MKN-45 and AGS cell viability. (C) MKN-45 and AGS cell colony formation determined by colony formation assay. (D) EdU assay of the DNA synthesis ability of MKN-45 and AGS cells. (E) TUNEL assay of the apoptotic rate of MKN-45 and AGS cells. (F) flow cytometry of MKN-45 and AGS cell cycle changes. (G) MKN-45 and AGS cell migration evaluated by wound healing assay. (H) Transwell assay to detect MKN-45 and AGS cell invasion. (I) HUVEC angiogenesis examined by tube formation assay in vitro. * p < 0.05 vs MKN-45 and AGS cells transfected with sh-SNHG22 + NC inhibitor according to two-way ANOVA. All experiments were repeated three times independently, and the results were averaged. |
Furthermore, wound healing and Transwell assays exhibited that miR-361-3p inhibitor restored the decreased migration and invasion of MKN-45 and AGS cells caused by sh-SNHG22 (Figure 6G and H). The supernatants of MKN-45 and AGS cells transfected with sh-SNHG22 + NC inhibitor and sh-SNHG22 + miR-361-3p inhibitor were collected for tube formation assay in vitro (Figure 6I). The results displayed that miR-361-3p inhibitor triggered HUVEC angiogenesis around MKN-45 and AGS cells in the presence of sh-SNHG22. Taken together, SNHG22 decreased miR-361-3p expression to promote cell proliferation, migration, invasion, and angiogenesis but inhibit apoptosis in GC.
Overexpression of miR-361-3p Inhibits the Promotive Effect of pcDNA-SNHG22 on the Malignant Biological Behavior of GC Cells
To validate the effect of miR-361-3p expression on the carcinogenic effect of SNHG22, we transfected miR-361-3p mimic into SNU-1 cells overexpressing SNHG22 and measured effective transfection by RT-qPCR (Figure 7A). By the CCK8, colony formation and EdU staining, we found that overexpression of miR-361-3p significantly inhibited the promoting effect of overexpression of SNHG22 on the proliferation, colony formation and DNA synthesis abilities of GC cells (Figure 7B-D). TUNEL staining showed that overexpression of miR-361-3p promoted apoptosis of GC cells in the presence of pcDNA-SNHG22. Moreover, miR-361-3p attenuated the promotive role of GC cell cycle entry by pcDNA-SNHG22 (Figure 7E and F). Overexpression of miR-361-3p significantly reversed the promoting effect of overexpression of SNHG22 on the migratory and invasive ability of GC cells by wound healing method and Transwell assay (Figure 7G and H). Finally, by tube formation in vitro assay, we observed a significant decline in the tube formation capacity of pcDNA-SNHG22-transfected GC cells after overexpression of miR-361-3p (Figure 7I).
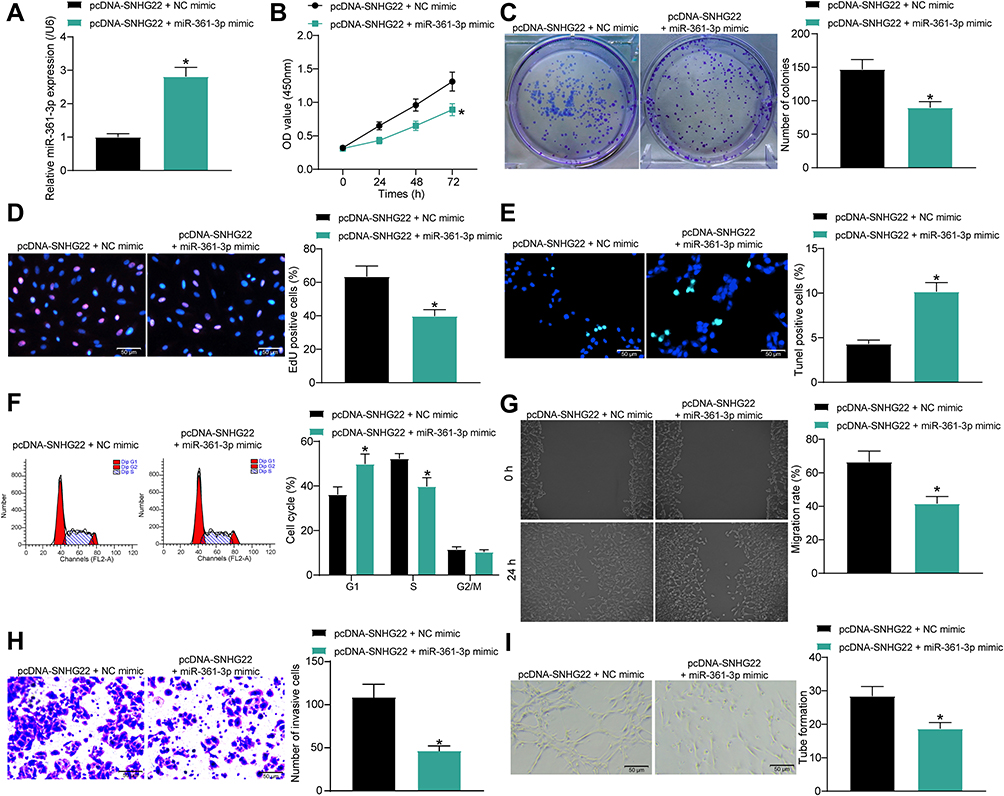 |
Figure 7 Overexpression of miR-361-3p abrogates the promotion of the malignant biological behavior of GC cells by pcDNA-SNHG22. SNU-1 cells were co-transfected with pcDNA-SNHG22 + NC mimic or pcDNA-SNHG22 + miR-361-3p mimic. (A) RT-qPCR determination of the cell transfection efficiency. (B) CCK-8 assay of SNU-1 cell viability. (C) SNU-1 cell colony formation determined by colony formation assay. (D) EdU assay of the DNA synthesis ability of SNU-1 cells. (E) TUNEL assay of the apoptotic rate of SNU-1 cells. (F) flow cytometry of SNU-1 cell cycle changes. (G) SNU-1 cell migration evaluated by wound healing assay. (H) Transwell assay to detect SNU-1 cell invasion. (I) HUVEC angiogenesis examined by tube formation assay in vitro. * p < 0.05 vs SNU-1 cells transfected with pcDNA-SNHG22 + NC mimic according to unpaired t test (panel (A, C, D, E, G, H and I) or two-way ANOVA (panel B and F)). All experiments were repeated three times independently, and the results were averaged. |
SNHG22 Activated the HMGA1/Wnt/β-Catenin Axis by Binding to miR-361-3p in GC Cells
miR-361-5p can inhibit GC progression through the Wnt/β-catenin axis,22 while HMGA1 can promote GC progression through Wnt/β-catenin pathway.23 In addition, it was predicted in Starbase that miR-361-3p targeted HMGA1 (Figure 8A). Therefore, we speculated that HMGA1 was a downstream target gene of miR-361-3p in GC.
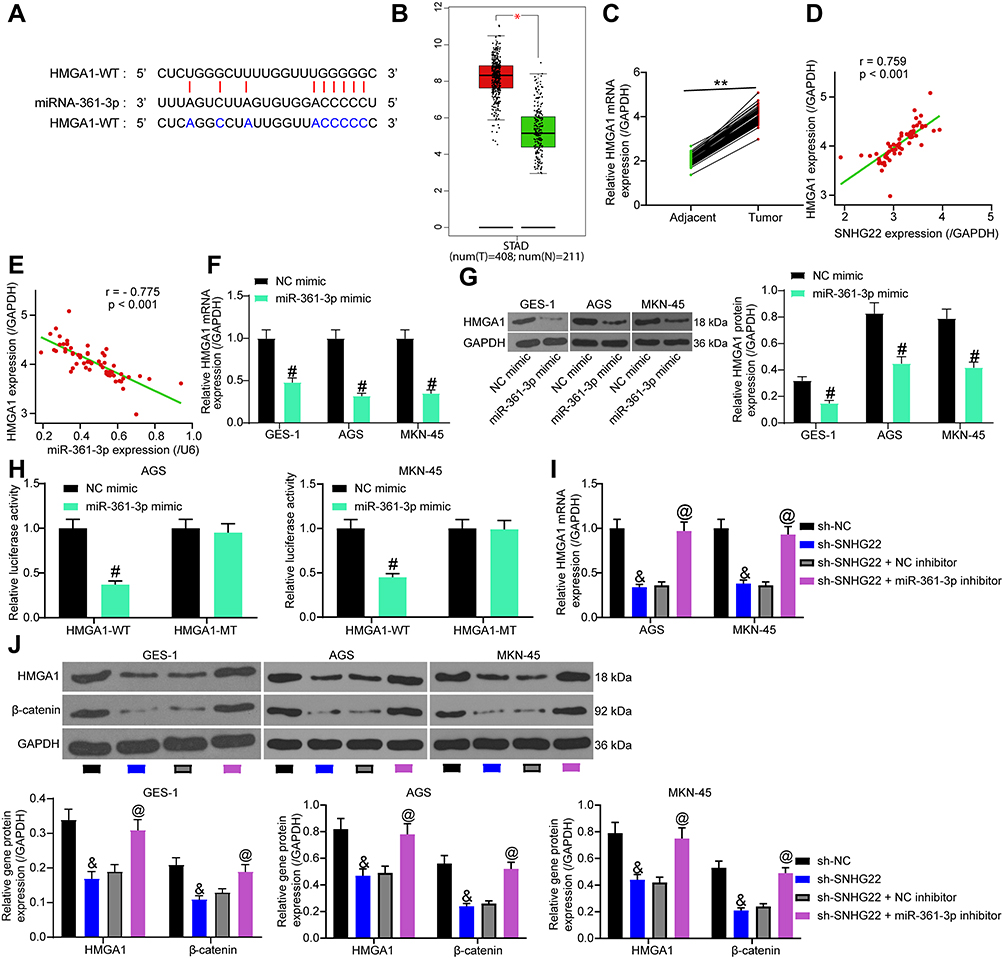 |
Figure 8 SNHG22 promotes Wnt/β-catenin pathway activation via HMGA1 upregulation by binding to miR-361-3p in GC cells. (A) Potential binding sites of miR-361-3p to HMGA1. (B) HMGA1 expression in GC patients predicted in GEPIA database. (C) HMGA1 expression in GC and adjacent normal tissues of GC patients detected by RT-qPCR. (D) Correlation between HMGA1 and SNHG22 expression in GC tissues (r = 0.759). (E) The correlation between HMGA1 and miR-361-3p expression in GC tissues (r = −0.775). (F) RT-qPCR to measure the effect of miR-361-3p mimic on HMGA1 mRNA expression. (G) Western blot analysis of the effect of miR-361-3p mimic on HMGA1 protein expression. (H) The effect of miR-361-3p mimic on the luciferase activity of HMGA1-WT/HMGA1-MT detected by dual-luciferase assay. (I) RT-qPCR detection on the effect of sh-SNHG22 and miR-361-3p inhibitor on HMGA1 mRNA expression. (J) Western blot analysis to assess the effect of sh-SNHG22 and miR-361-3p inhibitor on HMGA1 and β-catenin protein expression. In panel (A), **p < 0.01 according to two-way ANOVA; in panel (D) and (E), Pearce correlation analysis was conducted, p < 0.001; in panel (F–H) #p < 0.05 according to two-way ANOVA; in panel (I) and (J), and p < 0.05 vs MKN-45 and AGS cells transfected with sh-SNHG22, & p < 0.05 vs GES-1, MKN-45 and AGS cells transfected with sh-NC, & p<0.05 vs GES-1, AGS and MKN-45 cells transfected with sh-NC according to two-way ANOVA, @ p < 0.05 vs GES-1, MKN-45 and AGS cells transfected with sh-SNHG22 + NC inhibitor according to two-way ANOVA. All experiments were repeated three times independently, and the results were averaged. |
GEPIA predicted HMGA1 overexpression in GC patients (Figure 8B). HMGA1 expression was evaluated by RT-qPCR in GC and adjacent normal tissues collected from GC patients. As demonstrated in Figure 8C, higher HMGA1 expression was observed in GC tissues than in adjacent normal tissues. Furthermore, HMGA1 expression was positively correlated with SNHG22 expression (Figure 8D) and inversely associated with miR-361-3p expression (Figure 8E) in GC tissues. RT-qPCR and Western blot analysis found that miR-361-3p mimic treatment substantially diminished HMGA1 expression in MKN-45, AGS cells as well as normal gastric epithelial cell line GES-1 (Figure 8F and G). Dual-luciferase reporter assay exhibited that luciferase activity of HMGA1-WT was decreased by miR-361-3p mimic, whereas the luciferase activity of HMGA1-MT did not change (Figure 8H).
MKN-45 and AGS cells were delivered with sh-NC, sh-SNHG22, sh-SNHG22 + NC inhibitor, or sh-SNHG22 + miR-361-3p inhibitor. RT-qPCR results documented that silencing of SNHG22 appreciably restrained HMGA1 expression, and this inhibition was strikingly reversed after treatment with miR-361-3p inhibitor (Figure 8I). Western blot analysis in Figure 8J exhibited that sh-SNHG22 contributed to significant declines in the expression of HMGA1 and β-catenin, which was remarkably annulled by miR-361-3p inhibitor. The above findings suggested that SNHG22 downregulated miR-361-3p, which targeted HMGA1 to activate the Wnt/β-catenin pathway in GC cells.
Overexpression of HMGA1 Alleviated the Repressive Effect of SNHG22 Silencing on the Malignant Biological Behavior of GC Cells
The aforementioned results demonstrated that SNHG22 enhanced HMGA1 expression by binding to miR-361-3p, thereby promoting GC progression through the Wnt/β-catenin pathway. Next, we conducted a rescue experiment to explore whether HMGA1 overexpression affected the effect of sh-SNHG22 on GC cells. MKN-45 and AGS cells were co-transfected with sh-SNHG22 + pcDNA-HMGA1 or pcDNA3.1, followed by RT-qPCR detection of HMGA1 overexpression efficiency. The results manifested the obvious elevation of HMGA1 expression in MKN-45 and AGS cells after co-transfection (Figure 9A). Western blot analysis depicted that pcDNA-HMGA1 transfection neutralized the diminished HMGA1 and β-catenin expression induced by SNHG22 silencing in MKN-45 and AGS cells (Figure 9B).
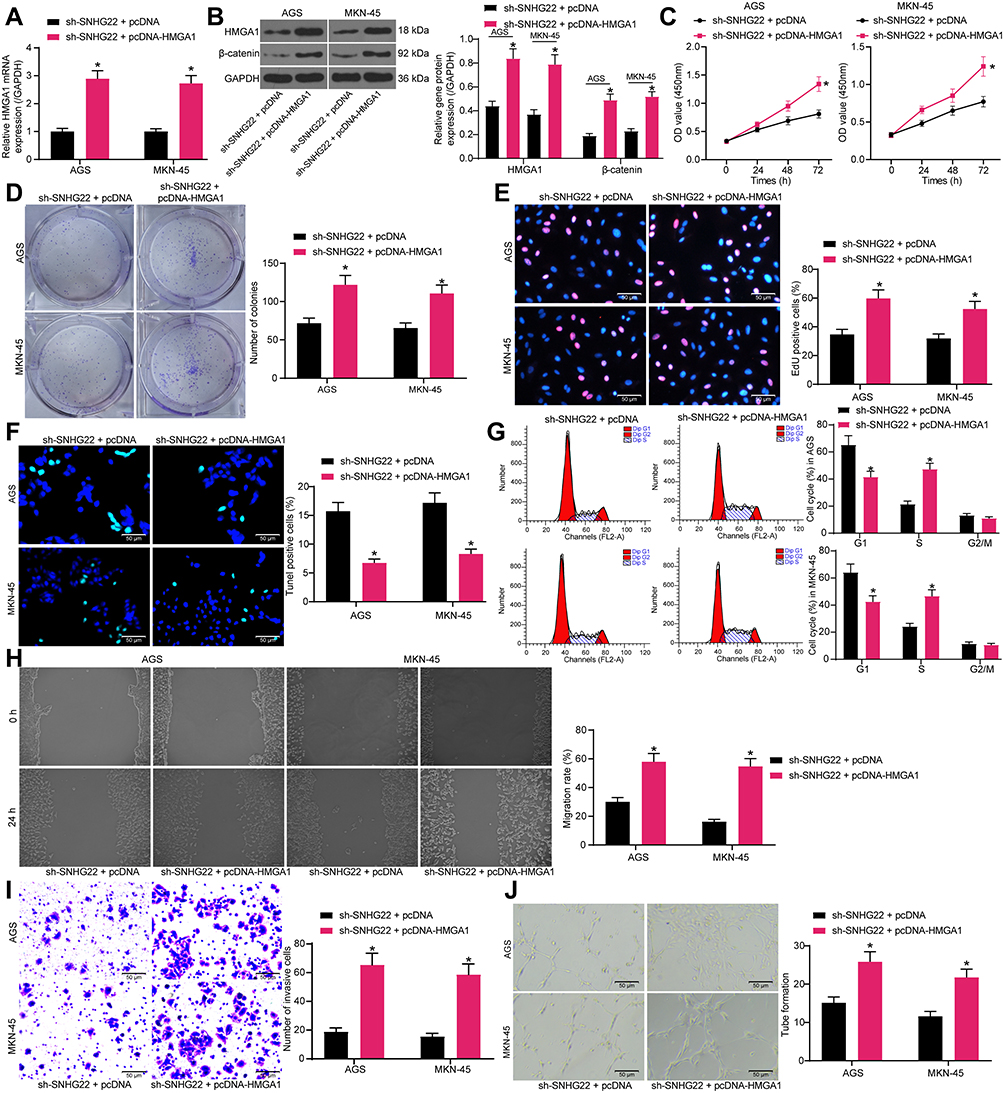 |
Figure 9 SNHG22 silencing restricts cell proliferation, migration, invasion, and angiogenesis but facilitates cell apoptosis in GC by downregulating HMGA1. MKN-45 and AGS cells were transfected with sh-SNHG22 + pcDNA-HMGA1 or sh-SNHG22 + PDNA3.1. (A) RT-qPCR determination of the transfection efficiency of pcDNA-HMGA1 in SNHG22-silenced MKN-45 and AGS cells. (B) HMGA1 and β-catenin expression in MKN-45 and AGS cells evaluated by Western blot analysis. (C) CCK-8 assay of MKN-45 and AGS cell viability. (D) MKN-45 and AGS cell colony formation determined by colony formation assay. (E) EdU assay of the DNA synthesis ability of MKN-45 and AGS cells. (F) TUNEL assay of the apoptotic rate of MKN-45 and AGS cells. (G) flow cytometry analysis of MKN-45 and AGS cell cycle changes. (H) MKN-45 and AGS cell migration evaluated by wound healing assay. (I) Transwell assay to detect MKN-45 and AGS cell invasion. (J) HUVEC angiogenesis examined by tube formation assay in vitro. * p < 0.05 vs MKN-45 and AGS cells transfected with sh-SNHG22 + PDNA3.1 according to two-way ANOVA. All experiments were repeated three times independently, and the results were averaged. |
CCK-8 (Figure 9C), colony formation (Figure 9D), and EdU (Figure 9E) assays described that in the presence of sh-SNHG22, MKN-45 and AGS cells had augmented viability, colony formation, and DNA synthesis after pcDNA-HMGA1 transfection. Meanwhile, TUNEL assay presented that the apoptosis of MKN-45 and AGS cells was enhanced by overexpressing HMGA1 even under the influence of sh-SNHG22 (Figure 9F). Flow cytometry analysis revealed that the cell cycle arrest caused by sh-SNHG22 could be significantly reversed by pcDNA-HMGA1 (Figure 9G).
According to wound healing and Transwell assays, HMGA1 overexpression resulted in elevation of migration and invasion of MKN-45 and AGS cells in the presence of sh-SNHG22 (Figure 7H and I). Besides, tube formation assay in vitro indicated that HMGA1 overexpression negated the reduction of HUVEC angiogenesis around MKN-45 and AGS cells induced by sh-SNHG22 (Figure 7J). To sum up, HMGA1 upregulation decreased the inhibitory effect of SNHG22 silencing on the malignant biological behavior of GC cells.
Discussion
In spite of significant improvements in surgical techniques, radiotherapy, chemotherapy, and neoadjuvant therapy, GC remains the second principle cause of cancer-related death across the globe.24 Hence, there is an urgent need to figure out the underlying molecular mechanism of GC for a more effective and safe treatment. Moreover, the correlation of altered ncRNAs with GC progression has been well-acknowledged.25 In our study, we conducted tissue and cell experiments to test whether SNHG22 and miR-361-3p orchestrated GC cell biological function. Our findings highlighted that downregulation of SNHG22 hampered GC cell proliferation, migration and invasion and HUVEC angiogenesis but accelerated GC cell apoptosis and cell cycle arrest by disrupting HMGA1-activated Wnt/β-catenin pathway via miR-361-3p.
Our initially finding manifested that SNHG22 was highly expressed in GC tissues and associated with disease progression and poor prognosis of GC. Similarly, there is a report showing the contribution of lncRNA SNHGs to tumorigenesis of digestive cancers by modulating cell proliferation, migration, invasion, and apoptosis.26 For instance, Zhou et al observed that SNHG16 was overexpressed in GC tissues, which was related to poor prognosis and malignant phenotype of GC.27 Also, the data provided by Yan et al indicated that the high SNHG6 expression in GC tissues and cell lines was associated with poor prognosis of GC.28 Similarly, another research illustrated that higher SNHG20 expression was observed in GC tissues than in adjacent normal tissues.29 In line with our results, SNHG22 was measured to be highly expressed in triple-negative breast cancer tissues by a prior investigation.20 These findings indirectly supported our results of SNHG22 overexpression in GC tissues. Moreover, our data suggested that SNHG22 silencing restrained cell proliferation, migration, invasion, and angiogenesis but facilitated cell apoptosis and cell cycle arrest in GC. Concurred with our results, an investigation on papillary thyroid cancer elucidated decreased proliferation, migration, and invasion but increased apoptosis of SNHG22-silenced papillary thyroid cancer cells.21
Another interesting finding in our study was that SNHG22 reduced miR-361-3p expression by binding to miR-361-3p in GC cells. A previous study documented that SNHG1 sponged miR-361-3p and negatively modulated miR-361-3p expression in non-small-cell lung cancer cells, which indirectly supported our finding.30 More importantly, the results in the present study demonstrated that miR-361-3p was downregulated in GC tissues and that GC cell proliferation, migration, and invasion and HUVEC angiogenesis were facilitated but GC cell apoptosis and cell cycle arrest were restricted after miR-361-3p knockdown in the presence of sh-SNHG22. Similarly, a previous research identified miR-361-3p poor expression in cervical cancer tissues and the repressive role of miR-361-3p upregulation on cervical cancer cell growth and migration.31 Also, another study clarified that the low miR-361-3p expression was found in thyroid cancer tissues, and that miR-361-3p upregulation caused declines in thyroid cancer cell proliferation, migration and invasion but elevation in cell apoptosis.32 Interestingly, the results obtained by Xin et al unveiled that GC cell proliferation was reduced but cell apoptosis was accelerated by upregulating miR-361-3p, which was agreed with our results.14
Intriguingly, it is widely acknowledged that miRs assumed a critical role in posttranscriptional control of target gene expression.33 In the subsequent assays of our study, we validated that HMGA1 was directly targeted by miR-361-3p in GC cells. Further investigation discovered that miR-361-3p blocked Wnt/β-catenin pathway by targeting HMGA1 to inhibit cell proliferation, migration, invasion, and angiogenesis but enhance cell apoptosis and cell cycle arrest in GC. Coincided with our results, HMGA1 was documented by a prior study to be highly expressed in GC tissues and to augment GC cell proliferation, invasion, and anti-apoptosis.34,35 More recently, it was determined that HMGA1 contributed to breast cancer angiogenesis, which supported our results.36 Furthermore, a research conducted by Han et al observed that HMGA1 induced activation of the Wnt/β-catenin pathway to promote endometrial cancer cell migration and invasion.37 More importantly, impairment of Wnt/β-catenin pathway could result in suppression of cell migration, invasion, and angiogenesis in GC.38
Conclusion
To sum up, our work proved that SNHG22 had a strong expression in GC and exerted carcinogenic effects in GC. Mechanistically, SNHG22 could facilitate GC progression by upregulating HMGA1 and then activating the Wnt/β-catenin pathway via miR-361-3p downregulation. Considering this mechanism, SNHG22 inhibition was suggested to serve as a promising molecular target for targeted therapy in GC in the future. Further in vivo studies are warranted to substantiate our assumption.
Disclosure
The authors declare no potential conflicts of interest.
References
1. Rocken C. Molecular classification of gastric cancer. Expert Rev Mol Diagn. 2017;17(3):293–301. doi:10.1080/14737159.2017.1286985
2. Matsuoka T, Yashiro M. Biomarkers of gastric cancer: current topics and future perspective. World J Gastroenterol. 2018;24(26):2818–2832. doi:10.3748/wjg.v24.i26.2818
3. Berger H, Marques MS, Zietlow R, Meyer TF, Machado JC, Figueiredo C. Gastric cancer pathogenesis. Helicobacter. 2016;21(Suppl 1):34–38. doi:10.1111/hel.12338
4. Molina-Castro S, Pereira-Marques J, Figueiredo C, Machado JC, Varon C. Gastric cancer: basic aspects. Helicobacter. 2017;22(Suppl):1.
5. Figueiredo C, Camargo MC, Leite M, Fuentes-Panana EM, Rabkin CS, Machado JC. Pathogenesis of gastric cancer: genetics and molecular classification. Curr Top Microbiol Immunol. 2017;400:277–304.
6. Wei L, Sun J, Zhang N, et al. Noncoding RNAs in gastric cancer: implications for drug resistance. Mol Cancer. 2020;19(1):62.
7. Liu L, Shi Y, Shi J, et al. The long non-coding RNA SNHG1 promotes glioma progression by competitively binding to miR-194 to regulate PHLDA1 expression. Cell Death Dis. 2019;10(6):463. doi:10.1038/s41419-019-1698-7
8. Saeinasab M, Bahrami AR, Gonzalez J, et al. SNHG15 is a bifunctional MYC-regulated noncoding locus encoding a lncRNA that promotes cell proliferation, invasion and drug resistance in colorectal cancer by interacting with AIF. J Exp Clin Cancer Res. 2019;38(1):172. doi:10.1186/s13046-019-1169-0
9. Xu R, Feng F, Yu X, Liu Z, Lao L. LncRNA SNHG4 promotes tumour growth by sponging miR-224-3p and predicts poor survival and recurrence in human osteosarcoma. Cell Prolif. 2018;51(6):e12515. doi:10.1111/cpr.12515
10. Zhang PF, Wang F, Wu J, et al. LncRNA SNHG3 induces EMT and sorafenib resistance by modulating the miR-128/CD151 pathway in hepatocellular carcinoma. J Cell Physiol. 2019;234(3):2788–2794. doi:10.1002/jcp.27095
11. Zhang PF, Wu J, Luo JH, et al. SNHG22 overexpression indicates poor prognosis and induces chemotherapy resistance via the miR-2467/Gal-1 signaling pathway in epithelial ovarian carcinoma. Aging (Albany NY). 2019;11(19):8204–8216.
12. He L, Chen Y, Hao S, Qian J. Uncovering novel landscape of cardiovascular diseases and therapeutic targets for cardioprotection via long noncoding RNA-miRNA-mRNA axes. Epigenomics. 2018;10(5):661–671. doi:10.2217/epi-2017-0176
13. Meng L, Jia X, Yu W, Wang C, Chen J, Liu F. Circular RNA UBAP2 contributes to tumor growth and metastasis of cervical cancer via modulating miR-361-3p/SOX4 axis. Cancer Cell Int. 2020;20:357. doi:10.1186/s12935-020-01436-z
14. Xin H, Yan Z, Cao J. Long noncoding RNA ABHD11-AS1 boosts gastric cancer development by regulating miR-361-3p/PDPK1 signaling. J Biochem. 2020. doi:10.1093/jb/mvaa065
15. Wang Y, Hu L, Zheng Y, Guo L. HMGA1 in cancer: cancer classification by location. J Cell Mol Med. 2019;23(4):2293–2302. doi:10.1111/jcmm.14082
16. Jin GH, Shi Y, Tian Y, Cao TT, Mao Y, Tang TY. HMGA1 accelerates the malignant progression of gastric cancer through stimulating EMT. Eur Rev Med Pharmacol Sci. 2020;24(7):3642–3647.
17. Chen M, Xu K, Li B, et al. HMGA1 regulates the stem cell-like properties of circulating tumor cells from GIST patients via wnt/beta-catenin pathway. Onco Targets Ther. 2020;13:4943–4956. doi:10.2147/OTT.S249063
18. Gao J, Zhao C, Liu Q, et al. Cyclin G2 suppresses Wnt/beta-catenin signaling and inhibits gastric cancer cell growth and migration through Dapper1. J Exp Clin Cancer Res. 2018;37(1):317. doi:10.1186/s13046-018-0973-2
19. Liu X, Ma R, Yi B, Riker AI, Xi Y. MicroRNAs are involved in the development and progression of gastric cancer. Acta Pharmacol Sin. 2020. doi:10.1038/s41401-020-00540-0
20. Fang X, Zhang J, Li C, Liu J, Shi Z, Zhou P. Long non-coding RNA SNHG22 facilitates the malignant phenotypes in triple-negative breast cancer via sponging miR-324-3p and upregulating SUDS3. Cancer Cell Int. 2020;20:252. doi:10.1186/s12935-020-01321-9
21. Gao H, Sun X, Wang H, Zheng Y. Long noncoding RNA SNHG22 increases ZEB1 expression via competitive binding with microRNA-429 to promote the malignant development of papillary thyroid cancer. Cell Cycle. 2020;19(10):1186–1199. doi:10.1080/15384101.2020.1749466
22. Tian L, Zhao Z, Xie L, Zhu J. MiR-361-5p inhibits the mobility of gastric cancer cells through suppressing epithelial-mesenchymal transition via the Wnt/beta-catenin pathway. Gene. 2018;675:102–109. doi:10.1016/j.gene.2018.06.095
23. Akaboshi S, Watanabe S, Hino Y, et al. HMGA1 is induced by Wnt/beta-catenin pathway and maintains cell proliferation in gastric cancer. Am J Pathol. 2009;175(4):1675–1685. doi:10.2353/ajpath.2009.090069
24. Tan Z. Recent advances in the surgical treatment of advanced gastric cancer: a review. Med Sci Monit. 2019;25:3537–3541. doi:10.12659/MSM.916475
25. Xie S, Chang Y, Jin H, et al. Non-coding RNAs in gastric cancer. Cancer Lett. 2020;493:55–70. doi:10.1016/j.canlet.2020.06.022
26. Yang H, Jiang Z, Wang S, et al. Long non-coding small nucleolar RNA host genes in digestive cancers. Cancer Med. 2019;8(18):7693–7704. doi:10.1002/cam4.2622
27. Zhou C, Zhao J, Liu J, et al. LncRNA SNHG16 promotes epithelial- mesenchymal transition via down-regulation of DKK3 in gastric cancer. Cancer Biomark. 2019;26(4):393–401. doi:10.3233/CBM-190497
28. Yan K, Tian J, Shi W, Xia H, LncRNA ZY. SNHG6 is associated with poor prognosis of gastric cancer and promotes cell proliferation and EMT through epigenetically silencing p27 and sponging miR-101-3p. Cell Physiol Biochem. 2017;42(3):999–1012. doi:10.1159/000478682
29. Cui N, Liu J, Xia H, Xu D. LncRNA SNHG20 contributes to cell proliferation and invasion by upregulating ZFX expression sponging miR-495-3p in gastric cancer. J Cell Biochem. 2019;120(3):3114–3123. doi:10.1002/jcb.27539
30. Li X, Zheng H. LncRNA SNHG1 influences cell proliferation, migration, invasion, and apoptosis of non-small cell lung cancer cells via the miR-361-3p/FRAT1 axis. Thorac Cancer. 2020;11(2):295–304. doi:10.1111/1759-7714.13256
31. Xu J, Yang B, Wang L, et al. LncRNA BBOX1-AS1 upregulates HOXC6 expression through miR-361-3p and HuR to drive cervical cancer progression. Cell Prolif. 2020;53(7):e12823. doi:10.1111/cpr.12823
32. Xia F, Chen Y, Jiang B, Bai N, Li X. Hsa_circ_0011385 accelerates the progression of thyroid cancer by targeting miR-361-3p. Cancer Cell Int. 2020;20:49. doi:10.1186/s12935-020-1120-7
33. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141(4):1202–1207. doi:10.1016/j.jaci.2017.08.034
34. Cao XP, Cao Y, Zhao H, Yin J, Hou P. HMGA1 promoting gastric cancer oncogenic and glycolytic phenotypes by regulating c-myc expression. Biochem Biophys Res Commun. 2019;516(2):457–465. doi:10.1016/j.bbrc.2019.06.071
35. Lin Y, Li J, Ye S, et al. LncRNA GACAT3 acts as a competing endogenous RNA of HMGA1 and alleviates cucurbitacin B-induced apoptosis of gastric cancer cells. Gene. 2018;678:164–171. doi:10.1016/j.gene.2018.08.037
36. Zanin R, Pegoraro S, Ros G, et al. HMGA1 promotes breast cancer angiogenesis supporting the stability, nuclear localization and transcriptional activity of FOXM1. J Exp Clin Cancer Res. 2019;38(1):313. doi:10.1186/s13046-019-1307-8
37. Han X, Cao Y, Wang K, Zhu G. HMGA1 facilitates tumor progression through regulating Wnt/beta-catenin pathway in endometrial cancer. Biomed Pharmacother. 2016;82:312–318. doi:10.1016/j.biopha.2016.05.004
38. Tang L, Wen JB, Wen P, Li X, Gong M, Li Q. Long non-coding RNA LINC01314 represses cell migration, invasion, and angiogenesis in gastric cancer via the Wnt/beta-catenin signaling pathway by down-regulating KLK4. Cancer Cell Int. 2019;19:94. doi:10.1186/s12935-019-0799-9
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.