")
Back to Journals » Cancer Management and Research » Volume 12
Long Non-Coding RNA UCA1 Modulates Paclitaxel Resistance in Breast Cancer via miR-613/CDK12 Axis
Authors Liu C, Jiang F, Zhang X, Xu X
Received 11 December 2019
Accepted for publication 2 April 2020
Published 23 April 2020 Volume 2020:12 Pages 2777—2788
DOI https://doi.org/10.2147/CMAR.S241969
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eileen O'Reilly
Chunhong Liu,1,* Feng Jiang,2,* Xueqin Zhang,3 Xiulong Xu1
1Department of Chinese Medicine, Yantai Hospital of Traditional Chinese Medicine, Yantai, Shandong, People’s Republic of China; 2Department of Pharmacy, Yantai Affiliated Hospital of Binzhou Medical University, Yantai, People’s Republic of China; 3Department of Internal Medicine, Shenxian Hospital of Traditional Chinese Medicine, Liaocheng, Shandong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiulong Xu
Department of Chinese Medicine, Yantai Hospital of Traditional Chinese Medicine, Yantai City, Shandong Province 264000, People’s Republic of China
Email [email protected]
Background: Paclitaxel (PTX) occupies a considerable status in the chemotherapies of breast cancer (BC), but the drug resistance keeps an obstructive factor of PTX treatment. This study was designed to explore the molecular mechanism of long non-coding RNA (lncRNA) urothelial carcinoma-associated 1 (UCA1) in PTX resistance of BC.
Methods: UCA1, microRNA-613 (miR-613) and cyclin-dependent kinase 12 (CDK12) expression was assayed through quantitative real-time polymerase chain reaction (qRT-PCR). Cell Counting Kit-8 (CCK-8) assay was implemented for evaluating the half inhibitory concentrations (IC50) of PTX and cell viability. Cell apoptosis was examined by flow cytometry. The target relationship was explored using dual-luciferase reporter assay and RNA immunoprecipitation (RIP) assay. CDK12 protein level was detected through Western blot. Xenograft tumor assay was applied for assessing the influence of UCA1 on PTX resistance of BC in vivo.
Results: UCA1 expressed highly in PTX-resistant BC tissues and cells and regulated PTX resistance in BC cells by affecting cell viability and apoptosis in part. UCA1 negatively interacted with miR-613 and modulated PTX resistance via sponging miR-613. CDK12 was a downstream gene of miR-613 and miR-613 exerted the modulation of PTX resistance via targeting CDK12. Furthermore, UCA1 regulated CDK12 level through interacting with miR-613. The regulatory role of UCA1 in PTX resistance of BC was achieved by miR-613/CDK12 axis in vivo.
Conclusion: UCA1 mediated PTX resistance in BC through the miR-613/CDK12 axis, manifesting that UCA1 might improve the PTX treatment of BC as a significant therapeutic biomarker.
Keywords: UCA1, paclitaxel resistance, breast cancer, miR-613, CDK12
Introduction
Breast cancer (BC), the most frequent gynecological cancer, is the major cause of cancer-associated death among females in the majority of the countries.1 Despite the modern therapeutic strategies that have been developed, conventional chemotherapy remains a crucial significance, including the drugs of paclitaxel (PTX), cisplatin, doxorubicin, and so on.2–4 PTX is an effective drug chemotherapy in clinical treatment for BC, and the combined therapy based on PTX is usually as first-line therapy.5–7 However, the acquired resistance might seriously affect the therapeutic effect to cause the eventual failure of treatment. Hence, the underlying molecular mechanism of PTX resistance needs to be expounded for alternative treatment.
As a class of non-coding RNAs (ncRNAs), long ncRNAs (lncRNAs) are deficient in the ability of coding proteins.8 Emerging studies have validated the participation of lncRNAs in the formation of PTX chemoresistance in BC. For example, lncRNA FTH1P3 was shown to expedite PTX resistance in BC via the miR-206/ABCB1 axis;9 Han et al illuminated that lncRNA H19 activated PTX resistance in BC through inhibiting apoptosis and the regulation of Akt pathway;10 Zheng et al asserted that CASC2 generated the promotion of PTX resistance of BC by regulating miR-18a-5p/CDK19.11 Recently, urothelial carcinoma-associated 1 (UCA1) was reported to be dysregulated in BC and involved in the multiple resistance regulation.12 But the role and functional mechanism of UCA1 in PTX resistance in BC are obscure.
MicroRNAs (miRNAs) belong to short ncRNAs and can regulate protein expression via combining with the 3ʹ-untranslated region (3ʹUTR) of the messenger RNA (mRNA) in mammalian cells.13 Wang et al announced that miR-335 suppressed PTX resistance in BC by targeting SETD8.14 Zhang et al proved that miR-155-3p reversed PTX resistance through the negative interaction with MYD88 in BC.15 Previous reports demonstrated that miR-613 was down-regulated in BC16 and impeded the production of PTX resistance in triple-negative BC.17 However, it needs further exploration of the molecular mechanism behind miR-613 in PTX resistance of BC.
Cyclin-dependent kinase 12 (CDK12), a member of CDK family, was up-regulated and actively involved in the regulation of chemoresistance in BC.18 A recent study claimed that CDK12 induced trastuzumab resistance by regulating WNT and IRS1-ErbB-PI3K signaling pathways in BC.19 Besides, CDK12 down-regulation inhibited de novo and PARP inhibitor resistance in triple-negative BC.20 Herein, we intended to investigate whether CDK12 affected PTX resistance in BC and the possible relation with UCA1 and miR-613. Our study emphasized on the regulatory mechanism among UCA1, miR-613 and CDK12 in PTX resistance of BC, hoping to offer a preferable understanding regarding the molecular mechanism of drug resistance in BC.
Materials and Methods
Tissues Collection
BC tissues were collected from 60 patients subjected to adenomammectomy in Yantai Hospital of Traditional Chinese Medicine. These tissues from BC patients with progressive disease during primary chemotherapy or recurrent disease within 6 months of completing primary chemotherapy were defined as PTX-resistant tissues (n=30) while PTX-sensitive tissues (n=30) were obtained from those patients suffered recurrence beyond 6 months or without recurrence, referring to the classification criteria in a previous study.21 These tissue samples were preserved in liquid nitrogen at once. All the BC tissue samples were collected with the signing of written informed consent and in accordance with the Declaration of Helsinki. This study was performed with the approval of the Ethical Committee of Yantai Hospital of Traditional Chinese Medicine.
Cell Culture and Treatment
All cell lines used in the current study were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Human BC cell line MCF-7 was maintained in Dulbecco’s Modified Eagle’s medium (DMEM; Corning Life Sciences, Corning, NY, USA) and breast epithelial cell line MCF-10A was cultivated in DMEM/F12 (1:1) mixed solution, adding with 10% fetal bovine serum (FBS; Serapro, Naila, Germany) and 1% penicillin-streptomycin mixture (Transgen, Beijing, China). Anabiotic cells were cultured in a 37°C incubator with abundant humidified air and 5% CO2. The PTX-resistant BC cell line (MCF-7/PTX) was established through treating MCF-7 cells with different concentrations of PTX (Sigma, Saint Louis, MO, USA) as previously reported.22
Cell Transfection
The vectors or oligonucleotides were transfected into BC cells with 60% confluence using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA). The vectors and oligonucleotides used in this study were bought from GenePharma (Shanghai, China) and listed as below: overexpression vector of UCA1 and CDK12 (OE-UCA1 and OE-CDK12), small interfering RNA (siRNA) targeting UCA1 and CDK12 (si-UCA1 and si-CDK12), short hairpin RNA (shRNA) against UCA1 (sh-UCA1), miR-613 mimic (miR-613), miR-613 inhibitor (anti-miR-613) and corresponding negative controls (Vector, si-NC, sh-NC, miR-NC and anti-NC).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
The qRT-PCR experiment was executed exploiting TB Green® Premix Ex Taq™ II Kit (Takara, Dalian, China) via the ABI Prism 7500 sequence detection system (Applied Biosystems, Foster City, CA, USA) according to the presentation of the last study.23 The expression levels of UCA1 and CDK12 were standardized by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) while small nuclear RNA U6 was used for normalizing miR-613, using the 2−∆∆Ct method.24 The sequences of primers are shown in Table 1.
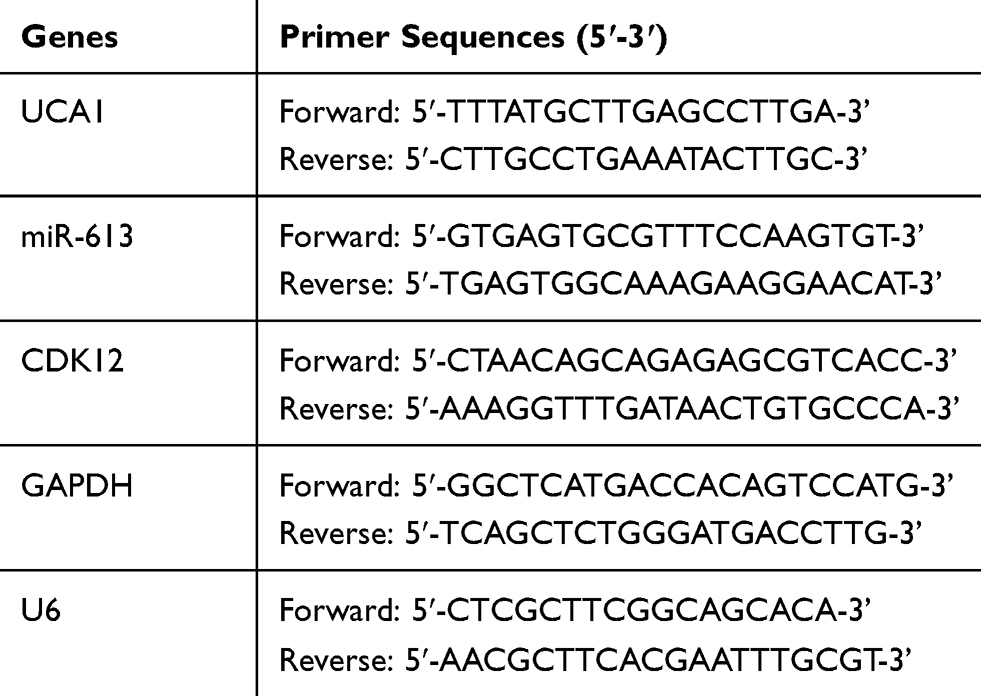 |
Table 1 Primers Used for qRT-PCR Analysis |
Cell Counting Kit-8 (CCK-8)
The CCK-8 assay was administered to examine the half inhibitory concentration (IC50) of PTX and cell viability. Firstly, the 96-well plates were seeding with a total number of 1 × 103 BC cells per well overnight. After treatment with PTX for 48 h, the plates were supplemented with CCK-8 reagent (Beyotime, Shanghai, China) with 20 μL/well. Following incubation for 2 h, the absorbance was recorded under a microplate reader at 450 nm. Particularly, the value of IC50 pointed to the concentration of PTX with cell activity of 50%. And at 24 h, 48 h and 72 h post-transfection, cell viability of MCF-7 and MCF-7/PTX cells was also examined as above.
Flow Cytometry
Annexin V-fluorescein isothiocyanate (Annexin V-FITC)/propidium iodide (PI) kit (Sigma) was exploited to determine cell apoptosis. Briefly, cells were collected through the digestion by 0.25% trypsin (Transgen) and centrifugation at 3000 rpm for 10 min. After washing by pre-cooled 1 × Phosphate buffered saline (PBS; Transgen), 500 μL 1 × binding buffer was used for the preparation of cell suspension, which was incubated with Annexin V-FITC (5 μL) and PI (5 μL) for staining in the dark. Twenty min later, apoptotic cells were quantified using the flow cytometer (BD Biosciences, San Diego, CA, USA).
Dual-Luciferase Reporter Assay
After the target prediction by Starbse3.0, the dual-luciferase reporter assay was applied for the further validation of the target combination. The sequences of wild-type (WT, with the binding sites for miR-613) and mutant-type control (MUT, with the mutant sites for miR-613) UCA1 were, respectively, cloned into the pGL3 vector (Promega, Madison, WI, USA) to construct UCA1-WT and UCA1-MUT. Likewise, CDK12-WT and CDK12-MUT were obtained through cloning the 3ʹUTR sequence into the pGL3 vector. Afterwards, these recombinant reporters were transfected into BC cells with miR-613 or miR-NC, separately. After lysing with 1 × passive lysis buffer (PLB; Promega), the relative luciferase activities of lysates were assayed following the producer’s instruction of the dual-luciferase reporter system (Promega). Renilla activity served as the internal reference for firefly activity.
RNA Immunoprecipitation (RIP) Assay
RIP assay was conducted applying with Magna RNA immunoprecipitation kit (Millipore, Bedford, MA, USA) following the user’s guideline. In short, harvested BC cells were incubated with magnetic beads pre-coated with antibodies against Argonaute2 (anti-Ago2; Abcam, Cambridge, United Kingdom, ab32381) using anti-Immunoglobulin G (anti-IgG; Abcam, ab205718) as the negative control. After the extraction of RNA, qRT-PCR was used for measuring RNA enrichment of UCA1 and miR-613. Cell lysates un-incubated with magnetic beads acted as the positive control (In-put).
Western Blot Assay
To begin with, total proteins were obtained from tissues and cells via Radio-Immunoprecipitation Assay (RIPA) lysis buffer (Sigma). Next, 40 μg proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for 90 min, and instantly transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore) for 1 h. Whereafter, PVDF membranes were blocked by 5% skim milk (Beyotime). After 3 h, primary antibodies anti-CDK12 (Abcam, ab37914, 1:1000) and anti-GAPDH (Abcam, ab181602, 1:3000) were incubated at the room temperature for 4 h. Then, goat anti-rabbit secondary antibody (Abcam, ab205718, 1:5000) was applied to conjugate with primary antibodies for 1 h. Finally, the immunoreactive signals were analyzed using the Image J software (NIH, Bethesda, MD, USA) after the detection of the enhanced chemiluminescence reagent (Sigma).
Xenograft Tumor Assay
Twenty-four BALB/c nude mice (seven-week-old) were bought from Vital River Laboratory Animal Technology (Beijing, China) and randomly divided into four groups with 6 mice/group. Mice were subcutaneously injected with MCF-7 cells (5×106 cells/mice) transfected with OE-UCA1 or Vector, as well as MCF-7/PTX cells stably expressed sh-UCA1 or sh-NC. Following injection for 5 d, mice were intraperitoneally injected with 5 mg/kg PTX every 5 d. Tumor volume was measured with the formula: length × width2 ×0.5. After 25 d, mice were executed with euthanasia and tumor tissues were weighed. And the expression levels of UCA1, miR-613 and CDK12 in excised tissues were detected by qRT-PCR or Western blot. Animal studies were performed in compliance with the ARRIVE guidelines and the Basel Declaration. Experimental procedures were approved by the Institutional and Local Committee on the Care and Use of Animals of Yantai Hospital of Traditional Chinese Medicine. All animals received humane care according to the National Institutes of Health (USA) guidelines.
Statistical Analysis
All experiments were administrated three times and data were presented as the mean ± standard deviation (SD). Statistical analysis of data was through SPSS 19.0 software and GraphPad Prism 7 was applied for graphing. The difference analysis was carried out by Student’s t-test or one-way analysis of variance followed by Tukey’s test. P < 0.05 represented a significant difference at the statistical level.
Results
The Up-Regulation of UCA1 Was Shown in PTX-Resistant BC Tissues and Cells
The demographic features of participated BC patients are shown in Table 2. Then, the expression of UCA1 collected tissues was initially analyzed by qRT-PCR. As illustrated in Figure 1A, the expression of UCA1 was considerably higher in PTX-resistant BC tissues than in PTX-sensitive tissues. Similarly, a conspicuous increase of UCA1 level was exhibited in MCF-7/PTX cells by comparison with parental MCF-7 cells and normal MCF-10A cells (Figure 1B). Through the analysis of CCK-8 assay, IC50 of PTX was distinctly elevated in MCF-7/PTX cells compared to MCF-7 cells, proving the formation of PTX resistance in MCF-7/PTX cells (Figure 1C). Significantly, UCA1 was overexpressed in PTX-resistant BC tissues and cells.
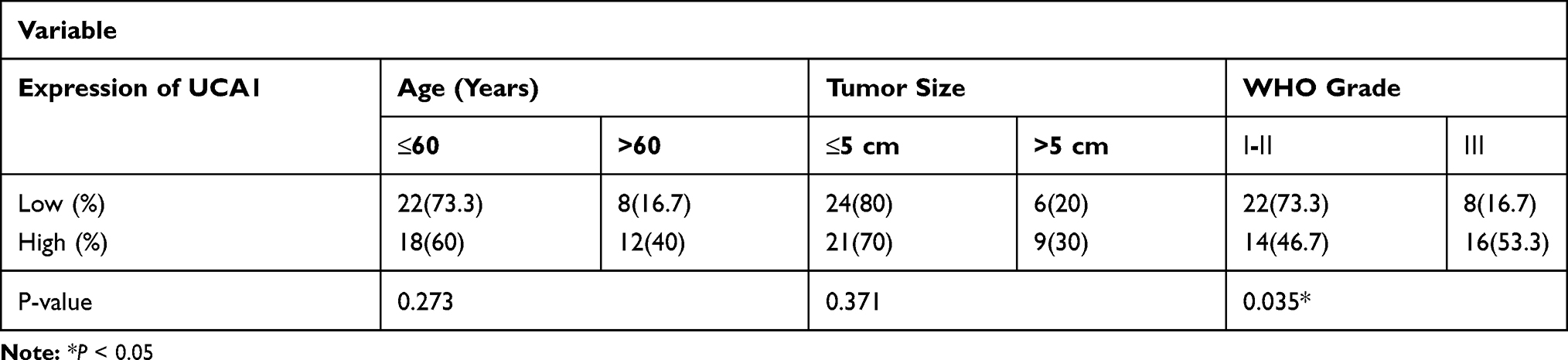 |
Table 2 Relationship of UCA1 Expression with Clinic Pathological Features in Breast Cancer Patients |
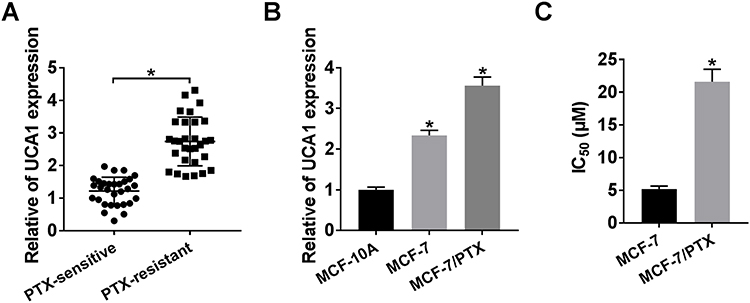 |
Figure 1 The up-regulation of UCA1 was shown in PTX-resistant BC tissues and cells. (A–B) The expression of UCA1 was assayed using qRT-PCR in PTX-resistant BC tissues (A) and cells (B) as well as their sensitive controls. (C) IC50 of PTX was evaluated through CCK-8 assay in MCF-7/PTX cells and parental MCF-7 cells. *P < 0.05. |
UCA1 Regulated PTX Resistance in BC Cells Partly by Affecting Cell Viability and Apoptosis
To investigate the effect of UCA1 on PTX resistance in BC, we transfected OE-UCA1 into MCF-7 cells and sh-UCA1 into MCF-7/PTX cells. Apparently, the overexpression efficiency of OE-UCA1 in MCF-7 cells and knockdown efficiency of sh-UCA1 in MCF-7/PTX cells were greatly contrasted to their controls (Figure 2A). IC50 of PTX was obviously boosted by UCA1 overexpression in MCF-7 cells and inhibited in MCF-7/PTX cells transfected with sh-UCA1 (Figure 2B). Regarding cell viability, as shown in Figure 2C, MCF-7 cells transfected with OE-UCA1 presented a significant promotion after treatment of 5 µM PTX compared to the Vector group, and MCF-7/PTX cells transfected with sh-UCA1 exhibited the decreased cell viability after treatment of 20 µM PTX relative to the sh-NC group. In addition, MCF-7 cells manifested a lower apoptosis rate as a result of UCA1 overexpression compared with PTX+Vector group, while knockdown of UCA1 had a stimulative effect on cell apoptosis in MCF-7/PTX cells treated with PTX (Figure 2D). Collectively, UCA1 could modulate PTX resistance of BC cells via affecting cell viability and apoptosis.
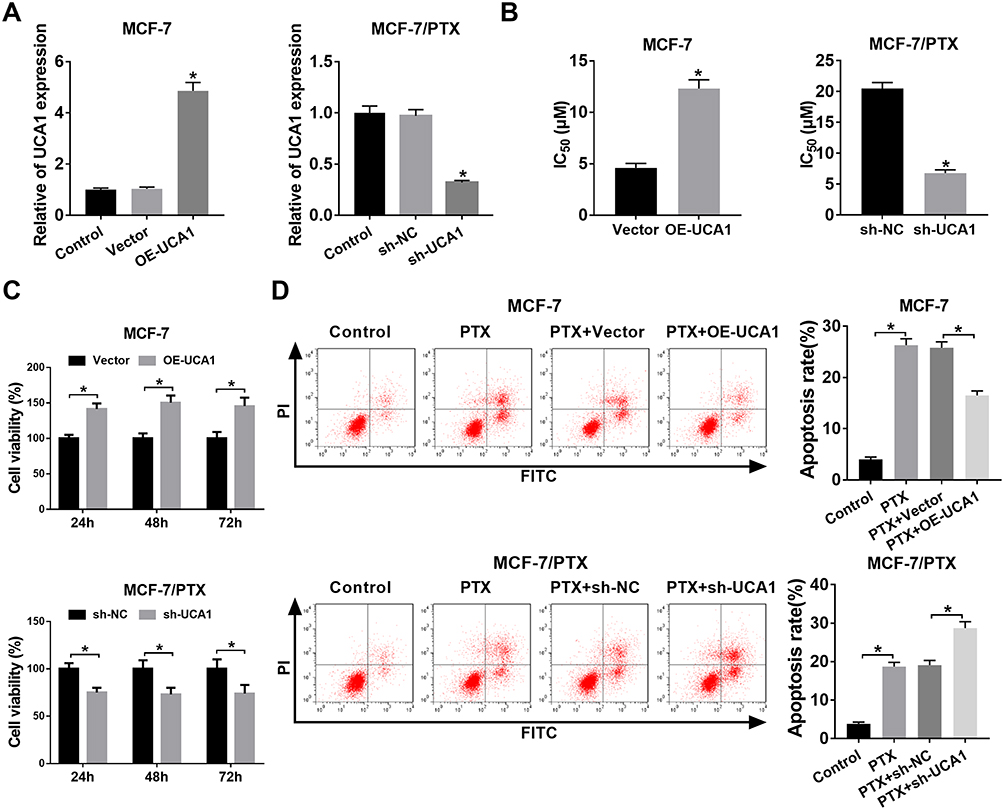 |
Figure 2 UCA1 regulated PTX resistance in BC cells by partly affecting cell viability and apoptosis. (A) Overexpression efficiency of OE-UCA1 in MCF-7 cells and knockdown effect of sh-UCA1 in MCF-7/PTX cells were assessed via qRT-PCR. (B) IC50 of PTX was analyzed by CCK-8 assay. (C) Cell viability was examined by CCK-8 in MCF-7 cells treated with 5 μM PTX and transfected with OE-UCA1 or Vector, as well as in MCF-7/PTX cells treated with 20 μM PTX and transfected with sh-UCA1 or sh-NC. (D) Cell apoptosis was measured by flow cytometry. *P < 0.05. |
UCA1 Directly Interacted with miR-613
We discovered the conjugated sites between UCA1 and miR-613 using Starbase3.0 (Figure 3A), having a conjecture that UCA1 might target miR-613. To affirm this prediction, UCA1-WT or UCA1-MUT was transfected into MCF-7 or MCF-7/PTX cells with miR-613 or miR-NC. As displayed in Figure 3B, the relative luciferase activity was clearly descended in UCA1-WT group after overexpression of miR-613, while it remained almost unchanged in UCA1-MUT transfection group. Also, RIP assay demonstrated that UCA1 and miR-613 were enriched in Ago2 pellet compared with IgG group (Figure 3C). Then, the level of miR-613 was determined by qRT-PCR. In comparison to PTX-sensitive BC tissues and cells, the miR-613 expression was much lower in PTX-resistant tissues and cells (Figure 3D). Moreover, the up-regulation of UCA1 caused the decrease of miR-613 expression in MCF-7 cells, and there was an overt elevation of miR-613 level in MCF-7/PTX cells transfected with sh-UCA1 oppositely (Figure 3E). The above results indicated that UCA1 negatively interacted with miR-613.
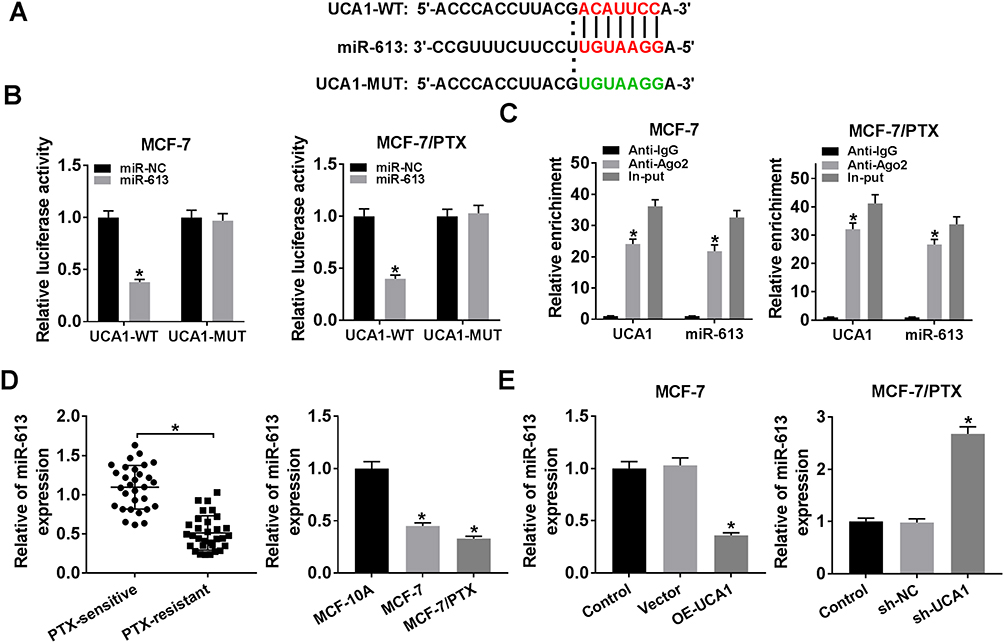 |
Figure 3 UCA1 directly interacted with miR-613. (A) Starbase3.0 was exploited to predict the binding sites between UCA1 and miR-613. (B) and (C) Dual-luciferase reporter assay and RIP assay were applied for analyzing the interaction between UCA1 and miR-613. (D) The expression of miR-613 was measured by qRT-PCR in PTX-resistant BC tissues and cells. (E) QRT-PCR was implemented to determine miR-613 level in MCF-7 cells transfected with OE-UCA1 or Vector and MCF-7/PTX cells transfected with sh-UCA1 or sh-NC. *P < 0.05. |
UCA1 Modulated PTX Resistance in BC Cells via Sponging miR-613
The regulatory mechanism between UCA1 and miR-613 was explored subsequently. We first examined the inhibitory effect of anti-miR-613 in MCF-7 cells and overexpression impact of miR-613 in MCF-7/PTX cells on the expression of miR-613, and qRT-PCR presented the successful outcomes (Figure 4A). MCF-7 cells were transfected with anti-miR-613, anti-miR-613+sh-UCA1 or relative controls and MCF-7/PTX cells were transfected with miR-613, miR-613+OE-UCA1 or matched controls. As for IC50 of PTX, the anti-miR-613-induced rising in MCF-7 cells and the lessening evoked by miR-613 in MCF-7/PTX cells were, respectively, mitigated by knockdown of UCA1 and up-regulation of UCA1 (Figure 4B). Notably, UCA1 depression alleviated the promotion of cell viability caused by miR-613 inhibitor in 5 µM PTX-treated MCF-7 cells and UCA1 overexpression also abated the miR-613-induced repression of cell viability of MCF-7/PTX cells treated with 20 µM PTX (Figure 4C). Besides, the transfection of sh-UCA1 relieved the inhibition of apoptosis rate by miR-613 inhibitor in MCF-7 cells; meanwhile, miR-613-motivated enhancement of cell apoptosis in MCF-7/PTX cells was ameliorated following the increase of UCA1 expression (Figure 4D). All in all, UCA1 regulated PTX resistance through sponging miR-613 in BC cells.
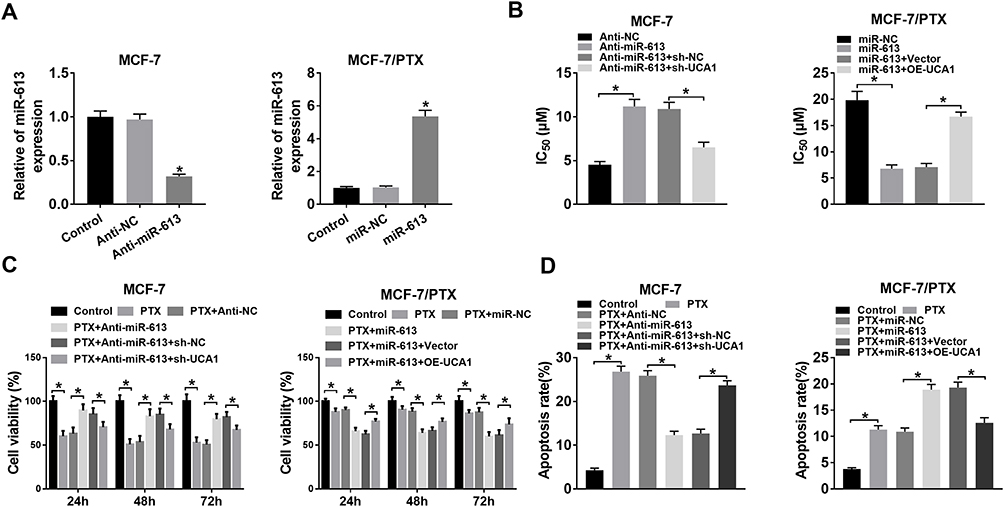 |
Figure 4 UCA1 modulated PTX resistance in BC cells via sponging miR-613. (A) The inhibition of anti-miR-613 and overexpression of miR-613 were severally measured in MCF-7 cells and MCF-7/PTX cells by qRT-PCR. (B) IC50 of PTX was assayed by CCK-8 in MCF-7 cells transfected with anti-miR-613, anti-miR-613+sh-UCA1 or matched controls and MCF-7/PTX cells transfected with miR-613, miR-613+OE-UCA1 or relative controls. (C) CCK-8 was used for detecting cell viability in MCF-7 cells treated with 5 μM PTX and MCF-7/PTX cells treated with 20 μM PTX, transfected with the above groups. (D) Flow cytometry was executed for assessing cell apoptosis. *P < 0.05. |
CDK12 Acted as a Target of miR-613
Through the bioinformatics analysis of Starbase3.0, the 3ʹ-UTR of CDK12 displayed the complementary points for miR-613 (Figure 5A). Then, dual-luciferase reporter assay showed that the introduction of miR-613 manifestly reduced the luciferase activity of CDK12-WT group compared to CDK12-MUT group in MCF-7 and MCF-7/PTX cells (Figure 5B). The combination between miR-613 and CDK12 was further verified by RIP assay, in which the enrichment of CDK12 and miR-613 was much higher in Ago-2 pellet that in IgG group (Figure 5C). And CDK12 was remarkably increased in PTX-resistant BC tissues and cells by contrast to PTX-sensitive tissues and cells (Figure 5D). Furthermore, there was a marked up-regulation of CDK12 protein expression in MCF-7 cells transfected with anti-miR-613, whereas the inverse phenomenon was shown in MCF-7/PTX cells after overexpression of miR-613 (Figure 5E). It is evidence-based that miR-613 targeted CDK12 in BC and PTX-resistant BC cells.
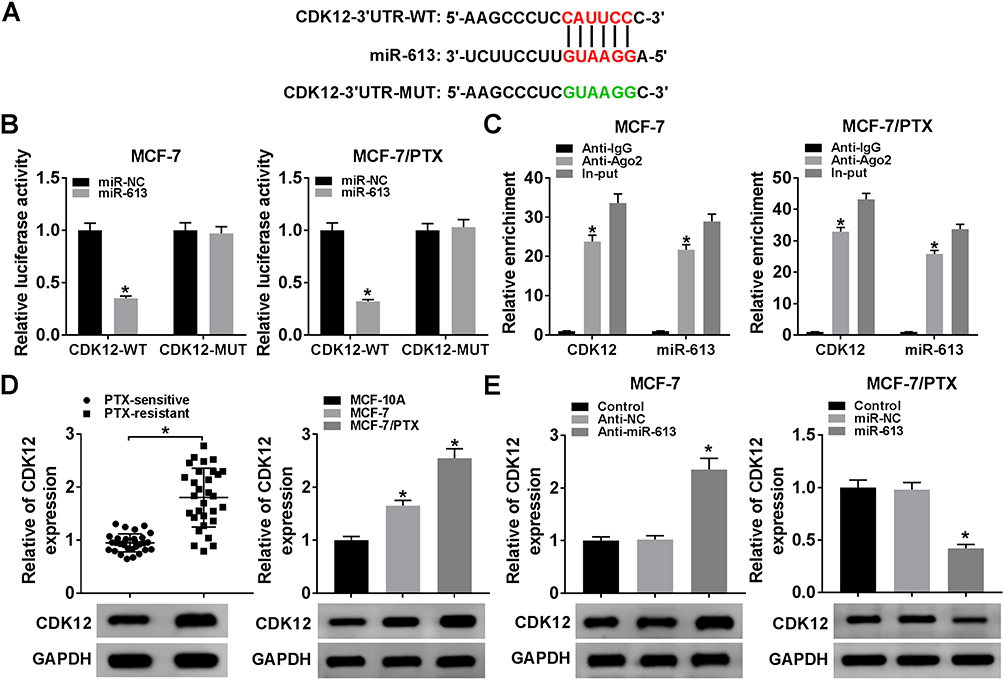 |
Figure 5 CDK12 acted as a target of miR-613. (A) The candidate target of miR-613 was predicted by Starbase3.0. (B) and (C) The target relation between miR-613 and CDK12 was analyzed by dual-luciferase reporter assay and RIP assay. (D) CDK12 expression was detected by Western blot in PTX-resistant BC tissues, cells and controls. (E) Western blot was performed to examine the CDK12 level in MCF-7 cells transfected with anti-miR-613 or anti-NC and MCF-7/PTX cells transfected with miR-613 or miR-NC. *P < 0.05. |
MiR-613 Negatively Mediated PTX Resistance in BC Cells via Targeting CDK12
To study the modulatory relation between miR-613 and CDK12 in PTX resistance of BC, the overexpression effect of OE-CDK12 and interference effect of sh-CDK12 on CDK12 level were evaluated firstly. As Figure 6A presented, CDK12 protein level was signally up-regulated in MCF-7 cells transfected with OE-CDK12 and the decline of CDK12 expression was noteworthy in sh-CDK12 group compared to sh-NC group. After transfection of OE-CDK12, OE-CDK12+miR-613 or respective controls in MCF-7 cells and transfection of sh-CDK12, sh-CDK12+anti-miR-613 or relative controls in MCF-7/PTX cells, IC50 of PTX were determined. The results manifested that miR-613 transfection weakened the promoted effect on IC50 of PTX evoked by CDK12 overexpression in MCF-7 cells; similarly, miR-613 inhibitor counteracted the sh-CDK12-mediated inhibition of IC50 of PTX in MCF-7/PTX cells (Figure 6B). After treatment with PTX, transfection of OE-CDK12 triggered the enhancement of cell viability and suppression of cell apoptosis in MCF-7 cells but CDK12 knockdown led to the restraint of cell viability and elevation of apoptosis rate in MCF-7/PTX cells, whereas these effects were, respectively, receded following overexpression of miR-613 in MCF-7 cells and miR-613 down-regulation in MCF-7/PTX cells (Figure 6C and D). Taken together, miR-613 exerted the regulation of PTX resistance in BC cells by targeting miR-613.
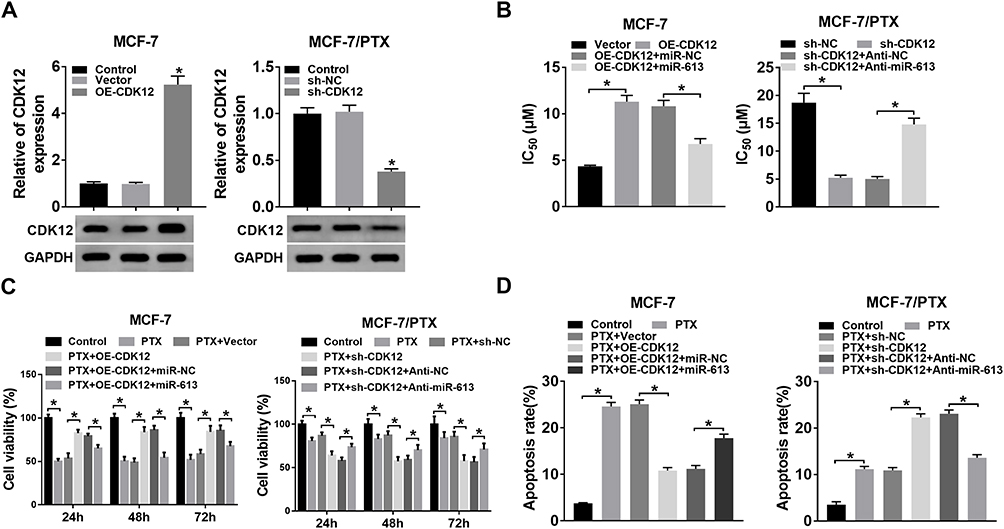 |
Figure 6 MiR-613 negatively mediated PTX resistance in BC cells via targeting CDK12. (A) Western blot was conducted to assess the overexpression efficiency of OE-CDK12 in MCF-7 cells and knockdown effect of sh-CDK12 in MCF-7/PTX cells. (B) CCK-8 was applied for detecting the IC50 of PTX in MCF-7 cells transfected with OE-CDK12, OE-CDK12+miR-613 or relative controls in MCF-7 cells and MCF-7/PTX cells transfected with sh-CDK12, sh-CDK12+anti-miR-613 or matched controls. (C–D) Cell viability (C) and apoptosis (D) were severally assayed by CCK-8 and flow cytometry in 5 μM PTX-treated MCF-7 cells and 20 μM PTX-treated MCF-7/PTX cells transfected with the above groups. *P < 0.05. |
UCA1 Could Regulate CDK12 Expression via the Negative Interaction with miR-613
To analyze whether CDK12 was affected by UCA1, MCF-7 cells were transfected with OE-UCA1, OE-UCA1+miR-613 and MCF-7/PTX cells were transfected with sh-UCA1, sh-UCA1+anti-miR-613, along with their negative controls. After the analysis of Western blot, the transfection of OE-UCA1 resulted in the up-regulation of CDK12 protein expression, which was rescued following overexpression of miR-613 in MCF-7 cells (Figure 7A). Simultaneously, we found that the sh-UCA1-induced descent of CDK12 protein level in MCF-7/PTX cells was also neutralized by co-transfection of sh-UCA1 and anti-miR-613 (Figure 7B). Therefore, UCA1 regulated the level of CDK12 through direct interaction with miR-613.
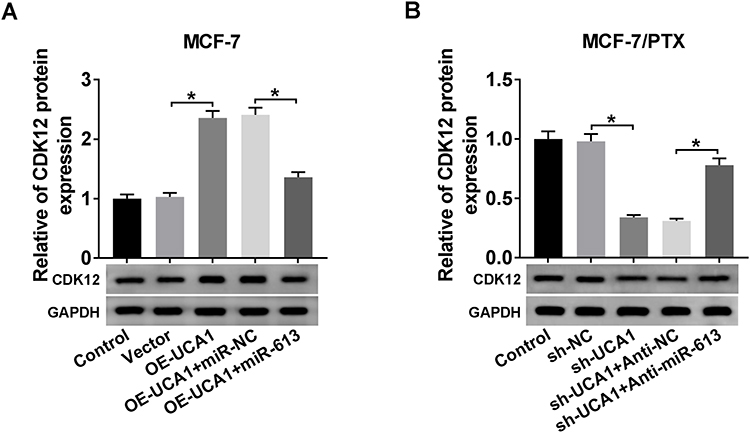 |
Figure 7 UCA1 could regulate CDK12 expression via the negative interaction with miR-613. (A–B) The protein expression of CDK12 was examined by Western blot in MCF-7 cells transfected with OE-UCA1, OE-UCA1+miR-613 or corresponding controls (A) as well as MCF-7/PTX cells transfected with sh-UCA1, sh-UCA1+anti-miR-613 or matched controls (B). *P < 0.05. |
The Modulation of UCA1 in PTX Resistance of BC Was Achieved by miR-613/CDK12 Axis in vivo
Xenograft tumor model was constructed to research the effect of UCA1 on PTX resistance in vivo. As shown in Figure 8A, the volume and weight of tumor from mice injected with MCF-7 cells were conspicuously increased in PTX+OE-UCA1 group in contrast to PTX+Vector group. The qRT-PCR and Western blot suggested the introduction of UCA1 promoted the UCA1 expression as well as CDK12 mRNA and protein levels but refrained the expression of miR-613 in excised tissues (Figure 8B and C). On the contrary, there were presented much lower tumor volume and weight in PTX+sh-UCA1 group than that in PTX-sh-NC group of MCF-7/PTX cells (Figure 8D). The analysis of qRT-PCR and Western blot suggested that the expression of UCA1 and CDK12 (mRNA and protein) was decreased while miR-613 was enhanced after knockdown of UCA1 in vivo (Figure 8E and F). The above data clarified that UCA1 modulated tumor growth of BC by affecting PTX resistance via miR-613/PTX axis in vivo.
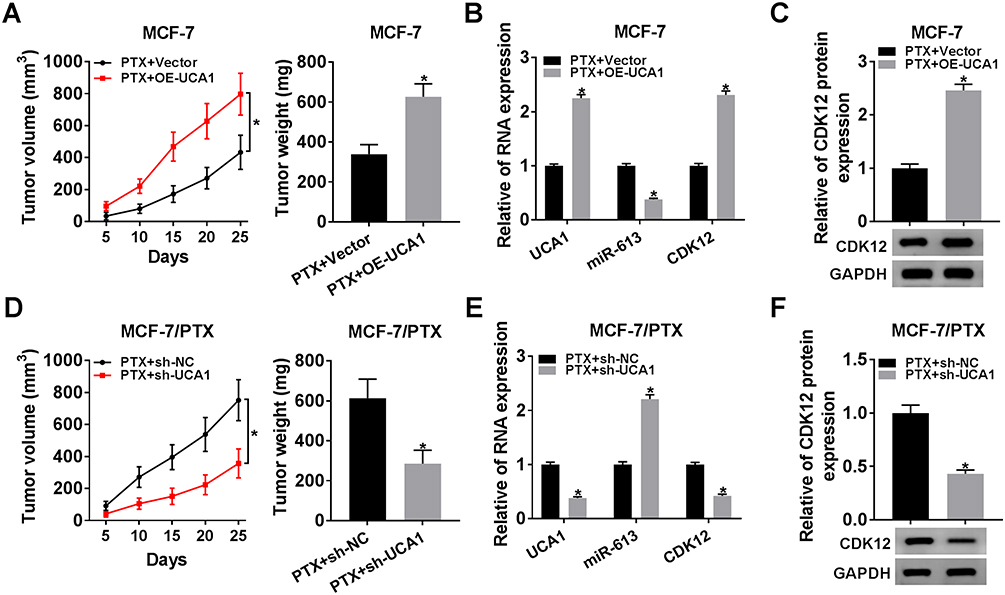 |
Figure 8 The modulation of UCA1 in PTX resistance of BC was achieved by miR-613/CDK12 axis in vivo. (A) Tumor volume and weight were measured in mice injected with MCF-7 cells of PTX+OE-UCA1 and PTX+Vector groups. (B) The expression of UCA1, miR-613 and CDK12 was determined using qRT-PCR in excised tissues. (C) CDK12 protein level was detected by Western blot. (D) After injection with MCF-7/PTX cells treated with PTX+sh-UCA1 and PTX+sh-NC, the volume and weight of tumors from mice were assayed. (E) QRT-PCR was performed for the examination of UCA1, miR-613 and CDK12 levels. (F) Western blot was administrated to test the protein expression of CDK12 in vivo. *P < 0.05. |
Discussion
In the current clinic therapeutic strategies, traditional chemotherapy is still an effective treatment for BC.25 Thus, it is imperative to seek biomarkers for alleviating the acquired drug resistance. In this study, we explored the role of lncRNA UCA1 in PTX-resistant BC and the molecular mechanism of PTX resistance generation behind UCA1 in BC.
According to the published articles, UCA1 exhibited the essential function in the regulation of chemoresistance of various types of cancers. For instance, Yang et al validated the stimulative effect of UCA1 on multidrug resistance of retinoblastoma cells by inhibiting miR-513a-5p.26 A study of ovarian cancer indicated that UCA1 contributed to cisplatin resistance via the signal network of miR-143/FOSL2.27 Li et al reported that UCA1 facilitated resistance to tamoxifen in BC via mediating the EZH2/p21 axis and the PI3K/AKT pathway.28 Our assays firstly indicated the up-regulation of UCA1 in PTX-resistant BC tissues and cells. Subsequently, we found that overexpression of UCA1 elevated PTX resistance and down-regulation of UCA1 refrained resistance to PTX in BC cells. The positive regulation of UCA1 on PTX resistance in BC was consistent with the above-mentioned reports.
LncRNAs are usually considered as miRNA sponges in various cancers.29–31 In this report, we identified miR-613 as a miRNA target through the bioinformatics analysis, and then dual-luciferase reporter assay and RIP assay affirmed that UCA1 targeted miR-613. MiR-613 was shown to reduce PTX resistance in triple-negative BC cells.17 In conformity with this finding, our results verified the inhibitory effect of miR-613 on PTX resistance in BC. Moreover, UCA1 regulated resistance to PTX through directly combining with miR-613. MiRNAs regulate the expression of downstream gene by interaction with its 3ʹUTR generally.32–34 CDK12 was testified to be a decided target gene of miR-613. And CDK12 could positively regulate PTX resistance in BC. Furthermore, the effect of miR-613 on PTX resistance was achieved by targeting CDK12.
Liu et al declared that UCA1 was conductive to the progression of nasopharyngeal carcinoma through the regulation of miR-124-3p/ITGB1 network.35 Wu et al attested that UCA1 promoted drug resistance via inhibiting autophagy by miR-582-5p/AG7 axis in bladder cancer.36 Additionally, UCA1 improved BC progression through enhancing PTP1B by sponging miR-206.37 These studies suggested the signal network of lncRNA-miRNA-mRNA in cancer regulation. Our data presented that UCA1 regulated the expression of CDK12 via the direct combination with miR-613. Therefore, we concluded that the function of UCA1 in regulating PTX resistance in BC was carried out by the miR-613/CDK12 axis. And the assays in vivo confirmed again that UCA1 affected the PTX resistance to regulate tumor growth of BC in vivo through miR-613/CDK12.
In summary, lncRNA UCA1 mediated PTX resistance in BC via the regulation of miR-613/CDK12 axis. The molecular modulatory network of UCA1/miR-613/CDK12 was in the elucidation of PTX resistance in BC for the first time, which exploited a potential pathway for restoring PTX sensitivity to BC. UCA1 might be a resultful indicator for PTX treatment in BC.
Disclosure
The authors declare that they have no financial conflicts of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Tang H, Chen J, Wang L, et al. Co-delivery of epirubicin and paclitaxel using an estrone-targeted PEGylatedliposomal nanoparticle for breast cancer. Int J Pharm. 2019:118806. doi:10.1016/j.ijpharm.2019.118806
3. Dunne M, Dou YN, Drake DM, et al. Hyperthermia-mediated drug delivery induces biological effects at the tumor and molecular levels that improve cisplatin efficacy in triple negative breast cancer. J Control Release. 2018;282:35–45. doi:10.1016/j.jconrel.2018.04.029
4. Lehuede C, Li X, Dauvillier S, et al. Adipocytes promote breast cancer resistance to chemotherapy, a process amplified by obesity: role of the major vault protein (MVP). Breast Cancer Res. 2019;21(1):7. doi:10.1186/s13058-018-1088-6
5. Cannita K, Paradisi S, Cocciolone V, et al. New schedule of bevacizumab/paclitaxel as first-line therapy for metastatic HER2-negative breast cancer in a real-life setting. Cancer Med. 2016;5(9):2232–2239. doi:10.1002/cam4.803
6. Kim SB, Dent R, Im SA, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, Phase 2 trial. Lancet Oncol. 2017;18(10):1360–1372. doi:10.1016/S1470-2045(17)30450-3
7. Bourgeois-Daigneault MC, St-Germain LE, Roy DG, et al. Combination of paclitaxel and MG1 oncolytic virus as a successful strategy for breast cancer treatment. Breast Cancer Res. 2016;18(1):83. doi:10.1186/s13058-016-0744-y
8. Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–641. doi:10.1016/j.cell.2009.02.006
9. Wang R, Zhang T, Yang Z, Jiang C, Seng J. Long non-coding RNA FTH1P3 activates paclitaxel resistance in breast cancer through miR-206/ABCB1. J Cell Mol Med. 2018;22(9):4068–4075. doi:10.1111/jcmm.13679
10. Han J, Han B, Wu X, et al. Knockdown of lncRNA H19 restores chemo-sensitivity in paclitaxel-resistant triple-negative breast cancer through triggering apoptosis and regulating Akt signaling pathway. Toxicol Appl Pharmacol. 2018;359:55–61. doi:10.1016/j.taap.2018.09.018
11. Zheng P, Dong L, Zhang B, et al. Long noncoding RNA CASC2 promotes paclitaxel resistance in breast cancer through regulation of miR-18a-5p/CDK19. Histochem Cell Biol. 2019;152(4):281–291. doi:10.1007/s00418-019-01794-4
12. Campos-Parra AD, Lopez-Urrutia E, Orozco Moreno LT, et al. Long non-coding RNAs as new master regulators of resistance to systemic treatments in breast cancer. Int J Mol Sci. 2018;19(9):2711. doi:10.3390/ijms19092711
13. Matoulkova E, Michalova E, Vojtesek B, Hrstka R. The role of the 3ʹ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 2012;9(5):563–576. doi:10.4161/rna.20231
14. Wang Y, Wang H, Ding Y, et al. N-peptide of vMIP- reverses paclitaxel-resistance by regulating miRNA-335 in breast cancer. Int J Oncol. 2017;51(3):918–930. doi:10.3892/ijo.2017.4076
15. Zhang L, Chen T, Yan L, et al. MiR-155-3p acts as a tumor suppressor and reverses paclitaxel resistance via negative regulation of MYD88 in human breast cancer. Gene. 2019;700:85–95. doi:10.1016/j.gene.2019.02.066
16. Wu J, Yuan P, Mao Q, et al. miR-613 inhibits proliferation and invasion of breast cancer cell via VEGFA. Biochem Biophys Res Commun. 2016;478(1):274–278. doi:10.1016/j.bbrc.2016.07.031
17. Xiong H, Yan T, Zhang W, et al. miR-613 inhibits cell migration and invasion by downregulating Daam1 in triple-negative breast cancer. Cell Signal. 2018;44:33–42. doi:10.1016/j.cellsig.2018.01.013
18. Quereda V, Bayle S, Vena F, et al. Therapeutic targeting of CDK12/CDK13 in triple-negative breast cancer. Cancer Cell. 2019;36(5):545–558 e547. doi:10.1016/j.ccell.2019.09.004
19. Choi HJ, Jin S, H C, et al. CDK12 drives breast tumor initiation and trastuzumab resistance via WNT and IRS1-ErbB-PI3K signaling. EMBO Rep. 2019;20(10):e48058. doi:10.15252/embr.201948058
20. Johnson SF, Cruz C, Greifenberg AK, et al. CDK12 inhibition reverses de novo and acquired parp inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 2016;17(9):2367–2381. doi:10.1016/j.celrep.2016.10.077
21. Zhao Z, Ji M, Wang Q, et al. Circular RNA Cdr1as upregulates SCAI to suppress cisplatin resistance in ovarian cancer via miR-1270 suppression. Mol Ther Nucleic Acids. 2019;18:24–33. doi:10.1016/j.omtn.2019.07.012
22. Cai J, Chen S, Zhang W, et al. Paeonol reverses paclitaxel resistance in human breast cancer cells by regulating the expression of transgelin 2. Phytomedicine. 2014;21(7):984–991. doi:10.1016/j.phymed.2014.02.012
23. Jin Y, Liu F, Huang W, Sun Q, Huang X. Identification of reliable reference genes for qRT-PCR in the ephemeral plant Arabidopsis pumila based on full-length transcriptome data. Sci Rep. 2019;9(1):8408. doi:10.1038/s41598-019-44849-1
24. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
25. Talima S, Kassem H, Kassem N. Chemotherapy and targeted therapy for breast cancer patients with hepatitis C virus infection. Breast Cancer. 2019;26(2):154–163. doi:10.1007/s12282-018-0904-2
26. Yang L, Zhang L, Lu L, Wang Y. lncRNA UCA1 increases proliferation and multidrug resistance of retinoblastoma cells through downregulating miR-513a-5p. DNA Cell Biol. 2020;39(1):69–77. doi:2019;doi:10.1089/dna.2019.5063
27. Li Z, Niu H, Qin Q, et al. lncRNA UCA1 mediates resistance to cisplatin by regulating the miR-143/FOSL2-signaling pathway in ovarian cancer. Mol Ther Nucleic Acids. 2019;17:92–101. doi:10.1016/j.omtn.2019.05.007
28. Li Z, Yu D, Li H, Lv Y, Li S. Long noncoding RNA UCA1 confers tamoxifen resistance in breast cancer endocrinotherapy through regulation of the EZH2/p21 axis and the PI3K/AKT signaling pathway. Int J Oncol. 2019;54(3):1033–1042. doi:10.3892/ijo.2019.4679
29. Wang Y, Chen F, Zhao M, et al. The long noncoding RNA HULC promotes liver cancer by increasing the expression of the HMGA2 oncogene via sequestration of the microRNA-186. J Biol Chem. 2017;292(37):15395–15407. doi:10.1074/jbc.M117.783738
30. Su P, Mu S, Wang Z. Long noncoding RNA SNHG16 promotes osteosarcoma cells migration and invasion via sponging miRNA-340. DNA Cell Biol. 2019;38(2):170–175. doi:10.1089/dna.2018.4424
31. Peng W, Deng W, Zhang J, Pei G, Rong Q, Zhu S. Long noncoding RNA ANCR suppresses bone formation of periodontal ligament stem cells via sponging miRNA-758. Biochem Biophys Res Commun. 2018;503(2):815–821. doi:10.1016/j.bbrc.2018.06.081
32. Wei YT, Guo DW, Hou XZ, Jiang DQ. miRNA-223 suppresses FOXO1 and functions as a potential tumor marker in breast cancer. Cell Mol Biol (Noisy-Le-Grand). 2017;63(5):113–118. doi:10.14715/cmb/2017.63.5.21
33. Li D, Hao X, Song Y. Identification of the key microRNAs and the miRNA-mRNA regulatory pathways in prostate cancer by bioinformatics methods. Biomed Res Int. 2018;2018:6204128. doi:10.1155/2018/6204128
34. Wei D, Yu G, Zhao Y. MicroRNA-30a-3p inhibits the progression of lung cancer via the PI3K/AKT by targeting DNA methyltransferase 3a. Onco Targets Ther. 2019;12:7015–7024. doi:10.2147/OTT.S213583
35. Liu C, Zhang H, Liu H. Long noncoding RNA UCA1 accelerates nasopharyngeal carcinoma cell progression by modulating miR-124-3p/ITGB1 axis. Onco Targets Ther. 2019;12:8455–8466. doi:10.2147/OTT.S215819
36. Wu J, Li W, Ning J, Yu W, Rao T, Cheng F. Long noncoding RNA UCA1 targets miR-582-5p and contributes to the progression and drug resistance of bladder cancer cells through ATG7-mediated autophagy inhibition. Onco Targets Ther. 2019;12:495–508. doi:10.2147/OTT.S183940
37. Li Y, Zeng Q, Qiu J, Pang T, Xian J, Zhang X. Long non-coding RNA UCA1 promotes breast cancer by upregulating PTP1B expression via inhibiting miR-206. Cancer Cell Int. 2019;19(1):275. doi:10.1186/s12935-019-0958-z
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.