")
Back to Journals » OncoTargets and Therapy » Volume 13
Long Non Coding RNA SNHG16 Facilitates Proliferation, Migration, Invasion and Autophagy of Neuroblastoma Cells via Sponging miR-542-3p and Upregulating ATG5 Expression
Authors Wen Y, Gong X, Dong Y, Tang C
Received 12 August 2019
Accepted for publication 2 December 2019
Published 10 January 2020 Volume 2020:13 Pages 263—275
DOI https://doi.org/10.2147/OTT.S226915
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjay Singh
Yi Wen, 1 Xiaohui Gong, 2 Yubin Dong, 1 Chenghe Tang 3
1Neonatal Pediatrics, Central Hospital of Zhoukou City, Zhoukou, Henan, People’s Republic of China; 2Neonatal Pediatrics, Shanghai Children’s Hospital, Shanghai, People’s Republic of China; 3Neonatal Pediatrics, First Affiliated Hospital of Xinxiang Medical College, Xinxiang, Henan, People’s Republic of China
Correspondence: Xiaohui Gong
Neonatal Pediatrics, Shanghai Children’s Hospital, No. 355, Luding Road, Putuo District, Shanghai 200062, People’s Republic of China
Tel +86 13838691130
Email [email protected]
Background: Neuroblastoma (NB) is a heterogeneous pediatric malignant tumor with many biological and clinical characteristics. Long non-coding RNA small nucleolar RNA host gene 16 (SNHG16) plays vital role in the development of NB. However, the potential mechanism of SNHG16 in the progression of NB is rarely reported.
Methods: The expression levels of SNHG16, miR-542-3p and autophagy-related gene 5 (ATG5) were measured with quantitative real-time polymerase chain reaction (qRT-PCR). The proliferation, migration and invasion of NB cells were determined using 3-(4, 5-dimethylthiazol-2-YI)-2, 5-diphenyltetrazolium bromide (MTT) or transwell assay. Protein levels of ATG5, microtubule-associated protein A1/1B-light chain3 (LC3-I/II) and p62 were detected by Western blot analysis. The interaction between miR-542-3p and SNHG16 or ATG5 was predicted by starBase and confirmed by dual luciferase reporter assay. Xenograft mice models were constructed to confirm the role of SNHG16 in vivo.
Results: SNHG16 was upregulated in NB tissues and cells and associated with clinical stage and poor prognosis of NB. Knockdown of SNHG16 impeded proliferation, migration, invasion and autophagy of NB cells in vitro, and suppressed tumor growth in vivo. Interestingly, SNHG16 mediated ATG5 expression through sponging miR-542-3p in NB cells. Moreover, miR-542-3p downregulation reversed the inhibitory effects of SNHG16 silencing on proliferation, migration, invasion and autophagy of NB cells. Besides, ATG5 overturned the regulatory effects on proliferation, migration, invasion and autophagy of NB cells induced by SNHG16 or miR-542-3p knockdown.
Conclusion: SNHG16 facilitated proliferation, migration, invasion and autophagy of NB cells via sponging miR-542-3p and upregulating ATG5 expression in NB.
Keywords: NB, SNHG16, miR-542-3p, ATG5
Introduction
Neuroblastoma (NB) is an aggressive pediatric malignancy originating in the neural crest of the sympathetic nervous system and usually presenting as a mass in the abdomen, chest or neck.1,2 NB has a wide of clinical manifestations from spontaneous tumor regression to the progress of drug resistance and eventual death due to its heterogeneity.3 Moreover, different ages, histological and biological characteristics of tumors give rise to different NB progression.4 High-risk NB with high-risk gene modification and histological defects is a metastatic disease.5 Despite intensive treatment, patients with advanced or metastatic NB often have a poor prognosis, with less than 40% long-term survival.6 Therefore, a more thorough understanding of the pathogenesis of NB is needed to provide new possible strategies for NB treatment.
Long non-coding RNAs (lncRNAs) are emerging regulators that regulate gene expression and cell fate and lack protein-coding functions.7 They play crucial roles in cell differentiation, immune response, apoptosis and other physiological processes.8 Long non-coding RNA small nucleolar RNA host gene 16 (SNHG16) was implicated in multiple cancers, such as colorectal cancer,9 gastric cancer,10 non-small cell lung cancer,11 bladder cancer12 and osteosarcoma.13 It was worth noting that SNHG16 was reported to play a vital role in the process of the occurrence and development of NB and might be a target for the treatment of NB.14 However, the potential molecular mechanisms of SNHG16 in the progression of NB are rarely reported.
MicroRNA (miRNA) regulates gene expression through translation inhibition or mRNA degradation.15,16 It is implicated in the basic processes of cell survival, death, proliferation and differentiation, and is supposed to be an emerging biomarker for the diagnosis of diseases and a possible target for the treatment of miscellaneous diseases.17 It was revealed that miR-542-3p was downregulated in a verity of cancers, such as epithelial ovarian cancer,18 hepatocellular cancer19 as well as non-small cell lung cancer.20 One report showed that in NB, miR-542-3p played an anti-tumor role.21 Nevertheless, the potential molecular mechanism of miR-542-3p in NB needs to be further elucidated.
Autophagy-related gene 5 (ATG5), as a valve for autophagy and apoptosis, exists crucial roles in the course of autophagy.22 Cumulating data indicated that ATG5 was a common target of different miRNAs for regulating autophagy.23–26 For example, Cheng et al revealed that miR-34a could repress the progression of NB via downregulating ATG5 expression.26 However, the exact role of ATG5 and its related mechanism in NB are poorly understood.
Hence, we assessed the expression pattern of SNHG16 in NB tissues and cells. Furthermore, we investigated the molecular mechanism of SNHG16 in NB cells in vitro. Moreover, the roles of SNHG16 in vitro and in vivo were explored. This study would provide new possible strategies for NB treatment.
Materials and Methods
Tissue Samples
This study was ratified by the Ethics Committee of Neonatal pediatrics, Central Hospital of Zhoukou City. Forty-five paired NB tissues and the corresponding non-tumors samples were converged from Neonatal pediatrics, Central Hospital of Zhoukou City. All samples in this study were promptly snap-frozen and stored at −80°C until it was used. The clinicopathologic features of NB patients were displayed in Table 1. The written informed consent was provided by all NBparents or legal guardians.
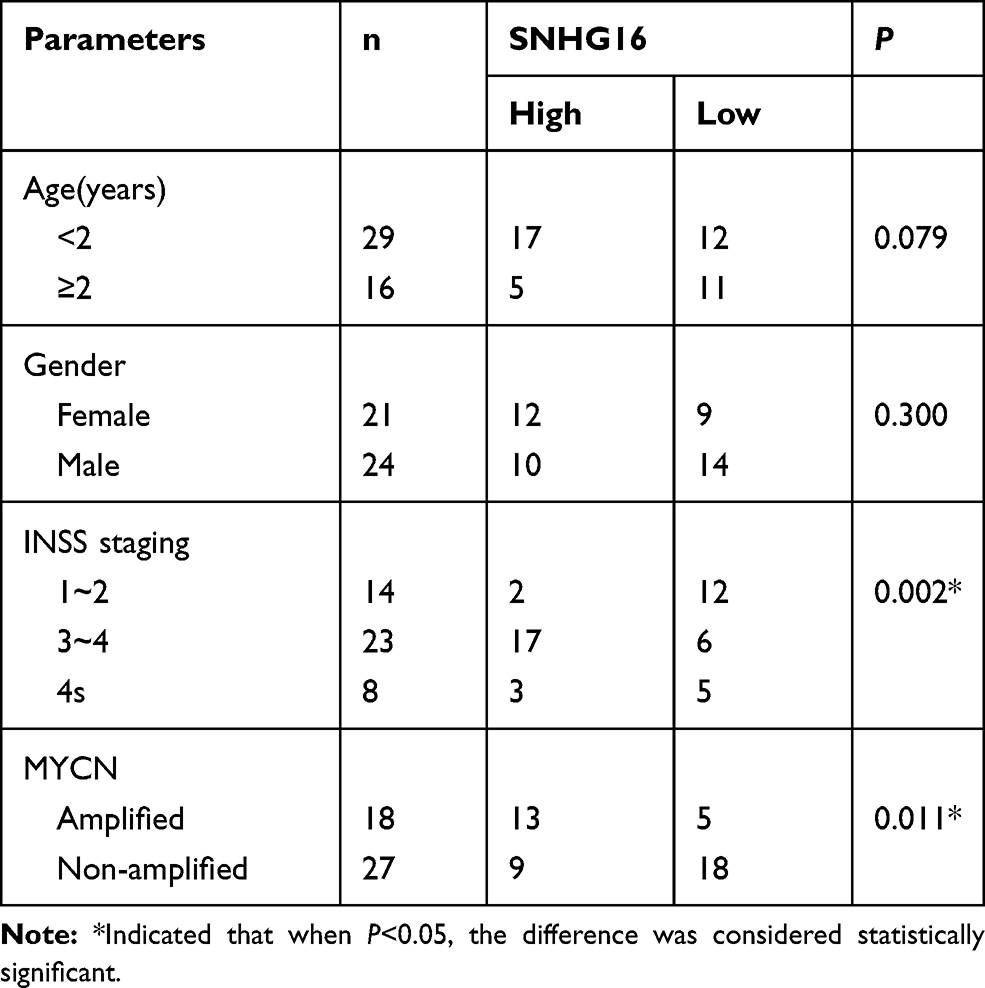 |
Table 1 High SNHG16 Expression and Low SNHG16 Expression Correlated with Clinicopathological Parameters of NB |
Cell Culture
LAN1 cells were obtained from BeNa Culture Collection (Beijing, China). SHEP cells were procured from Abmgoodchina (Zhenjiang, China). Human NB cells SK-N-SH and IMR-32 as well as human umbilical vein endothelial cells HUVEC were acquired from the American Type Culture Collection (ATCC, Manassas, VA, USA). Roswell Park Memorial Institute (RPMI) 1640 medium (Life Technologies, Carlsbad, CA) was employed to cultivate above cell lines. The medium was supplemented with fetal bovine serum (10%, FBS, Life Technologies) at 37°C constant temperature in an incubator with 5% CO2.
Cell Transfection
Three specific small interference RNA (siRNA) against SNGH16 (si-SNGH16#1, si-SNGH16#2 and si-SNGH16#3), SNGH16 smell hairpin (shRNA)(sh-SNGH16), shRNA negative control (sh-NC), and siRNA negative control (si-NC) were obtained from Genepharma (Shanghai, China). The pcDNA-ATG5 overexpression vector (ATG5) and pcDNA were acquired from Invitrogen (Carlsbad, CA, USA). Transfection was conducted using Lipofectamine 3000 reagent (Invitrogen) based on the instructions of the manufacturer. Also, miRNA mimic targeting miR-542-3p (miR-542-3p), miR-542-3p mimic negative control (miR-NC), miRNA inhibitor targeting miR-542-3p (anti-miR-542-3p) and miR-542-3p inhibitor negative control (anti-miR-NC) were purchased from Ribobio Co (Guangzhou, China).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
TRIzol reagent (Invitrogen) was employed to isolate total RNA from paired NB tissues or cells. The SuperScript Reverse Transcriptase Kit (Vazyme, Nanjing, China) was used to generate cDNA for SNHG16, miR-542-3p and ATG5. Also, SNHG16, miR-542-3p and ATG5 expression were analyzed by SYBR Green PCR Master Mix (Vazyme). Relative expression levels of miR-542-3p and ATG5 were figured through the 2−ΔΔCt method. Primers were exhibited as below: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward, 5ʹ-GAAGGTGAAGGTCGGAGTC-3ʹ; reverse, 5ʹ-GAAGATGGTGATGGGATTTC-3ʹ), SNHG16 (forward, 5ʹ-CAGTCAGCCTCAGTTTCCAA-3ʹ; reverse, 5ʹ-AGGCAGGGCTGTGCTGAT-3ʹ), miR-542-3p (forward, 5ʹ-UGUGACAGAUUGAUAACUGAAA-3ʹ; reverse, 5ʹ-GTGCAGGGTCCGAGGT-3ʹ) ATG5 (forward, 5ʹ-CCATCAATCGGAAACTCATGGA-3ʹ; reverse, 5ʹ-ATCTGCAGCCACAGGACGAA-3ʹ), U6 small nuclear RNA (snRNA) (forward, 5ʹ-GCTCGCTTCGGCAGCACA-3ʹ; reverse, 5ʹ-GAGGTATTCGCACCAGAGGA-3ʹ). SNHG16, ATG5 and miR-542-3p were normalized to GAPDH or U6 snRNA.
3-(4, 5-Dimethylthiazol-2-YI)-2, 5-Diphenyltetrazolium Bromide (MTT) Assay
The MTT kit from Promega (Madison, WI, USA) was applied to assess proliferation NB cells. In short, the transfected NB cells (3×103 cells/well) were inoculated into 96-well plates (Corning Costar, Corning, NY, USA) and cultured at 37°C constant temperature with 5% CO2 for 24 h, 48 h and 72 h. Then, each well inoculated with transfected NB cells was added with 20 μL of MTT solution (5 mg/mL) at the appointed time and maintained for 4 h. The supernatants were discarded and dimethyl sulfoxide solution (100 μL) was replenished for the dissolution of the formazan crystal. In the end, the Microplate Absorbance Reader from Thermo Fisher Scientific (Waltham, MA, USA) was conducted to determine the color reaction at 490 nm.
Transwell Assay
The migration and invasion of transfected NB cells were evaluated with transwell chamber (8 μm pore filter) (BD Biosciences, San Jose, CA, USA). Briefly, the RPMI medium with FBS (10%) was then added to the lower chamber as a chemoattractant. Synchronously, transfected NB cells (1×104) were added to the upper chamber of containing RPMI medium with FBS. For the invasion assay, the matrigel matrix (BD Biosciences) was used and placed in the upper chamber. Subsequently, cells were maintained in a 5% CO2 atmosphere at 37°C constant temperature. After that, methanol (100%) was used for the fixation of the NB cells on the lower surface of the membrane. Following this, 0.1% crystal violet was employed to stain. At last, an inverted microscope (Olympus, Tokyo, Japan) was utilized to figure migrated and invasive NB cells.
Western Blot Analysis
Total proteins of transfected NB cells were extracted with RIPA lysis buffer (Beyotime, Shanghai, China). Following this, the BCA assay kit (Pierce, Rockford, IL) was applied to detect the concentration of the total protein. 8–12% of sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to segregate the total proteins. Next, segregated proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After that, tris buffered saline tween (TBST) buffer with 5% skim milk was used for blocking the PVDF membranes. Subsequently, the PVDF membranes with primary antibodies were incubated overnight at 4°C. In addition, β-actin was regarded as a loading control. Primary antibodies were exhibited as below: anti-LC3-I and LC3-II (#2775), anti-β-actin (#4970) and anti-ATG5 (#12994), anti-p62 (#5114) at 4°C overnight. After that, the membranes were washed and incubated with HRP-conjugated secondary antibody (#7074). Above antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). The ImageJ software from National institutes of health (Bethesda, MD, USA) was performed to visualize the bands.
Dual-Luciferase Reporter Assay
The binding sites between miR-542-3p and SNHG16 or ATG5 were predicted by bioinformatics database starBase. Partial sequences of SNHG16 or ATG5 3ʹ untranslated regions (UTR) or their mutant encompassing potential miR-542-3p-binding sites were amplified. Following this, the partial sequences of SNHG16 or ATG5 3ʹUTR was embedded into a pGL3 basic vector (Promega, Fitchburg, WI, USA) for the construction of the wild-type reporter vector of SNHG16 (SNHG16 wt) or ATG5 3ʹUTR (ATG5 wt 3ʹUTR) as well as mutant reporter vector of SNHG16 (SNHG16 mut) or ATG5 3ʹUTR (ATG5 mut 3ʹUTR). After that, NB cells co-transfected miR-NC or miR-542-3p and the luciferase reporter vectors using Lipofectamine 3000 transfection reagent (Invitrogen). Finally, the luciferase activity of NB cells transfected with luciferase reporter vectors was assessed by dual-luciferase Reporter assay kit (Promega).
Xenograft Mice Assay
The animal experiments were approved by the Ethics Committee of Neonatal pediatrics, Central Hospital of Zhoukou City, and were conducted based on the institutional guidelines. BALB/c nude mice (five-week-old, five mice every group) were selected from Shanghai Experimental Animal Center (Shanghai, China) and assigned to two groups: the NC group (injected with SK-N-SH cells transfected with sh-NC) and the sh-SNHG16 group (injected with SK-N-SH cells transfected with stably sh-SNHG16). In short, a number of 5×105 SK-N-SH cells transfected with sh-SN or sh-SNHG16 were suspended in 0.1 mL phosphate buffered saline. Then, transfected SK-N-SH cells were injected subcutaneously into each nude mouse. A digital caliper was used to measure tumor size once per week. After five weeks, the mice were euthanized under anesthesia for tumor resection and weight, and other analyses. The size of tumor was calculated with the equation: Volume = (length ×width2)/2.
Statistical Analysis
The data for this study were derived from at least 3 independent experiments and were displayed as mean ± standard deviation. Statistical data were analyzed using SPSS 17.0 (SPSS, Chicago, IL, USA) and Prism 6.0 (GraphPad, USA). Kaplan-Meier method was applied to assess the overall survival of NB patients with high or low SNHG16 expression. Student’s t-test or one-way ANOVA analysis was employed to compare between two or more groups. Pearson’s correlation analysis was used to assess the relationship between miR-542-3p and SNHG16 or ATG5, and the criterion of statistical significant was P<0.05.
Results
High SNHG16 Expression in NB Tissues and Cells Was Associated with Clinical Stage and Poor Prognosis
To begin with, we utilized qRT-PCR for the investigation of the expression pattern of SNHG16 and its role in NB tumorigenesis and progression. The results displayed that SNHG16 was aberrantly upregulated in 45 NB tissues compared to the corresponding adjacent normal samples (Figure 1A). Furthermore, we divided cases of 45 NB into two groups according to the median expression level of SNHG16: high SNHG16 expression group (n=22) and low SNHG16 expression group (n=23). As indicated in Table 1, the expression level of SNHG16 was connected with international neuroblastoma staging system (INSS) stage (P=0.002) and MYCN (V-myc myelocytomatosis viral-related oncogene, neuroblastoma derived) status (P=0.001), but not associated with age and gender (P>0.05). In addition, Kaplan-Meier survival analysis presented that patients with high expression of SNHG16 had worse overall survival when compared to those with low SNHG16 expression (P=0.03) (Figure 1B). Besides, results of qRT-PCR displayed that an obvious elevation of SNHG16 was observed in NB cell lines (SK-N-SH, IMR-32, SHEP and LAN1) than that in HUVEC cells (Figure 1C). Together, these results indicated that SNHG16 was upregulated in NB tissues and cells, and high SNHG16 expression might be associated with clinical stage and poor prognosis.
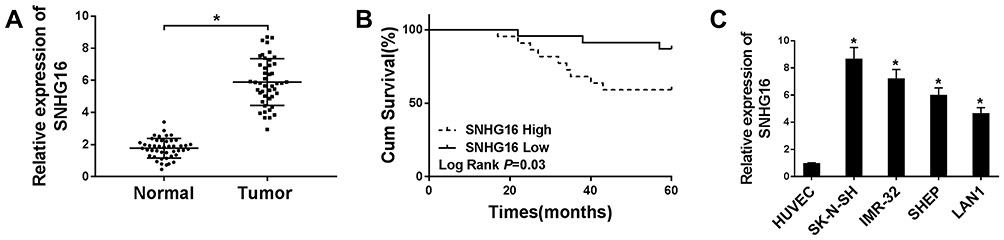 |
Figure 1 High SNHG16 expression was associated with clinical stage and poor prognosis. (A) QRT-PCR was performed to measure SNHG16 expression level in NB tissues and corresponding adjacent normal tissues. (B) Kaplan-Meier was applied to analyze the survival according to the median level of SNHG16 expression. (C) qRT-PCR was used to detect the expression of SNHG16 in NB cell lines and HUVEC cells. *P<0.05. |
Silencing of SNHG16 Suppressed Proliferation, Migration and Invasion of NB Cells
In view of the above results, SK-N-SH and IMR-32 cells with relative higher SNHG16 expression were selected for the exploration of the biological function of SNHG16. In the first place, the specific siRNA for SNHG16 (si-SNHG16#1, si-SNHG16#2 and si-SNHG16#3) was used to silence the expression of SNHG16 in SK-N-SH and IMR-32 cells. As shown in Figure 2A, the expression of SNHG16 was remarkably reduced in SK-N-SH and IMR-32 cells respectively transfected with si-SNHG16#1, si-SNHG16#2 and si-SNHG16#3. Subsequently, SK-N-SH and IMR-32 cells transfected with si-SNHG16#2 were selected for further study. MTT assay was then performed and the results exhibited that knockdown of SNHG16 dramatically inhibited the proliferation of both SK-N-SH and IMR-32 cells (Figure 2B and C). As expected, transwell assay showed that the migration ability of SK-N-SH and IMR-32 cells was conspicuously repressed by the knockdown of SNGH16 (Figure 2D). Moreover, transwell assay for invasion also presented that the invasion ability of SK-N-SH and IMR-32 cells was prominently restrained by SNGH16 silencing (Figure 2E). These results indicated that silencing of SNHG16 suppressed cell proliferation, migration and invasion in NB cells.
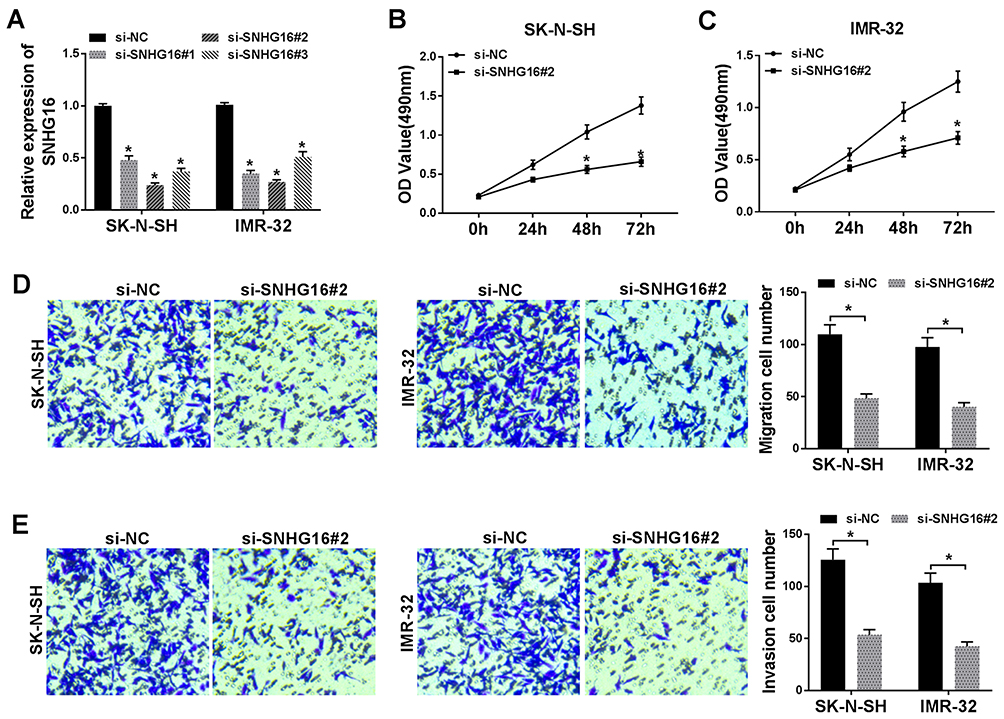 |
Figure 2 Silencing of SNHG16 suppressed proliferation, migration and invasion in NB cells. (A) The transfection efficiency of si-SNHG16#1, si-SNHG16#2 and si-SNHG16#3 in SK-N-SH and IMR-32 cells was determined by qRT-PCR. (B and C) The proliferation of SK-N-SH and IMR-32 cells transfected with si-SNHG16#2 was assessed with MTT assay. (D and E) The migration and invasion of SK-N-SH and IMR-32 cells transfected with si-SNHG16#2 was evaluated by transwell assay. *P<0.05. |
Knockdown of SNGH16 Impeded the Autophagy of NB Cells
To better understand the regulation of SNGH16 on autophagy in NB cells, the expression levels of autophagy relative marker protein ATG5, p62 and microtubule-associated protein A1/1B-light chain3 (LC3-I and LC3-II) were evaluated using Western blot analysis. First, the results of qRT-PCR exhibited that the mRNA levels of ATG5 and LC3 were abnormally decreased while p62 mRNA was enhanced in SK-N-SH and IMR-32 cells transfected with si-SNHG16#2 than the control group (Figure 3A and C). As expected, ATG5 and the ratio of LC3-II/I were markedly reduced while p62 protein was strikingly increased in SK-N-SH and IMR-32 cells by SNHG16 knockdown (Figure 3B and D). These results suggested that knockdown of SNGH16 could constrain the autophagy of NB cells.
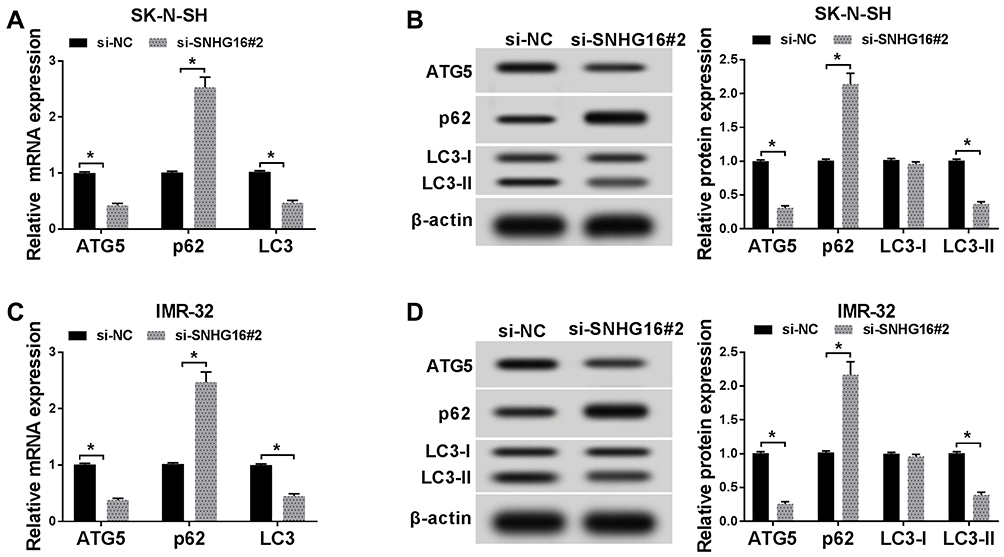 |
Figure 3 Knockdown of SNGH16 impeded the autophagy of NB cells. (A and B) The mRNA and protein expression of ATG5, p62 and LC3 in SK-N-SH cells transfected with si-SNHG16#2 were detected using qRT-PCR or Western blot analysis. (C and D) The mRNA and protein levels of ATG5, p62 and LC3 in IMR-32 cells transfected with si-SNHG16#2 were surveyed with qRT-PCR or Western blot analysis. *P<0.05. |
SNHG16 Acted as a Sponge of miR-542-3p in NB Cells
In order to understand how SNHG16 worked, we explored the potential molecular mechanisms of SNHG16 in the progression of NB. Firstly, an online bioinformatics database starBase was utilized to predict potential target for SNHG16. As shown in Figure 4A, miR-542-3p had potential binding sites for SNHG16. Then, the SNHG16 wild type (SNHG16 wt) luciferase plasmid containing the binding sites of miR-542-3p and SNHG16 mutant (SNHG16 mut) luciferase plasmid were constructed for the execution of the dual-luciferase reporter assay. The results of dual-luciferase reporter assay exhibited that an evident reduction of the luciferase activity of SNHG16 wt in SK-N-SH and IMR-32 cells transfected with miR-542-3p mimics compared with the control group (Figure 4B and C). However, there was no difference in the luciferase activity of SNHG16 mut in SK-N-SH and IMR-32 cells transfected with miR-542-3p mimics (Figure 4B and C). Also, results of qRT-PCR showed that the expression of miR-542-3p was conspicuously upregulated in both SK-N-SH and IMR-32 cells transfected with si-SNHG16#2 when compared to the control group (Figure 4D). Moreover, miR-542-3p expression was apparently downregulated in NB tissues compared with the corresponding adjacent normal samples (Figure 4E). Furthermore, Pearson’s correlation analysis showed that the expression of SNHG16 and miR-542-3p was negatively correlated in NB tissues (Figure 4F). These results indicated that miR-542-3p was negatively regulated by SNHG16 in NB cells.
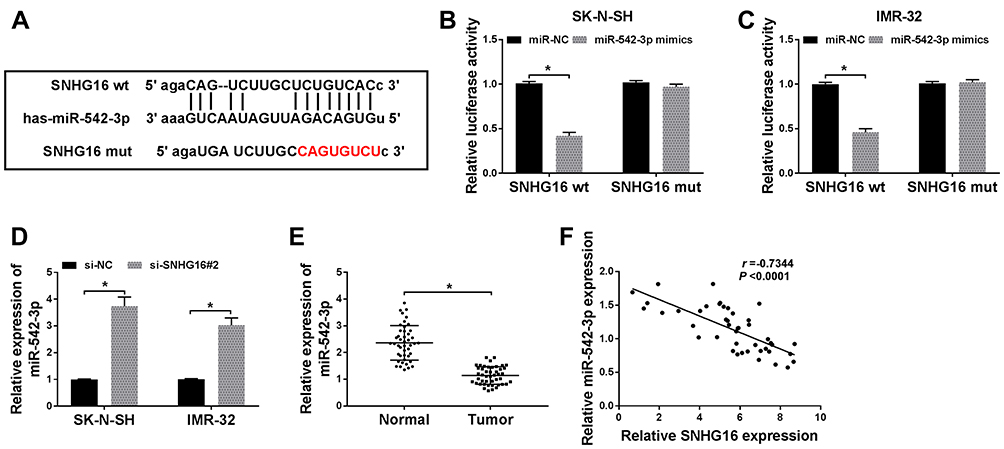 |
Figure 4 SNHG16 acted as a sponge of miR-542-3p in NB cells. (A)The binding sites between SNHG16 and miR-542-3p were predicted by starBase database. The red sequence indicated that the mutation in TUG1 was at the predicted miR-542-3p binding sites. (B and C) The luciferase activity in SK-N-SH and IMR-32 cells co-transfected miR-542-3p mimics or miR-NC and SNHG16 wt or SNHG16 mut was determined with dual-luciferase reporter assay. (D) The expression of SNHG16 mRNA in SK-N-SH and IMR-32 cells transfected with si-SNHG16#2 or si-NC was detected using qRT-PCR. (E) QRT-PCR was used to assess the expression of miR-542-3p in NB tissues and corresponding adjacent normal tissues. (F) Pearson’s correlation analysis was performed for the assessment of the relationship between SNHG16 and miR-542-3p in NB tissues. *P<0.05. |
Downregulation of miR-542-3p Reversed the Effects of SNHG16 Silencing on Proliferation, Migration, Invasion and Autophagy of NB Cells
To further analyze whether SNHG16 affected the proliferation, migration, invasion and autophagy of NB cells via miR-542-3p, we transfected miR-NC, miR-542-3p mimics, si-SNHG16#2+anti-NC or si-SNHG16#2+anti-miR-542-3p into SK-N-SH and IMR-32 cells. QRT-PCR was then conducted and the results exhibited that the expression of miR-542-3p was specially augmented by transfecting with miR-542-3p mimics in both SK-N-SH and IMR-32 cells (Figure 5A and B). Besides, knockdown of SNHG16 apparently reinforced miR-542-3p expression in SK-N-SH and IMR-32 cells, while this increase was reversed by miR-542-3p downregulating (Figure 5A and B). Subsequently, the effects of miR-542-3p inhibition on SNHG16 reduction-medicated proliferation, migration, invasion and autophagy and miR-542-3p of NB cells were further explored. Results of MTT assay revealed that a significant inhibition of the proliferation of SK-N-SH and IMR-32 cells by the overexpression of miR-542-3p (Figure 5C and D). Moreover, the inhibitory effect of SNHG16 knockdown on proliferation was overturned by the downregulation of miR-542-3p (Figure 5C and D). Then, we found that the migration and invasion of SK-N-SH and IMR-32 cells were distinctly constrained by miR-542-3p overexpression using transwell assay, and miR-542-3p downregulation receded the inhibitory effects of SNHG16 knockdown on migration and invasion of SK-N-SH and IMR-32 cells (Figure 5E and F). In addition, Western blot analysis showed that the ratio of LC3-II/I and the protein expression of ATG5 in SK-N-SH and IMR-32 cells transfected with miR-542-3p mimics were markedly reduced, whereas p62 protein was distinctly enhanced (Figure 5G and H). The effect of SNHG16 silencing on autophagy of NB cells was restored by miR-542-3p knockdown (Figure 5G and H). Taken together, these findings suggested that downregulation of miR-542-3p reversed the inhibitory effects of SNHG16 silencing on proliferation, migration, invasion and autophagy of NB cells.
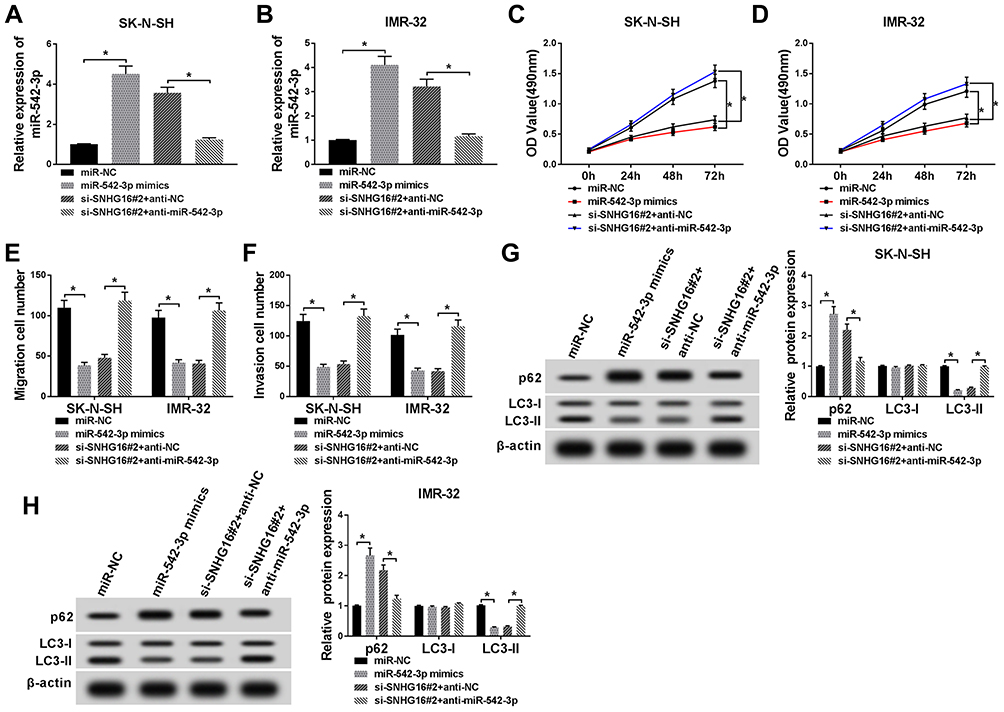 |
Figure 5 Downregulation of miR-542-3p reversed the effects of SNHG16 knockdown on NB cells. SK-N-SH and IMR-32 cells were transfected with miR-NC, miR-542-3p mimics, si-SNHG16#2+anti-NC or si-SNHG16#2+anti-miR-542-3p, respectively. (A and B) QRT-PCR was applied for the determination of the expression miR-542-3p in SK-N-SH and IMR-32 cells. (C and D) MTT assay was conducted to evaluate the proliferation ability of SK-N-SH and IMR-32 cells. (E and F) Transwell assay was used to determine the migration and invasion of SK-N-SH and IMR-32 cells. (G and H) Western blot analysis was utilized to measure the protein expression of LC3-I, LC3-II and p62 in SK-N-SH and IMR-32 cells. *P<0.05. |
SNHG16 Regulated ATG5 Expression via Binding to miR-542-3p
Based on the competition principle of competing endogenous RNA (ceRNA), we concluded that SNHG16 involved in the progression of NB by binding to miR-542-3p and regulating the expression of the target of miR-542-3p. As shown in Figure 6A, results of bioinformatics database starBase showed that miR-542-3p harbored potential binging sites in the 3ʹ-UTR of ATG5. To verify whether ATG5 was a target of miR-542-3p, the dual-luciferase reporter assay was conducted on SK-N-SH and IMR-32 cells. The results disclosed that in the miR-542-3p mimics group, the luciferase activity driven by ATG5 wt 3ʹUTR was dramatically reduced compared with the control group. However, the luciferase activity driven by ATG5 mut 3ʹUTR was no difference, indicating that ATG5 was a target of miR-542-3p (Figure 6B and C). To determine whether ATG5 was regulated by miR-542-3p, SK-N-SH and IMR-32 cells were transfected with miR-NC or miR-542-3p mimics. Western blot analysis exhibited that the expression level of ATG5 protein was apparently downregulated in both SK-N-SH and IMR-32 cells by miR-542-3p overexpression (Figure 6D). To further confirm if SNHG16 regulated ATG5 protein expression via miR-542-3p, we transfected si-NC, si-SNHG16#2, si-SNHG16#2+anti-NC or si-SNHG16#2+anti-miR-542-3p into SK-N-SH and IMR-32 cells. Results of Western blot analysis presented that knockdown of SNHG16 distinctly decreased the protein expression of ATG5, while this reduction was abolished by the downregulation of miR-542-3p (Figure 6E). Then, the mRNA level of ATG5 in NB tissues and the corresponding adjacent normal samples was assessed by qRT-PCR. We found that ATG5 mRNA was markedly increased in NB tissues than that in the corresponding adjacent normal samples (Figure 6F). Besides, Pearson’s correlation analysis suggested that in NB tissues, ATG5 expression was positively correlated with SNHG16 and negatively correlated with miR-542-3p (Figure 6G and H). These data demonstrated that SNHG16 could regulate ATG5 expression via binding to miR-542-3p.
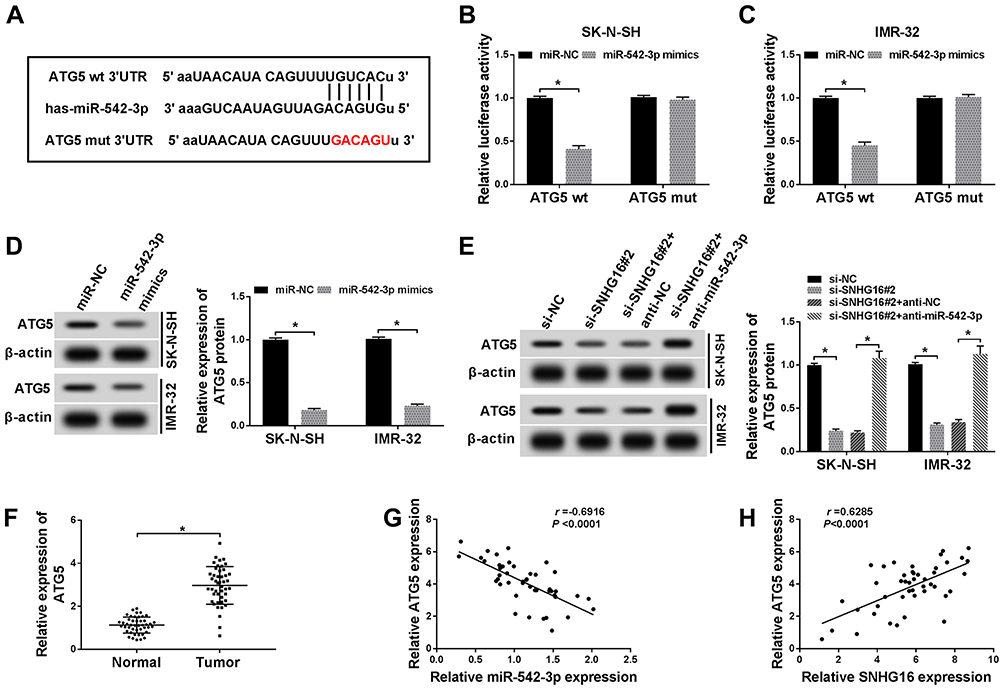 |
Figure 6 SNHG16 regulated ATG5 expression via binging to miR-542-3p. (A) The binding sites between ATG5 and miR-542-3p were predicted by starBase database. The red sequence indicated that the mutation in ATG5 was at the predicted miR-542-3p binding sites. (B and C) The luciferase activity was assessed by dual-luciferase reporter assay in SK-N-SH and IMR-32 cells co-transfected miR-542-3p mimics or miR-NC and ATG5 wt 3ʹUTR or ATG5 mut 3ʹUTR. (D) Western blot analysis was applied to detect ATG5 protein level in SK-N-SH and IMR-32 cells transfected with miR-NC or miR-542-3p mimics. (E) Western blot analysis of ATG5 protein expression in SK-N-SH and IMR-32 cells transfected with si-NC, si-SNHG16#2, si-SNHG16#2+anti-NC or si-SNHG16#2+anti-miR-542-3p, respectively. (F) QRT-PCR was utilized for the measurement of the expression level of ATG5 mRNA in NB tissues and corresponding adjacent normal tissues. (G and H) Pearson’s correlation analysis was used to determine the relationship between miR-542-3p and ATG5 and SNHG16 and ATG5 in NB tissues. *P<0.05. |
ATG5 Overturned the Regulatory Effects on Proliferation, Migration, Invasion and Autophagy of NB Cells by SNHG16 or miR-542-3p Knockdown
Since SNHG16 binds to miR-542-3p to regulate ATG5 expression, we further analyzed the role of ATG5 in the progression of NB. First, the expression of ATG5 protein was measured using Western blot analysis in SK-N-SH and IMR-32 cells transfected with si-SNHG16#2+pcDNA, si-SNHG16#2+pcDNA-ATG5, anti-miR-542-3p+anti-NC or anti-miR-542-3p+anti-ATG5. The results showed that ATG5 protein expression was remarkably elevated in both SK-N-SH and IMR-32 cells transfected with si-SNHG16#2+pcDNA-ATG5 than the control group (Figure 7A and B). Besides, compared with the control group, SK-N-SH and IMR-32 cells transfected with anti-miR-542-3p+anti-ATG5 significantly reduced ATG5 protein expression (Figure 7A and B). Effects of ATG5 on SNHG16 or miR-542-3p knockdown-mediated proliferation, migration, invasion and autophagy of NB cells were explored with MTT, transwell or Western blot assay. As a result, overexpression of ATG5 recovered SNHG16 knockdown-mediated the inhibitory effect on proliferation of SK-N-SH and IMR-32 cells (Figure 7C and D). Moreover, knockdown of ATG5 reversed the repressive effect of miR-542-3p downregulation on proliferation of SK-N-SH and IMR-32 cells (Figure 7C and D). Meanwhile, increased ATG5 expression regained the effect of SNHG16 knockdown on migration and invasion of SK-N-SH and IMR-32 cells. However, interference of ATG5 reversed miR-542-3p downregulation-induced migration and invasion of SK-N-SH and IMR-32 cells (Figure 7E and F). In addition, compared with the si-SNHG16#2+pcDNA group, SK-N-SH and IMR-32 cells transfected with si-SNHG16#2+pcDNA-ATG5 strikingly enhanced LC3-II/I ratio and reduced p62 protein level (Figure 7G and H). However, ATG5 interference distinctly decreased the ratio of LC3-II/I and increased p62 protein level in both SK-N-SH and IMR-32 cells (Figure 7G and H). These data indicated that overexpression or knockdown ATG5 could overturn the regulatory effects on proliferation, migration, invasion and autophagy of NB cells by SNHG16 and miR-542-3p knockdown.
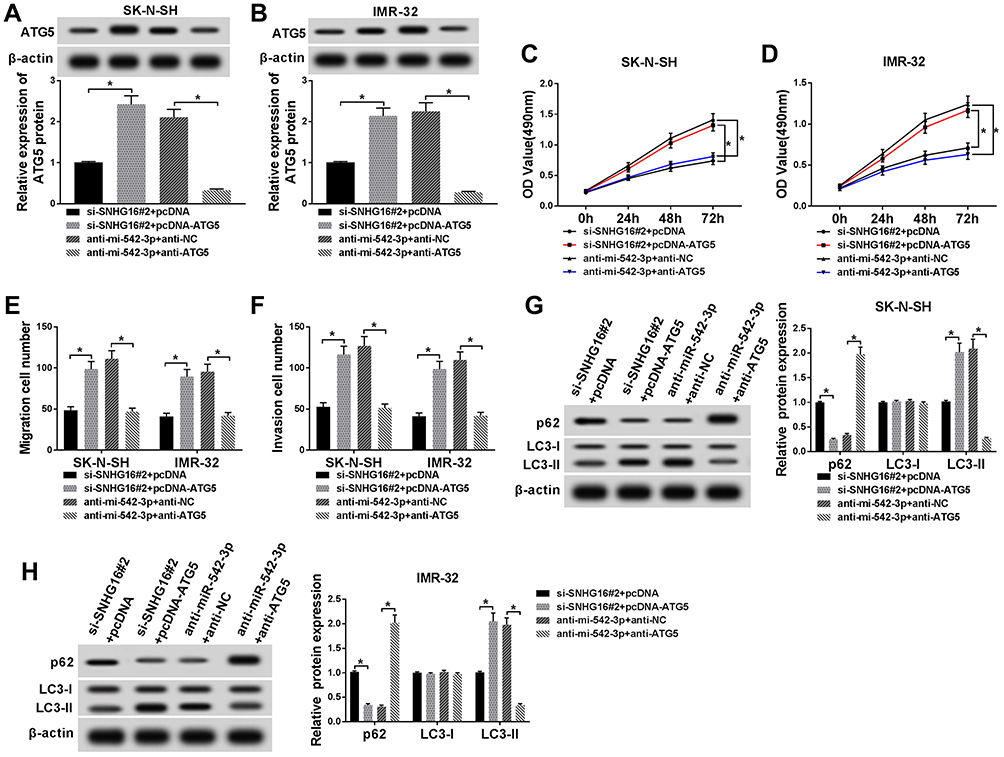 |
Figure 7 Effects of ATG5 on SNHG16 or miR-542-3p knockdown-mediated proliferation, migration, invasion and autophagy of NB cells. SK-N-SH and IMR-32 cells were transfected with si-SNHG16#2+pcDNA, si-SNHG16#2+pcDNA-ATG5, anti-miR-542-3p+anti-NC or anti-miR-542-3p+anti-ATG5, respectively. (A and B) ATG5 protein expression level in SK-N-SH and IMR-32 cells was assessed by Western blot analysis. (C and D) MTT assay was conducted for the determination of the proliferation of SK-N-SH and IMR-32 cells. (E and F) Transwell assay was performed for the assessment of the migration and invasion of SK-N-SH and IMR-32 cells. (G and H) Western blot analysis was used for the detection of the protein expression levels of LC3-I, LC3-II and p62 in SK-N-SH and IMR-32 cells. *P<0.05. |
Knockdown of SNHG16 Suppressed Tumor Growth of NB in vivo
Given that SNHG16 involved in NB via targeting miR-542-3p/ATG5, we constructed xenograft mice models to confirm the role of SNHG16 in vivo. SK-N-SH cells stably transfected with sh-SNHG16 or sh-NC were injected subcutaneously into each nude mouse (5 per group). The results displayed that the volume and weight were specially reduced in SK-N-SH cells transfected with sh-SNHG16 compared to that in the sh-NC group (Figure 8A and B). Moreover, compared to the sh-NC group, SK-N-SH cells transfected with sh-SNHG16 significantly downregulated SNHG16 expression while increased miR-542-3p expression (Figure 8C and D). Besides, the mRNA level of ATG5 and LC3 were markedly decreased while p62 was enhanced by SNHG16 knockdown in SK-N-SH cells (Figure 8E). Consistently, knockdown of SNHG16 significantly reduced the ratio of LC3-II/I and ATG5 protein level while elevated p62 protein expression (Figure 8F). Therefore, knockdown of SNHG16 suppressed tumor growth of NB in vivo.
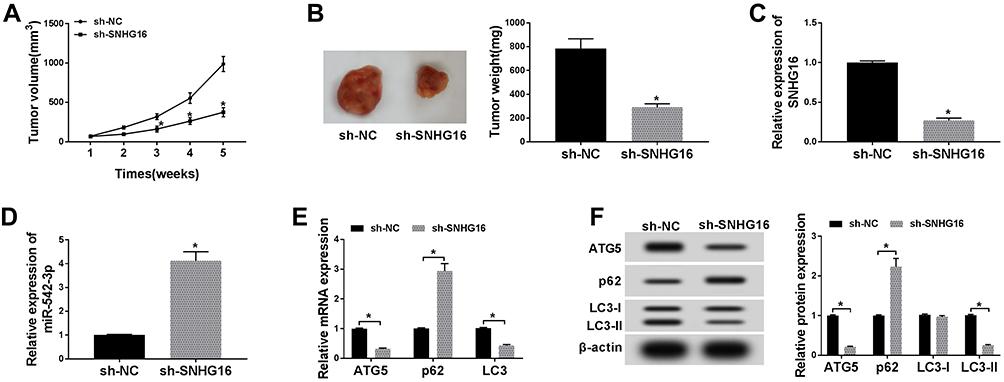 |
Figure 8 Knockdown of SNHG16 suppressed tumor growth of NB in vivo. (A) Tumor volume curves for the sh-NC and sh-SNHG16 groups. (B) Tumor weight and picture of the sh-NC and sh-SNHG16 groups. (C and D) The expression of SNHG16 and miR-542-3p was measured with qRT-PCR in the sh-SNHG16 group and control group. (E) QRT-PCR was utilized for the measurement of the mRNA expression levels of ATG5, LC3 and p62 in tumor tissues of sh-SNHG16 group and control group. (F) Western blot analysis was utilized to detect the protein expression levels of ATG5, LC3-I, LC3-II and p62 in tumor tissues of sh- SNHG16 group and control group. *P<0.05. |
Discussion
NB is the most common heterogeneous solid tumors in children. At present, although great progress has been made in treating NB at low to moderate risk, advanced patients still have a poor prognosis. LncRNA is a vital regulator in a variety of tumors, including NB. For instance, lncRNA XIST promoted the migration, growth and invasion of NB cells by regulating H3 histone methylation.27 Pan et al revealed that knockdown of lncRNA RMRP inactivated ERK1/2 pathway and regulated the miR-206/TACR1 axis to hinder the progression of NB.28 In the current study, the effect of SNHG16 in NB and its related molecular mechanism were explored.
Increasing evidence showed the essential roles of aberrant SNHG16 expression in the development of diverse tumors. For example, SNHG16 was elevated in hepatocellular carcinoma tissues and cell lines, and overexpression of SNHG16 promoted cell invasion, proliferation and migration in hepatocellular carcinoma cells by inhibiting miR-186 expression.29 Another study showed that silencing of SNHG16 could inhibit cell migration, proliferation and invasion, and induced cell apoptosis in osteosarcoma cells by sponging miR-340.13 In this present study, we found that a marked elevation of SNHG16 in NB tissues and cells. Furthermore, high SNHG16 expression was associated with poor prognosis and clinical stage. Moreover, knockdown of SNHG16 suppressed tumor growth in vivo and impeded proliferation, migration and invasion of NB cells in vitro. Yu et al revealed that the high SNHG16 expression was positively correlated with poor clinical prognosis, and knockdown of SNHG16 promoted cell cycle arrest in G0/G1 phase, and constrained migration and proliferation in NB cells.14
Autophagy is a highly conserved biological mechanism that breaks down unnecessary or dysfunctional components of cells for survival in conditions of hypoxia, starvation, nutrient deficiency.30 It has been reported that autophagy not only prevents malignant transformation but also enhances the adaptation of tumor cells to the undesirable microenvironment and promotes tumor progression.31 The transformation of LC3-I to LC3-II was positively correlated with the number of autophagosomes.32 Ubiqutination substrates can be promoted to transfer to autophagosomes by p62, which can be used as a marker of autophagy degradation.33 Qin et al revealed that knockdown of ATG5 weakened the activation of autophagy in papillary thyroid cancer cells.34 In this study, ATG5 expression and LC3-II/I ratio was markedly reduced while p62 protein was strikingly increased by SNHG16 knockdown in NB cells, it indicated that knockdown of SNGH16 impeded the autophagy of NB cells.
Additionally, it was indicated that lncRNAs function as ceRNA to modulate the expression of the target of miRNAs.35 In the present study, SNHG16 acted as a sponge of miR-542-3p was confirmed by dual-luciferase reporter assay. Moreover, miR-542-3p was downregulated in NB tissues. Besides, the expression of SNHG16 was negatively correlated with miR-542-3p in NB tissues. Furthermore, miR-542-3p silencing reversed the effects of SNHG16 knockdown on proliferation, migration, invasion and autophagy of NB cells. Report of Althoff et al revealed that miR-542-3p played an anti-tumor role in NB, and miR-542-3p downregulated survivin expression to inhibit activity and proliferation, and induce cell apoptosis in NB cells.21 In addition, miR-542-3p was markedly reduced in hepatocellular carcinoma tissues and cells, and inhibited miR-542-3p via activating TGF-β/Smad signaling to accelerate the metastasis of tumor.19 Li et al also claimed that miR-542-3p played tumor suppressor role via targeting CDK14 in ovarian cancer.18 In the current study, these results demonstrated that miR-542-3p played a tumor suppressor gene in NB, which was consistent with the above studies.
Furthermore, we found miR-542-3p targeted ATG5 by starBase database and confirmed this prediction using dual-luciferase reporter assay. ATG5, an autophagy-regulated protein, exist carcinogenesis in different cancers. For instance, ATG5 was regulated by lncRNA DICER1-AS1 and miR-30b, and lncRNA DICER1-AS1 was connected with cell invasion, autophagy and proliferation in osteosarcoma.23 ATG5 taken part in autophagy and chemosensitize of pancreatic cancer cells and was regulated by miR-137.36 In the present study, ATG5 was upregulated in NB tissues, and knockdown of SNHG16 decreased ATG5 protein expression while this reduction was abolished by miR−542−3p silencing. Additionally, ATG5 overturned the regulatory effects on proliferation, migration, invasion and autophagy of NB cells by SNHG16 or miR−542−3p knockdown. Cheng et al revealed that ATG5 was elevated in NB tissues and cells, and overexpression of ATG5 overturned the effects on proliferation, migration, invasion and autophagy of NB cells by miR−34a addition, our results were in line with them.26 Therefore, we concluded that SNHG16 upregulated ATG5 via sponging miR-542-3p to boost cell proliferation, migration, invasion and autophagy in NB cells (Figure 9).
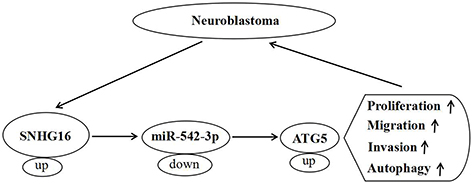 |
Figure 9 A schematic diagram of SNHG16 in the progression of ESCC. |
In conclusion, SNHG16 was upregulated in NB tissues and cells, and knockdown of SNHG16 impeded proliferation, migration, invasion and autophagy of NB cells in vitro, and suppressed tumor growth in vivo. Interestingly, SNHG16 regulated ATG5 expression via sponging miR-542-3p. Moreover, downregulation of miR-542-3p reversed the inhibitory effect of SNHG16 silencing on proliferation, migration, invasion and autophagy of NB cells. Besides, ATG5 overturned the regulatory effects on proliferation, migration, invasion and autophagy of NB cells induced by SNHG16 or miR-542-3p knockdown. Therefore, we concluded that SNHG16 facilitated proliferation, migration, invasion and autophagy of NB cells via sponging miR-542-3p and upregulating ATG5 expression, providing a new possible strategy for the detection and treatment of NB.
Disclosure
The authors have no conflict of interest to declare.
References
1. Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362(23):2202–2211. doi:10.1056/NEJMra0804577
2. Ritenour LE, Randall MP, Bosse KR, et al. Genetic susceptibility to neuroblastoma: current knowledge and future directions. Cell Tissue Res. 2018;372(2):287–307. doi:10.1007/s00441-018-2820-3
3. Schleiermacher G, Janoueix-Lerosey I, Delattre O. Recent insights into the biology of neuroblastoma. Int J Cancer. 2014;135(10):2249–2261. doi:10.1002/ijc.29077
4. Swift CC, Eklund MJ, Kraveka JM, et al. Updates in diagnosis, management, and treatment of neuroblastoma. Radiographics. 2018;38(2):566–580. doi:10.1148/rg.2018170132
5. Newton TC, Wolcott K, Roberts SS. Comparison of the side populations in pretreatment and postrelapse neuroblastoma cell lines. Transl Oncol. 2010;3(4):246–251. doi:10.1593/tlo.09301
6. Oh JM, Lee J. Ginsenoside Rk1 induces apoptosis in neuroblastoma cells through loss of mitochondrial membrane potential and activation of caspases. Int J Mol sci. 2019;20(5).
7. Salehi S, Taheri MN, Azarpira N, et al. State of the art technologies to explore long non-coding RNAs in cancer. J Cell Mol Med. 2017;21(12):3120–3140. doi:10.1111/jcmm.13238
8. Kunej T, Obsteter J, Pogacar Z, et al. The decalog of long non-coding RNA involvement in cancer diagnosis and monitoring. Crit Rev Clin Lab Sci. 2014;51(6):344–357. doi:10.3109/10408363.2014.944299
9. Christensen LL, True K, Hamilton MP, et al. SNHG16 is regulated by the Wnt pathway in colorectal cancer and affects genes involved in lipid metabolism. Mol Oncol. 2016;10(8):1266–1282. doi:10.1016/j.molonc.2016.06.003
10. Lian D, Amin B, Du D, et al. Enhanced expression of the long non-coding RNA SNHG16 contributes to gastric cancer progression and metastasis. Cancer Biomark. 2017;21(1):151–160. doi:10.3233/CBM-170462
11. Han W, Du X, Liu M, et al. Increased expression of long non-coding RNA SNHG16 correlates with tumor progression and poor prognosis in non-small cell lung cancer. Int J Biol Macromol. 2019;121:270–278. doi:10.1016/j.ijbiomac.2018.10.004
12. Feng F, Chen A, Huang J, et al. Long noncoding RNA SNHG16 contributes to the development of bladder cancer via regulating miR-98/STAT3/Wnt/beta-catenin pathway axis. J Cell Biochem. 2018;119(11):9408–9418. doi:10.1002/jcb.27257
13. Su P, Mu S, Wang Z. Long noncoding RNA SNHG16 promotes osteosarcoma cells migration and invasion via sponging miRNA-340. DNA Cell Biol. 2019;38(2):170–175. doi:10.1089/dna.2018.4424
14. Yu Y, Chen F, Yang Y, et al. lncRNA SNHG16 is associated with proliferation and poor prognosis of pediatric neuroblastoma. Int J Oncol. 2019;55(1):93–102. doi:10.3892/ijo.2019.4813
15. Cai Y, Yu X, Hu S, et al. A brief review on the mechanisms of miRNA regulation. Genomics Proteomics Bioinf. 2009;7(4):147–154. doi:10.1016/S1672-0229(08)60044-3
16. Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi:10.1146/annurev-biochem-060308-103103
17. Li J, Tan S, Kooger R, et al. MicroRNAs as novel biological targets for detection and regulation. Chem Soc Rev. 2014;43(2):506–517. doi:10.1039/C3CS60312A
18. Li J, Shao W, Feng H. miR-542-3p, a microRNA targeting CDK14, suppresses cell proliferation, invasiveness, and tumorigenesis of epithelial ovarian cancer. Biomed Pharmacother. 2019;110:850–856. doi:10.1016/j.biopha.2018.11.104
19. Zhang T, Liu W, Meng W, et al. Downregulation of miR-542-3p promotes cancer metastasis through activating TGF-beta/Smad signaling in hepatocellular carcinoma. Onco Targets Ther. 2018;11:1929–1939. doi:10.2147/OTT.S154416
20. Liu B, Li J, Zheng M, et al. miR-542-3p exerts tumor suppressive functions in non-small cell lung cancer cells by upregulating FTSJ2. Life Sci. 2017;188:87–95. doi:10.1016/j.lfs.2017.08.018
21. Althoff K, Lindner S, Odersky A, et al. miR-542-3p exerts tumor suppressive functions in neuroblastoma by downregulating survivin. Int J Cancer. 2015;136(6):1308–1320. doi:10.1002/ijc.29091
22. Liao Z, Dai Z, Cai C, et al. Knockout of Atg5 inhibits proliferation and promotes apoptosis of DF-1 cells. In Vitro Cell Dev Biol Anim. 2019;55(5):341–348. doi:10.1007/s11626-019-00342-7
23. Gu Z, Hou Z, Zheng L, et al. LncRNA DICER1-AS1 promotes the proliferation, invasion and autophagy of osteosarcoma cells via miR-30b/ATG5. Biomed Pharmacother. 2018;104:110–118. doi:10.1016/j.biopha.2018.04.193
24. Luo M, Wu L, Zhang K, et al. miR-216b enhances the efficacy of vemurafenib by targeting Beclin-1, UVRAG and ATG5 in melanoma. Cell Signal. 2018;42:30–43. doi:10.1016/j.cellsig.2017.09.024
25. Zheng Y, Tan K, Huang H. Long noncoding RNA HAGLROS regulates apoptosis and autophagy in colorectal cancer cells via sponging miR-100 to target ATG5 expression. J Cell Biochem. 2019;120(3):3922–3933. doi:10.1002/jcb.v120.3
26. Cheng X, Xu Q, Zhang Y, et al. miR-34a inhibits progression of neuroblastoma by targeting autophagy-related gene 5. Eur J Pharmacol. 2019;850:53–63. doi:10.1016/j.ejphar.2019.01.071
27. Zhang J, Li WY, Yang Y, et al. LncRNA XIST facilitates cell growth, migration and invasion via modulating H3 histone methylation of DKK1 in neuroblastoma. Cell Cycle. 2019;18(16):1882–1892. doi:10.1080/15384101.2019.1632134
28. Pan J, Zhang D, Zhang J, et al. LncRNA RMRP silence curbs neonatal neuroblastoma progression by regulating microRNA-206/tachykinin-1 receptor axis via inactivating extracellular signal-regulated kinases. Cancer Biol Ther. 2019;20(5):653–665. doi:10.1080/15384047.2018.1550568
29. Chen H, Li M, Huang P. LncRNA SNHG16 promotes hepatocellular carcinoma proliferation, migration and invasion by regulating miR-186 expression. J Cancer. 2019;10(15):3571–3581. doi:10.7150/jca.28428
30. Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25(19):1999–2010. doi:10.1101/gad.17558811
31. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi:10.1016/j.cell.2010.01.028
32. Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10(3):431–441. doi:10.4161/auto.27344
33. Qin Y, Sun W, Zhang H, et al. LncRNA GAS8-AS1 inhibits cell proliferation through ATG5-mediated autophagy in papillary thyroid cancer. Endocrine. 2018;59(3):555–564. doi:10.1007/s12020-017-1520-1
34. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–352. doi:10.1038/nature12986
35. Wang ZC, Huang FZ, Xu HB, et al. MicroRNA-137 inhibits autophagy and chemosensitizes pancreatic cancer cells by targeting ATG5. Int J Biochem Cell Biol. 2019;111:63–71. doi:10.1016/j.biocel.2019.01.020
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.