")
Back to Journals » International Journal of General Medicine » Volume 14
LncRNA UCA1 Accelerates the Progression of Ulcerative Colitis via Mediating the miR-331-3p/BRD4 Axis
Authors Rao J, Shao L, Lin M, Huang J, Fan L
Received 1 February 2021
Accepted for publication 6 April 2021
Published 10 June 2021 Volume 2021:14 Pages 2427—2435
DOI https://doi.org/10.2147/IJGM.S304837
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Jun Rao, Lihua Shao, Min Lin, Jin Huang, Li Fan
Department of Gastroenterology, The Affiliated Changzhou No.2 People’s Hospital of Nanjing Medical University, Changzhou, People’s Republic of China
Correspondence: Li Fan
Department of Gastroenterology, The Affiliated Changzhou No.2 People’s Hospital of Nanjing Medical University, No. 68 Gehu Road, Wujin District, Changzhou, Jiangsu, 213000, People’s Republic of China
Email [email protected]
Background: Ulcerative colitis (UC) has become one of the fastest-growing severe diseases worldwide with high morbidity. This research aimed to explore the function of lncRNA UCA1 in UC progression.
Methods: RT-qPCR analysis was used to examine the expression of UCA1 level in colonic mucosa tissues of UC patients. Then, fetal human cells (FHCs) were stimulated by LPS to induce inflammatory injury. CCK-8, flow cytometry and ELISA were adopted to determine the influence of UCA1 depletion on cell viability, apoptosis and pro-inflammatory factors levels in LPS-induced FHCs. The interaction between UCA1 and miR-331-3p or BRD4 was confirmed by luciferase reporter assay. The expressions of key factors involved in NF-κB pathway were assessed by Western blotting.
Results: LncRNA UCA1 level was elevated in colonic mucosa tissues of UC patients. LPS stimulation restrained cell viability and promoted the apoptosis and inflammatory factors levels, thus inducing FHCs inflammatory injury, while these effects were partially abolished by UCA1 knockdown. Moreover, it was found that UCA1 silence improved LPS-triggered cell injury via miR-331-3p. In addition, BRD4 was directly targeted by miR-331-3p, and BRD4 deficiency neutralized the effects of miR-331-3p repression on LPS-triggered injury in LPS-treated FHCs.
Conclusion: Our data determined that UCA1 knockdown attenuated UC development via targeting the miR-331-3p/BRD4/NF-κB pathway.
Keywords: UCA1, ulcerative colitis, miR-331-3p, BRD4
Introduction
Ulcerative colitis (UC) is a chronic idiopathic inflammatory disease of the rectal and colonic mucosa,1,2 and the incidence of UC is rising rapidly in Asian countries, especially China.3,4 Despite several pathogenic factors, such as viral and bacterial infection, autoimmunity, and vascular injury, have been indicated to contribute to the occurrence and development of UC,5,6 the molecular and pathophysiological mechanisms of UC are not fully understood.
Long noncoding RNAs (lncRNAs) are a subset of ncRNAs with a transcription length of >200 nts and lack protein-coding capacity.7 Abundant evidence has revealed that lncRNAs are involved in UC development. For example, lncRNA TUG1 attenuated intestinal epithelial cell apoptosis and UC progression through modulating miR-29b-3p/CDK2 axis.8 LncRNA MEG3 supplementation relieved the serve ulceration of UC rat colons via increasing IL-10 level by targeting miR-98-5p.9 UCA1 is located in chromosome 19p13, which was first uncovered as a new lncRNA gene significantly upregulated in bladder carcinoma using RT-qPCR by Wang.10 Recent research has reported that UCA1 is upregulated by NT/NTR1 activation in human colonocytes and overexpressed in the colonic mucosa from UC patients.11 Nevertheless, the mechanism of UCA1 in UC remains unknown.
MiRNAs are another category of ncRNAs of ~23 nts in length that serve as a regulatory role in cells via directly interacting with the 3′-UTR of target mRNAs, leading to mRNA degradation or posttranscriptional suppression.12 Several studies suggested that miRNAs play crucial roles in the incidence and development of UC. For instance, miR-142-5p improved the intestinal inflammation of active-UC patients by decreasing SOCS1 expression.13 miRNA-650 contributed to inflammation-induced cell apoptosis in UC by regulating NLRP6.14 miR-331-3p was uncovered to be involved in the inflammatory response processes of multiple diseases. Nie et al exhibited that miR-331-3p inhibits the levels of inflammatory factors TNF-α and IL-6 after cerebral hemorrhage via targeting NLRP6.15 However, the function of miR-331-3p in the regulation of inflammatory injury in UC is unclear.
The current research explored the role of UCA1 in LPS-triggered injury in FHCs to deepen the understanding of the molecular mechanism of UC.
Materials and Methods
Patient Tissue Samples
This study was approved by The Affiliated Changzhou No.2 People’s Hospital of Nanjing Medical University Ethics Committee and conducted in accordance with the Declaration of Helsinki. Colonic mucosa pinch biopsies were obtained from the sigmoid colons of 46 UC patients (severe condition; gender: 28 males and 18 females; age: 44 to 61 years; mean± SD: 50.5±8.7 years) and 46 healthy controls (gender: 29 males and 17 females; age: 45 to 54 years; mean± SD: 52.3±11.3 years) as described previously.13 Biopsies were deposited at −80 °C. Before collection, informed consent from all patients was gained.
Cell Culture
Human colorectal FHCs were obtained from the ATCC (Manassas, VA, USA) and cultured in DMEM (Solarbio, Beijing, China) supplemented with 10% FBS (Gibco, Carlsbad, CA), 100 mg/mL streptomycin and 100U/mL penicillin at 37°C with 5% CO2. LPS (1, 3, 6, 8, and 10 ng/mL, Solarbio, Beijing, China) was used to stimulate FHCs for 24 h.
Cell Transfection
Short-hairpin RNA against UCA1 (shUCA1; 5′-GCCCCUCAGCAGUCCAGCCAU-3′) or BRD4 (shBRD4; 5′-AGCAUUUUGCUUCUUGAUGUU-3′) and their negative control (shNC; 5′-AACAGUCGCGUUUGCGACUGG-3′), miR-331-3p mimics (5′-ACAGCAGGCACAGACAGGCAGU-3′) or NC mimics (5′-CGAUCGCAUCAGCAUCGAUUGC-3′) and miR-331-3p inhibitor (5′-ACUGAAUGUCUGUGCCUGCUGU-3′) or NC inhibitor (5′-CUAACGCAUGCACAGUCGUACG-3), pcDNA3.1, pcDNA3.1/UCA1 were synthesized by GenePharma (Shanghai, China). Cell transfection was performed using Lipofectamine® 3000 (Invitrogen).
CCK-8 Assay
FHCs (1×106/mL) were seeded in 96-well plates. After LPS treatments, FHCs in each well were incubated with 100 μL/mL medium, CCK-8 reagent (Sigma, USA) was added into each well for 2 h. Then, the OD value at 450 nm was assessed via a microplate reader (Bio-Rad).
RT-qPCR Assay
Total RNA from patient tissues or FHCs was harvested was extracted using TRIzol® reagent (Invitrogen) and then reverse-transcribed into cDNA using SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific, Inc.). RT-qPCR was conducted using a TaqMan® Universal PCR Master mix II (Thermo Fisher Scientific). The relative expression of RNAs (normalized to GAPDH or U6) were analyzed using the 2−ΔΔCq method.16 The following primer sequences were used: UCA1 forward, 5ʹ-CTCTCCATTGGGTTCACCATTC-3ʹ and reverse, 5ʹ- GCGGCAGGTCTTAAGAGATGAG-3ʹ; miR-331-3p forward, 5ʹ‑CGTTTCTTCCTTGTAAGGA ‑3ʹ and reverse, 5ʹ‑CCCAAGCTTGTTGGAGAACAGCAGCGAGGAC ‑3ʹ; BRD4 forward, 5ʹ- CACTGGGTTGGACCTTCGA-3ʹ and reverse, 5ʹ- CTTTGGTGGCTCAGGCTCT-3ʹ; GAPDH forward, 5ʹ‑GACTCCACTCACGGCAAATTCA‑3ʹ and reverse, 5ʹ‑TCGCTCCTGGAAGATGGTGAT‑3ʹ; U6 forward, 5ʹ‑GCTCGCTTCGGCAGCACA ‑3ʹ and reverse, 5ʹ‑GAGGTATTCGCACCAGAGGA‑3ʹ.
Cell Apoptosis Assay
FHCs were seeded in 6-well plates. After 48 h of transfection and LPS treatment, FHCs were incubated with Annexin V-FITC and propidium iodide (PI) provided from the Annexin V-FITC/PI Apoptosis Detection kit (Sigma-Aldrich, USA) for 30 min. Apoptosis was measured using flow cytometry (BD Biosciences, USA).
ELISA
The productions of inflammatory cytokines IL-1β, IL-6 and TNF-α were tested by ELISA kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Western Blotting
FHCs were lysed in protein lysis buffer (Sigma-Aldrich). Total protein was resolved by 10% SDS-PAGE and transferred to PVDF membranes (Millipore). Membranes were blocked with 5% fat-free milk and then incubated at 4°C overnight with primary antibodies against BRD4, phosphorylated P65 (p-P65), P65, IκB-α, and phosphorylated IκB-α (p-IκB-α). Then, the membranes were incubated with secondary antibodies. ECL (Sigma-Aldrich, USA) was for visualized the protein bands and analyzed using Image Lab ™ Software (Bio-Rad).
Luciferase Reporter Assay
The binding sites of UCA1-wild-type (wt), UCA1-mutant (mut), BRD4-wt and BRD4-mut were cloned into pmirGLO luciferase vectors (Promega Corporation). Then, FHCs were co-transfected with the above vectors and miR-331-3p mimics using Lipofectamine™ 3000 (Invitrogen). After 48h transfection, luciferase activities were evaluated by the Dual-Luciferase Reporter Assay System (Promega).
Statistical Analysis
Data are presented as mean ± SD, and the SPSS 19.0 statistical program was used for statistical analysis. The correlation between miR-331-3p and UCA1 levels was determined using Pearson’s correlation analysis. Differences between groups were compared using Student t-test or the ANOVA followed by the Tukey post hoc test. P < 0.05 was defined as statistically significant.
Results
UCA1 Expression is Elevated in UC Patients and LPS-Caused Injury in FHCs
To explore whether UCA1 participated in UC progression, we assessed the level of UCA1 in colonic mucosa tissues of UC patients. As displayed in Figure 1A, UCA1 was upregulated in UC tissue samples, suggesting that UCA1 might promote UC development. Moreover, it was observed that LPS treatment increased UCA1 expression in FHCs (Figure 1B). Then, LPS-induced FHCs were performed to simulate UC-triggered inflammatory injury, the results indicated that the viability of FHCs was inhibited, and cell apoptosis were promoted by LPS stimulation (Figure 1C and D). Moreover, ELISA results demonstrated that LPS treatment enhanced the levels of inflammatory cytokines IL-1β, IL-6 and TNF-α in FHCs (Figure 1E). These data implied that UCA1 level was enhanced in UC patients and LPS treatment could induce injury in FHCs. Due to 8 ng/mL LPS repressed ~50% FHCs viability,17 this dose was used for subsequent experiments.
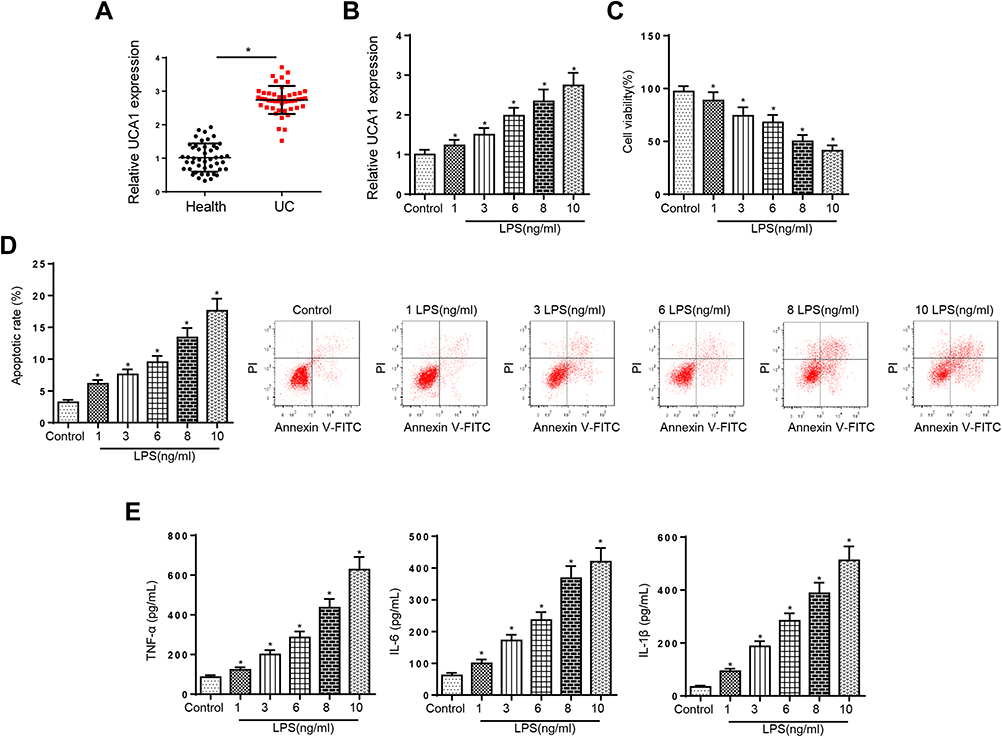 |
Figure 1 UCA1 expression was elevated in UC patients and LPS-caused injury in FHCs. (A) RT-qPCR showed the relative UCA1 expression in colonic mucosa tissues of UC patients compared with healthy controls. (B) RT-qPCR showed the expression of UCA1 in FHCs stimulated by LPS. (C and D) CCK-8 and flow cytometry were used to determine the viability and apoptosis in FHCs stimulated by LPS. (E) ELISA showed the production of inflammatory cytokines including TNF-a, IL-6 and IL-1β after LPS treatments. *p<0.05. |
UCA1 Deletion Accelerates Viability, Suppresses Apoptosis and Cytokines Production in LPS-Treated FHCs
To further determine the function of UCA1 in UC, LPS-stimulated FHCs were transfected with shUCA1 and the knockdown efficiency was exhibited in Figure 2A. Then, CCK-8 and flow cytometry assays demonstrated that LPS treatment repressed FHCs viability and induced cell apoptosis, and these effects were reversed by UCA1 silence (Figure 2B and C). Moreover, LPS induced the upregulation of IL-1β, IL-6 and TNF-α was counteracted by UCA1 deletion (Figure 2D). In sum, UCA1 knockdown relieved LPS-caused FHCs injury.
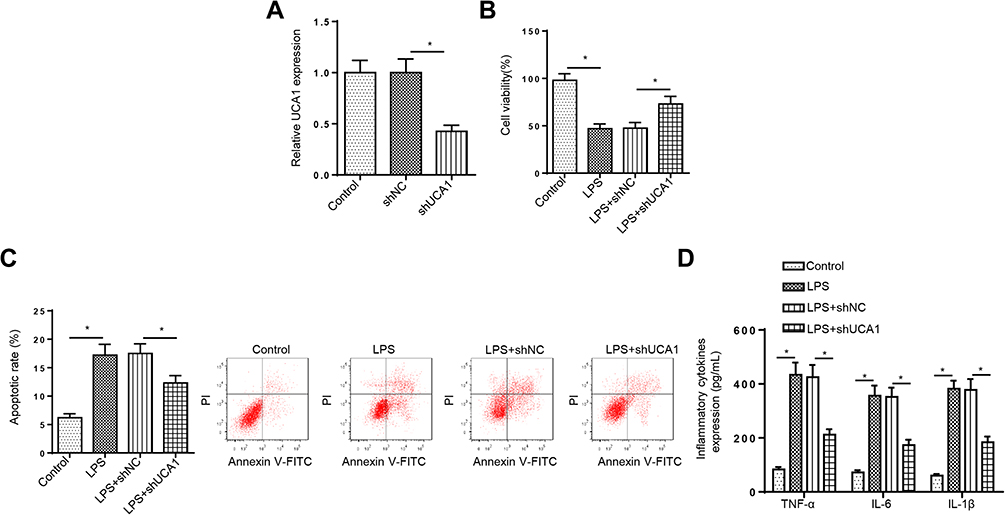 |
Figure 2 UCA1 deletion accelerates viability, suppresses apoptosis and cytokines production in LPS-treated FHCs. (A) RT-qPCR was used to determine the relative UCA1 expression in FHCs transfected with shUCA1. (B and C) FHCs were treated with different concentrations of LPS (1, 3, 6, 8, and 10 ng/mL). CCK-8 and flow cytometry were used to analyze the viability and apoptosis of FHCs stimulated by LPS, LPS+shNC, LPS+shUCA1 after different LPS treatments. (D) ELISA was used to determine the levels of IL-1β, IL-6 and TNF-α in FHCs stimulated by LPS, LPS+shNC, LPS+shUCA1 after different LPS treatments. *p<0.05. |
UCA1 Inhibition Improves LPS-Triggered Cell Injury via Sponging miR-331-3p
Through searching the starBase software, UCA1 was predicted to bind with miR-331-3p (Figure 3A). Then, luciferase reporter assays determined that the luciferase activity in UCA1-wt group was decreased by miR-331-3p addition in FHCs, while the activity in UCA1-mut group had no change (Figure 3B). Moreover, RT-qPCR results elaborated that miR-331-3p was downregulated in UC tissues (Figure 3C). miR-331-3p level was decreased by UCA1 overexpression, and was upregulated by UCA1 deletion in FHCs (Figure 3D). In addition, miR-331-3p expression was negatively correlated with UCA1 in UC tissues (Figure 3E). Furthermore, we demonstrated that miR-331-3p expression was elevated by miR-331-3p supplementation, which was reduced by miR-331-3p silence in FHCs (Figure 3F). Besides, UCA1 knockdown abrogated the effects of LPS treatment on FHC viability, apoptosis and inflammatory cytokines levels, while miR-331-3p deletion reversed these effects (Figure 3G–I). These results implied that repression of UCA1 improved LPS-stimulated injury via regulating miR-331-3p.
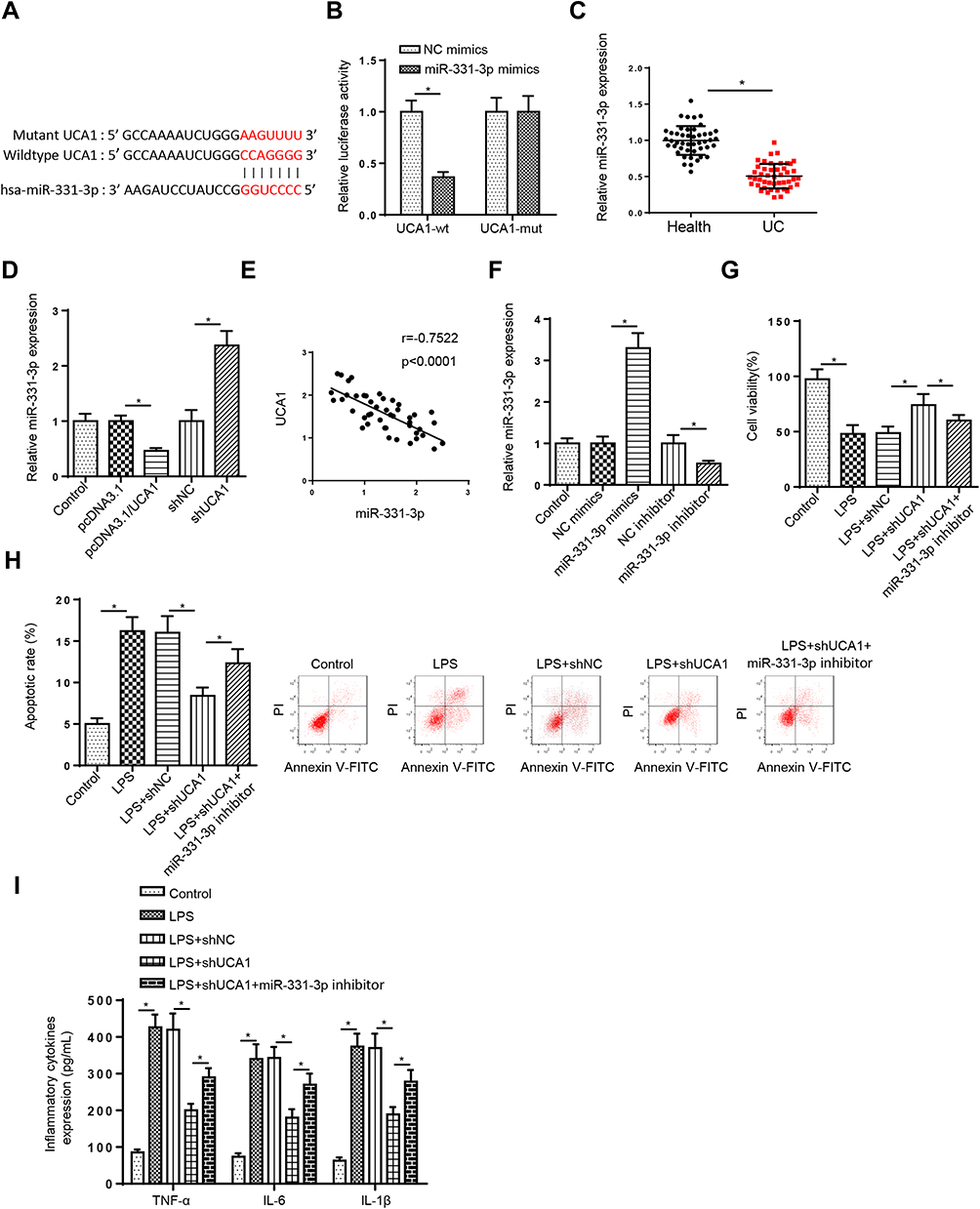 |
Figure 3 UCA1 inhibition improves LPS-triggered cell injury via sponging miR-331-3p. (A) StarBase website was used to predict the binding site between UCA1 and miR-331-3p. (B) Luciferase reporter assay showed luciferase activity of UCA1-wt or UCA1-mut in FHCs transfected with NC mimics or miR-331-3p mimics. (C) RT-qPCR showed the relative miR-331-3p expression in colonic mucosa tissues of UC patients compared with healthy controls. (D) RT-qPCR showed the relative miR-331-3p expression in FHCs transfection with pcDNA3.1, pcDNA3.1/UCA1, shNC or shUCA1. (E) Pearson’s correlation analysis showed the correlation between miR-331-3p and UCA1 in colonic mucosa tissues of UC patients. (F) RT-qPCR showed the relative miR-331-3p expression in FHCs transfected with NC mimics or miR-331-3p mimics and NC inhibitor or miR-331-3p inhibitor. (G–I) CCK-8, flow cytometry and ELISA were used to analyze the viability, apoptosis and inflammatory cytokines levels IL-1β, IL-6 and TNF-α of FHCs stimulated by LPS, LPS+shNC, LPS+shUCA1, and LPS+shUCA1+miR-331-3p inhibitor. *p<0.05. |
MiR-331-3p Regulates LPS-Triggered Injury in LPS-Induced FHCs by Modulating BRD4
To analyze miR-331-3p potential mechanism in UC, starBase software was used to screen the potential targets of miR-331-3p in UC. The binding sites in the 3ʹUTR correspondent to miR-331-3p were exhibited in Figure 4A. Then, RT-qPCR and Western blotting manifested that the mRNA and protein levels of BRD4 were decreased by miR-331-3p addition, while its expression was enhanced by miR-331-3p suppression (Figure 4B), determining that miR-331-3p negatively modulated BRD4 level. Moreover, luciferase reporter assay elaborated that the activity of BRD4-wt was attenuated by miR-331-3p supplementation, but the activity of BRD4-mt was no obvious changed (Figure 4C). Furthermore, to explore whether miR-331-3p modulated LPS-stimulated FHCs injury by targeting BRD4. FHCs were stimulated by LPS and co-transfected with NC inhibitor, miR-331-3p inhibitor and miR-331-3p inhibitor+shBRD4. First, RT-qPCR and Western blotting indicated that BRD4 was lowly expressed by BRD4 depletion (Figure 4D). Then, BRD4 silence in LPS+miR-331-3p inhibitor+shBRD4 group reversed the effects of miR-331-3p inhibition on LPS-treated cell injury in LPS-induced FHCs by facilitating cell viability, restraining the apoptosis, and reducing the levels of TNF-a, IL-6 and IL-1β (Figure 4E–G). Collectively, miR-331-3p deficiency might aggravate LPS-triggered injury through modulating BRD4.
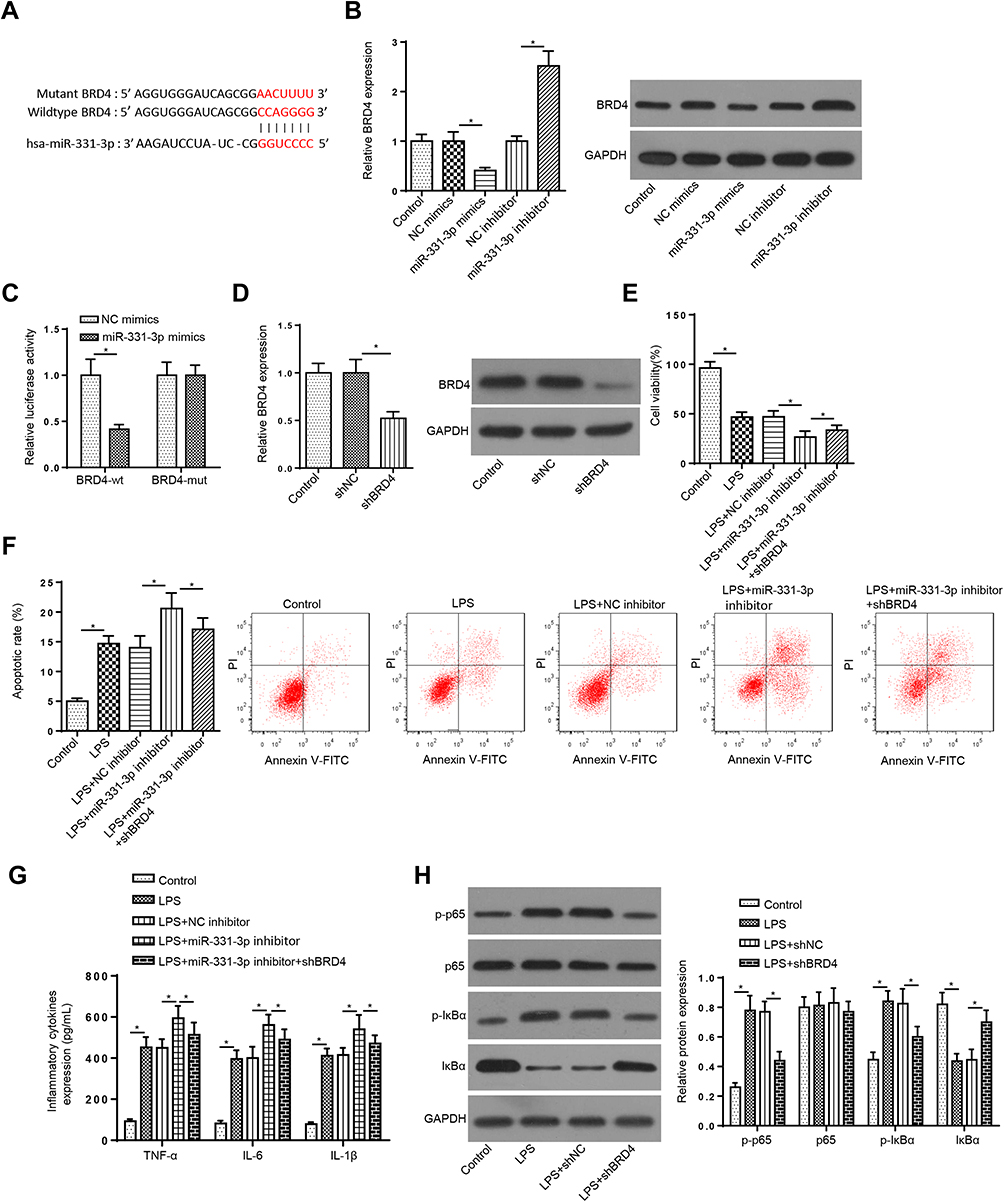 |
Figure 4 MiR-331-3p regulates LPS-triggered injury in LPS-induced FHCs by modulating BRD4 (A) StarBase website was used to predict the binding site between BRD4 and miR-331-3p. (B) RT-qPCR and Western blotting assays showed the mRNA and protein levels of BRD4 in FHCs transfected with NC mimics or miR-331-3p mimics and NC inhibitor or miR-331-3p inhibitor. (C) Luciferase reporter assay showed luciferase activity of BRD4-wt or BRD4-mut in FHCs transfected with NC mimics or miR-331-3p mimics. (D) RT-qPCR and Western blotting assays showed BRD4 expression of TIMP3 in FHCs treated with shBRD4. (E–G) CCK-8, flow cytometry and ELISA assays showed the viability, apoptosis and inflammatory cytokines levels of FHCs stimulated by LPS, LPS+NC inhibitor, LPS+miR-331-3p inhibitor, and LPS+miR-331-3p inhibitor+ shBRD4. (H) RT-qPCR and Western blotting assays determined the mRNA and protein levels of phosphorylated-p65 (p-p65), p65, phosphorylated-IκBα (p-IκBα), and IκBα of FHCs stimulated by LPS, LPS+shNC and LPS+shBRD4. *p<0.05. |
Since NF-kB is a vital transcription factor in the regulation of immune response, and its role in UC pathogenesis has been confirmed,18,19 we explored whether the NF-κB signaling was one of the downstream targets of BRD4 in UC. We found that BRD4 silence abrogated the increased levels of p-p65 and p-IκBα caused by LPS treatment (Figure 4H), which revealed BRD4 could activate the NF-κB signaling in LPS-treated FHCs. These discoveries elaborated that UCA1 aggravated LPS-triggered cell injury by modulating the miR-331-3p/NF-κB axis in UC.
Discussion
UC has become one of the fastest-growing serious diseases and with a high incidence rate in the world.20 Despite UC is not fatal, it is a long-term chronic disease and thus lead to life-threatening complications.21,22 The discovery of key lncRNAs involved in UC is helpful to provide new strategies for UC treatment. In this research, we demonstrated that deficiency of UCA1 suppressed LPS-triggered injury in FHCs by modulating miR-331-3p/BRD4 axis.
Previous studies have found several lncRNAs participate in UC pathogenesis. Yu et al reported UC-related lncRNA PMS2L2 decreased miR-24 level via methylation to restrain cell apoptosis in UC.23 Tian et al indicated lncRNA CDKN2B-AS1 improved UC inflammation through absorbing miR-16 and miR-195.24 UCA1 was reported to modulate cell viability and metastasis in many types of tumors, such as thyroid cancer and endometrial cancer.25,26 Moreover, UCA1 was found to act as a vital function in inflammatory diseases. For instance, lncRNA UCA1 expedited atherosclerosis treated by ox-LDL by targeting miR-206 in THP-1 cells.27 LncRNA UCA1 attenuated LPS-managed inflammatory damage by inactivating the miR-499b-5p/TLR4 axis.28 In this study, UCA1 level was elevated in colonic mucosa tissues of UC patients. LPS treatment suppressed cell viability and induced cell apoptosis as well as increased the levels of TNF-a, IL-6 and IL-1β. However, these effects were reversed by UCA1 knockdown in LPS-managed FHCs. Therefore, we confirmed that UCA1 was involved in UC development.
Extensive evidence has reported that lncRNAs exerted their functions by acting as ceRNAs and modulating the expression of miRNA targets in multiple diseases.29 Herein, we revealed that miR-331-3p targeted and inversely modulate by UCA1. Previous literature displayed that miR-331-3p participated in inflammatory responses.30,31 Moreover, miR-331-3p was displayed to attenuate the viability and facilitate apoptosis of the gastric cancer cells.32 Here, we manifested that miR-331-3p inhibition abrogated the influence of UCA1 silence on LPS-stimulated cell damage in LPS-treated FHCs, suggesting that miR-331-3p might play a crucial role in UC.
Increasing evidence reported that miRNAs exert their function by binding to target genes.33 To investigate the mechanism of miR-331-3p in UC development, BRD4 was uncovered as a direct target of miR-331-3p. BRD4 was reported as an important regulator in inflammatory diseases. Zhou et al indicated that BRD4 repression inhibited cerebral ischemia-induced brain injury via blocking glial activation by suppressing inflammatory response.34 BRD4 accelerated inflammatory response in microglia and blocking spinal cord injury recovery.35 Our study elaborated that BRD4 was regulated by miR-331-3p. Depletion of BRD4 reversed the effects of miR-331-3p repression on cell viability, apoptosis and inflammation in LPS-treated FHCs. NF-κB is a transcription factor that controls a series of pro-inflammatory genes involved in the cascade of inflammatory signals, which plays an essential function in UC pathogenesis.36 Several studies showed BRD4 was a crucial regulator of the NF-κB pathway in regulating inflammation.37,38 This research confirmed the NF-κB signaling was activated by BRD4 in LPS-managed FHCs. Thus, our observations implied that BRD4 could promote LPS-triggered cell injury by activating NF-κB signaling in UC.
In conclusion, we illustrated that UCA1 deficiency may attenuate UC development through modulating the miR-331-3p/TAB3/NF-κB pathway, indicating that UCA1 might be a novel therapeutic target for UC.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
All authors declare no conflicts of interest.
References
1. Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347(6):417–429. doi:10.1056/NEJMra020831
2. Farrell RJ, Peppercorn MA. Ulcerative colitis. Lancet. 2002;359(9303):331–340. doi:10.1016/S0140-6736(02)07499-8
3. Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2018;390(10114):2769–2778. doi:10.1016/S0140-6736(17)32448-0
4. Kaplan GG, Ng SC. Globalisation of inflammatory bowel disease: perspectives from the evolution of inflammatory bowel disease in the UK and China. Lancet Gastroenterol Hepatol. 2016;1(4):307–316. doi:10.1016/S2468-1253(16)30077-2
5. Eckburg PB, Relman DA. The role of microbes in Crohn’s disease. Clin Infect Dis. 2007;44(2):256–262. doi:10.1086/510385
6. Weinstock JV. Helminths and mucosal immune modulation. Ann N Y Acad Sci. 2006;1072:356–364. doi:10.1196/annals.1326.033
7. Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62. doi:10.1038/nrg.2015.10
8. Tian Y, Wang Y, Li F, et al. LncRNA TUG1 regulates the balance of HuR and miR-29b-3p and inhibits intestinal epithelial cell apoptosis in a mouse model of ulcerative colitis. Hum Cell. 2021;34(1):37–48. doi:10.1007/s13577-020-00428-5
9. Wang Y. Long Non-coding RNA MEG3 alleviated ulcerative colitis through upregulating miR-98-5p-Sponged IL-10. Inflammation. 2021.
10. Wang XS, Zhang Z, Wang H-C, et al. Rapid identification of UCA1 as a very sensitive and specific unique marker for human bladder carcinoma. Clin Cancer Res. 2006;12(16):4851–4858. doi:10.1158/1078-0432.CCR-06-0134
11. Ghafouri-Fard S, Eghtedarian R, Taheri M. The crucial role of non-coding RNAs in the pathophysiology of inflammatory bowel disease. Biomed Pharmacother. 2020;129:110507. doi:10.1016/j.biopha.2020.110507
12. Zhu M, Zhang N, He S, et al. Exosomal miR-106a derived from gastric cancer promotes peritoneal metastasis via direct regulation of Smad7. Cell Cycle. 2020;19(10):1200–1221. doi:10.1080/15384101.2020.1749467
13. Han J, Li Y, Zhang H, et al. MicroRNA-142-5p facilitates the pathogenesis of ulcerative colitis by regulating SOCS1. Int J Clin Exp Pathol. 2018;11(12):5735–5744.
14. Xu X, Zhu X, Wang C, et al. microRNA-650 promotes inflammation induced apoptosis of intestinal epithelioid cells by targeting NLRP6. Biochem Biophys Res Commun. 2019;517(4):551–556. doi:10.1016/j.bbrc.2019.06.077
15. Nie H, Hu Y, Guo W, et al. miR-331-3p inhibits inflammatory response after intracerebral hemorrhage by directly targeting NLRP6. Biomed Res Int. 2020;2020:6182464. doi:10.1155/2020/6182464
16. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
17. Qiao C, Yang L, Wan J, et al. Long noncoding RNA ANRIL contributes to the development of ulcerative colitis by miR-323b-5p/TLR4/MyD88/NF-kappaB pathway. Biochem Biophys Res Commun. 2019;508(1):217–224. doi:10.1016/j.bbrc.2018.11.100
18. Murano M, Maemura K, Hirata I, et al. Therapeutic effect of intracolonically administered nuclear factor κ B (p65) antisense oligonucleotide on mouse dextran sulphate sodium (DSS)-induced colitis. Clin Exp Immunol. 2000;120(1):51–58. doi:10.1046/j.1365-2249.2000.01183.x
19. Wei S-C, Rosenberg IM, Cao Z, et al. Tribbles 2 (Trib2) is a novel regulator of toll-like receptor 5 signaling. Inflamm Bowel Dis. 2012;18(5):877–888. doi:10.1002/ibd.22883
20. Molodecky NA. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):
21. de Silva S, Ma C, Proulx M, et al. Postoperative complications and mortality following colectomy for ulcerative colitis. Clin Gastroenterol Hepatol. 2011;9(11):972–980. doi:10.1016/j.cgh.2011.07.016
22. Bewtra M, Kaiser LM, TenHave T, et al. Crohn’s disease and ulcerative colitis are associated with elevated standardized mortality ratios: a meta-analysis. Inflamm Bowel Dis. 2013;19(3):599–613. doi:10.1097/MIB.0b013e31827f27ae
23. Yu T, Meng F, Xie M, et al. LncRNA PMS2L2 downregulates miR-24 through methylation to suppress cell apoptosis in ulcerative colitis. Dig Dis. 2020. doi:10.1159/000513330
24. Tian Y, Cui L, Lin C, et al. LncRNA CDKN2B-AS1 relieved inflammation of ulcerative colitis via sponging miR-16 and miR-195. Int Immunopharmacol. 2020;88:106970. doi:10.1016/j.intimp.2020.106970
25. Gao H. Long noncoding RNA UCA1 promotes proliferation and metastasis of thyroid cancer cells by sponging miR-497-3p. Eur Rev Med Pharmacol Sci. 2020;24(14):7555.
26. Liu T. Long Noncoding RNA UCA1 facilitates endometrial cancer development by regulating KLF5 and RXFP1 Gene Expressions. Cancer Biother Radiopharm. 2020.
27. Hu X, Ma R, Fu W, et al. LncRNA UCA1 sponges miR-206 to exacerbate oxidative stress and apoptosis induced by ox-LDL in human macrophages. J Cell Physiol. 2019;234(8):14154–14160. doi:10.1002/jcp.28109
28. Zhao YJ. LncRNA UCA1 remits LPS-engendered inflammatory damage through deactivation of miR-499b-5p/TLR4 axis. IUBMB Life. 2020.
29. Wang LX. Integrative analysis of long noncoding RNA (lncRNA), microRNA (miRNA) and mRNA expression and construction of a competing endogenous RNA (ceRNA) Network in Metastatic Melanoma. Med Sci Monit. 2019;25:2896–2907. doi:10.12659/MSM.913881
30. Li R. LncRNA SOX2OT knockdown alleviates lipopolysaccharide-induced damage of PC12 Cells by Regulating miR-331-3p/Neurod1 Axis. World Neurosurg. 2020.
31. Kong L, Wu P, Li J. miR-331 inhibits CLDN2 expression and may alleviate the vascular endothelial injury induced by sepsis. Exp Ther Med. 2020;20(2):1343–1352. doi:10.3892/etm.2020.8854
32. Li XM, Jiao -Y-Y, Luan B-H, et al. Long non-coding RNA MIAT promotes gastric cancer proliferation and metastasis via modulating the miR-331-3p/RAB5B pathway. Oncol Lett. 2020;20(6):355. doi:10.3892/ol.2020.12219
33. Adlakha YK, Saini N. Brain microRNAs and insights into biological functions and therapeutic potential of brain enriched miRNA-128. Mol Cancer. 2014;13:33. doi:10.1186/1476-4598-13-33
34. Zhou Y, Gu Y, Liu J. BRD4 suppression alleviates cerebral ischemia-induced brain injury by blocking glial activation via the inhibition of inflammatory response and pyroptosis. Biochem Biophys Res Commun. 2019;519(3):481–488. doi:10.1016/j.bbrc.2019.07.097
35. Wang J, Chen J, Jin H, et al. BRD4 inhibition attenuates inflammatory response in microglia and facilitates recovery after spinal cord injury in rats. J Cell Mol Med. 2019;23(5):3214–3223. doi:10.1111/jcmm.14196
36. Wang H, Gu J, Hou X, et al. Anti-inflammatory effect of miltirone on inflammatory bowel disease via TLR4/NF-kappaB/IQGAP2 signaling pathway. Biomed Pharmacother. 2017;85:531–540. doi:10.1016/j.biopha.2016.11.061
37. Gao L, Zhao H, Sun H, et al. TAB3 overexpression promotes NF-kappaB activation and inflammation in acute pancreatitis. Am J Blood Res. 2020;10(4):118–123.
38. Yang Q, Zhang D, Li Y, et al. Paclitaxel alleviated liver injury of septic mice by alleviating inflammatory response via microRNA-27a/TAB3/NF-kappaB signaling pathway. Biomed Pharmacother. 2018;97:1424–1433. doi:10.1016/j.biopha.2017.11.003
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.