")
Back to Journals » OncoTargets and Therapy » Volume 10
LncRNA MEG3 enhances cisplatin sensitivity in non-small cell lung cancer by regulating miR-21-5p/SOX7 axis
Authors Wang P, Chen D, Ma H, Li Y
Received 14 July 2017
Accepted for publication 11 September 2017
Published 25 October 2017 Volume 2017:10 Pages 5137—5149
DOI https://doi.org/10.2147/OTT.S146423
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ingrid Espinoza
Pei Wang,* Dong Chen,* Hongbing Ma, Yong Li
Department of Cardiothoracic Surgery, Huaihe Hospital of Henan University, Kaifeng, People’s Republic of China
*These authors contributed equally to this work
Background: Long noncoding RNAs (lncRNAs) have been revealed to play essential role in drug resistance of multiple cancers. LncRNA MEG3 was previously reported to be associated with cisplatin (DDP) resistance in non-small cell lung cancer (NSCLC) cells. However, the molecular mechanism of MEG3 affecting DDP resistance in NSCLC remains to be further illustrated. In this study, we attempted to discuss whether MEG3 also could function as a competing endogenous RNA to regulate DDP resistance in NSCLC.
Materials and methods: The expression of MEG3, miR-21-5p, and sex-determining region Y-box 7 (SOX7) in NSCLC tissues or cells was examined by quantitative real-time polymerase chain reaction (qRT-PCR). 3-(4,5-Dimethylthazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), flow cytometry, and caspase-3 activity analysis were applied to assess the DDP sensitivity of NSCLC cells. The interaction between MEG3, miR-21-5p, and SOX7 was explored by luciferase reporter assay, RNA immunoprecipitation (RIP) assay, qRT-PCR, and Western blot. Mouse NSCLC transplanted tumor was established to verify the functional role of MEG3 in DDP resistance in vivo.
Results: MEG3 was downregulated in DDP-resistant NSCLC cells. Overexpression of MEG3 enhanced DDP sensitivity of NSCLC cells in vitro. MEG3 directly interacted with miR-21-5p and suppressed its expression. miR-21-5p significantly abolished the effects of MEG3 on DDP resistance via modulating cell proliferation and apoptosis. SOX7 was identified as a direct target of miR-21-5p and MEG3 positively regulated SOX7 expression by suppressing miR-21-5p. Moreover, MEG3 knockdown-induced pro-proliferative and anti-apoptotic effects were reversed in DDP-resistant NSCLC cells by upregulating SOX7. Furthermore, upregulation of MEG3 induced sensitivity of NSCLC cells to DDP in vivo.
Conclusion: MEG3 overexpression induced DDP sensitivity of NSCLC cells by regulating miR-21-5p/SOX7 axis, shedding light on the molecular mechanism of MEG3 involved in the development of DDP resistance of NSCLC cells.
Keywords: MEG3, miR-21-5p/SOX7, DDP resistance, NSCLC
Introduction
Lung cancer, one of the most common devastating malignancies, is considered as the main cause of cancer-associated mortality and morbidity around the world.1 Non-small cell lung cancer (NSCLC) occupies about 85% of all lung cancer cases, with a 5-year survival rate as low as 15%.2 Currently, platinum-based chemotherapy has been applied as a standard adjunctive treatment strategy in advanced NSCLC patients following surgical resection. Cisplatin (DDP), a platinum-containing compound, is a first-line drug for chemotherapeutic administration in NSCLC.3 Unfortunately, the emergence of acquired drug resistance results in a limitation in the clinical application of DDP and prognosis of patients. Therefore, investigation of the molecular mechanisms underlying DDP resistance in NSCLC may be of great significance for improving the outcome of NSCLC patients.
Long noncoding RNAs (lncRNAs) are defined as a novel class of transcripts with more than 200 nucleotides and without protein-coding capacity, being able to regulate gene expression at transcriptional, posttranscriptional, and epigenetic levels. Up to now, great efforts have been made to elucidate the function and mechanism of lncRNA in development and disease.4 LncRNAs prompt lots of vital cancer phenotypes by interacting with other cellular molecules, such as DNA, protein, and RNA.5 Moreover, many lncRNAs have also been shown to be implicated in the chemoresistance of a wide range of tumors by promoting cellular proliferation and reducing apoptosis.6 For instance, lncRNA RP11-838N2.4 increased temozolomide sensitivity by serving as an endogenous sponge to suppress miR-10a function on an epigenetic level.7 Knockdown of lncRNA HOTAIR induced cell sensitivity to antitumor drugs by enhancing apoptosis and cell cycle arrest via modulation of HOXA1 methylation.8 MEG3, an imprinted lncRNA within DLK1-MEG3 locus located at human chromosome 14q32, is expressed in a number of normal tissues.9 MEG3 has been demonstrated to exert an antitumor activity in several cancers, such as bladder cancer,10 glioma,11 colorectal cancer,12 and NSCLC.13 MEG3 was previously reported to be downregulated in DDP-resistant lung cancer cells, and the overexpression of MEG3 enhanced DDP sensitivity in NSCLC.14,15 However, the molecular mechanism of MEG3 involved in DDP resistance in lung cancer remains to be further illustrated.
Up to now, many investigations have focused on the interplay between lncRNAs and microRNAs, as well as the importance of such interactions during tumorigenic process.16 One popular hypothesis points out that lncRNAs could serve as competing endogenous RNAs (ceRNAs) to segregate miRNAs away from target mRNAs, or lncRNAs could control target mRNA expression by competitively combining with miRNAs. In this study, we attempted to discuss whether MEG3 also could function as a ceRNA to regulate DDP resistance in NSCLC.
In the current study, we demonstrated the downregulation of MEG3 in DDP-resistant NSCLC cells. Functional analysis disclosed that the overexpression of MEG3 enhanced DDP sensitivity in NSCLC. Mechanistic analyses revealed that MEG3 functioned as a ceRNA to suppress the expression and activities of miR-21-5p, thus leading to derepression of miR-21-5p target sex-determining region Y-box 7 (SOX7). Moreover, the overexpression of SOX7 reversed si-MEG3-induced pro-proliferative and anti-apoptotic effect in NSCLC cells. Therefore, our findings provide novel insight into the molecular mechanism of MEG3 involved in DDP resistance in NSCLC.
Materials and methods
Patient samples
Our study was approved by the ethics committee on human research of Huaihe Hospital of Henan University. Written informed consents were obtained from all participants before the study. A total of 46 resected tumor tissues were acquired from advanced NSCLC patients who received DDP-based chemotherapy between January 2014 and December 2016 and were undergoing surgical resection at Huaihe Hospital of Henan University. All tissue samples were immediately snap frozen and kept at −80°C in liquid nitrogen. These samples were divided into two groups: 25 DDP sensitive and 23 DDP resistant, according to the objective responses assessed by medical image analysis and detection of serum tumor markers after two courses of DDP-based chemotherapy.
Cell culture
NSCLC cell lines (A549 and H1299) were obtained from China Center for Type Culture Collection (CCTCC, Shanghai, China). To construct DDP-resistant NSCLC cells (A549-DDP and H1299-DDP), A549 and H1299 cells were treated with a stepwise increasing concentration of DDP (Sigma-Aldrich Co., St Louis, MO, USA) until the survived cells exhibited normal morphology and activity, as described previously.17 All cells were cultured in RPMI-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified incubator with 5% CO2. To maintain the drug-resistant phenotype, 2 μM DDP (Sigma-Aldrich Co.) was added to the culture media of A549-DDP and H1299-DDP cells.
Cell transfection
To enhance MEG3 and SOX7 endogenous expressions, the full length of MEG3 and SOX7 sequences was synthesized and inserted into pCDNA3.1 empty plasmid (GenePharma, Shanghai, China), termed as pcDNA-MEG3 (MEG3) and pcDNA-SOX7 (SOX7), respectively. To attenuating MEG3 expression, siRNA against MEG3 (si-MEG3) was synthesized by GenePharma, with nonspecific oligonucleotides (si-NC) as a control. miR-21-5p mimic (miR-21-5p), scrambled oligonucleotides (miR-NC), miR-21-5p antagomirs (anti-miR-21-5p), and antagomirs negative control (anti-miR-NC) were synthesized by GenePharma. Cell transfection with oligonucleotides or plasmids into A549-DDP and H1299 cells was performed using Lipofectamine 2000 (Thermo Fisher Scientific). Cells were collected at 48 h post transfection for further investigations.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated from cultured cells or resected tissues using TRIzol reagent (Thermo Fisher Scientific), and RNA concentration was quantified using the Nanodrop 2000 (Thermo Fisher Scientific). Reverse transcription into the first strand of cDNA was performed using 2 μg of total RNA with a PrimeScript™ RT-PCR Kit (TaKaRa, Kusatsu, Japan). For the detection of expressions of MEG3 and miR-21-5p, RT-PCR was performed on the ABI 7300 Thermocycler (Thermo Fisher Scientific) using SYBR Premix Ex Taq kit (Thermo Fisher Scientific) and TaqMan miRNA assay (TaKaRa), respectively. GAPDH and U6 snRNA were used as an internal loading control for lncRNA and miRNA. The relative fold change of gene expression was calculated by the 2−ΔΔCt method.
Western blot analysis
Cultured cells were harvested and lysed in radioimmunoprecipitation assay lysis buffer in the presence of protease inhibitors (Hoffman-La Roche Ltd., Basel, Switzerland). Protein concentrations were quantified by Bradford Protein Assay Kit (Thermo Fisher Scientific). Equal amount of total protein (30 μg) was resolved by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a polyvinylidene fluoride (PVDF) membrane (EMD Millipore, Billerica, MA, USA). After blocking with 5% nonfat milk for 1 h, the membranes were probed with the primary antibody against SOX7 (R&D Systems, Inc., Minneapolis, MN, USA) or β-actin (Santa Cruz Biotechnology Inc., Dallas, TX, USA) overnight at 4°C, followed by incubation with horseradish peroxidase (HRP)-linked secondary antibody for 2 h. The signal intensity was visualized by an electrochemiluminescence kit (Pierce Biotechnology, Rockford, IL, USA).
3-(4,5-Dimethylthazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
MTT assay was performed to determine the sensitivity of cells to DDP treatment. In brief, transfected cells at the logarithmic phase were seeded into 96-well plates at a density of 4×104 cells per well. After 12 h, A549, A549-DDP, H1299, and H1299-DDP cells were treated with different concentrations of DDP (0, 2, 4, 8, 16, 32, and 64 μM) for 48 h. Subsequently, 20 μL of MTT reagent (5 mg/mL; Sigma-Aldrich Co.) was added and incubated for another 4 h at 37°C. The optical density at 490 nm was measured using a Bio-Rad 680 microplate reader (Bio-Tek Instruments, Winooski, VT, USA). The IC50 (resistance index) value was estimated according to the relative survival curve.
In addition, transfected A549-DDP and H1299-DDP cells (1×103 cells/well) were plated into 96-well plates and treated with DDP, followed by MTT analysis to evaluate cell proliferation at indicated time point (0, 24, 48, and 72 h).
Analysis of apoptosis by flow cytometry
Transfected A549-DDP and H1299-DDP cells were seeded into six-well plates at a density of 5×105 cells/well and incubated with DDP for 48 h. Then, cells were harvested and stained with annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) Apoptosis Detection Kit (Neobioscience, Inc., Shenzhen, China). The proportion of apoptotic cells was measured using a FACScan flow cytometer (Becton Dickinson, Mountain View, CA, USA) equipped with CellQuest software (BD Biosciences, San Jose, CA, USA).
Luciferase reporter assay
The fragments of MEG3 and 3′-untranslated region (UTR) of SOX7 containing the predicted wild-type (WT) binding sites of miR-21-5p or mutated miR-21-5p binding sites (MUT) were amplified by PCR and inserted into a pMIR-REPORT luciferase reporter vector (Ambion, Austin, TX, USA), named as WT-MEG3, MUT-MEG3, WT-SOX7-3′UTR, and MUT-SOX7-3′UTR. When the luciferase reporter assay is carried out, cells were cotransfected with 200 ng constructed luciferase reporter vectors, 25 ng pRL-TK (expressing renilla luciferase as the internal control) and 20 μM miR-21-5p or miR-NC using Lipofectamine 2000. A luciferase reporter assay system (Promega Corporation, Fitchburg, WI, USA) was applied to analyze the luciferase activities at 48 h post transfection.
RNA immunoprecipitation (RIP) assay
To confirm the endogenous relationship between MEG3 and miR-21-5p, RIP assays were carried out using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (EMD Millipore). A549-DDP and H1299-DDP cells were collected and lysed using RIP lysis buffer containing protease inhibitor and RNase inhibitor. Whole-cell extracts were incubated with RIP buffer containing magnetic beads conjugated with antihuman argonaute 2 (Ago2) antibody (EMD Millipore) for 1 h. Following digesting protein with proteinase K, the immunoprecipitated RNA was extracted for qRT-PCR analysis. Total RNA (input controls) and mouse IgG (negative controls) were detected synchronously to certify that the detected signals were RNAs specifically binding to Ago2.
Caspase-3 activity assay
The activity of caspase-3, a key enzyme in the regulation of apoptotic cascades, was measured in transfected A549-DDP and H1299-DDP cells using a Caspase-3 Colorimetric Assay kit (Promega Corporation).
In vivo chemosensitivity assay
All animal experiments were carried out according to the guidelines for Institutional Animal Care and Use, with approval of Animal Research Ethics Committee of Huaihe Hospital. Five-week-old BALB/c athymic nude mice were maintained under pathogen-free condition. A549-DDP cells (6×106/100 μL PBS) transfected with pcDNA-MEG3 (MEG3) or pcDNA-NC (vector) were subcutaneously injected into the right flank of nude mice. At 7 days, nude mice began to be intraperitoneally administered with 4 mg/kg DDP or PBS every 4 days. At 31 days after inoculation, mice were killed and transplanted tumors were removed for subsequent analysis.
Statistical analysis
All data were expressed as mean ± standard deviation (SD) of at least three independent experiments. Comparisons between groups were carried out using Student’s t-test or one-way analysis of variance (ANOVA) using SPSS 19.0 software (IBM Corporation, Armonk, NY, USA). A P-value of <0.05 was considered to indicate statistical significance.
Results
Upregulation of MEG3 enhanced DDP sensitivity of DDP-resistant NSCLC cells
First, the expression of MEG3 in NSCLC tumor tissues of 25 DDP-sensitive and 23 DDP-resistant NSCLC patients was evaluated by RT-PCR, and the results showed that MEG3 expression was markedly downregulated in DDP-resistant patients when compared with that in DDP-sensitive patients (Figure 1A). We then established two DDP-resistant NSCLC cells (A549-DDP and H1299-DDP). MTT assay ascertained the greater resistance to DDP in A549-DDP and H1299-DDP cells compared with corresponding parental cells A549 and H1299, presented as higher IC50 values (Figure 1B). To confirm the involvement of MEG3 in DDP resistance of NSCLC cells, MEG3 expression in parental and DDP-resistant NSCLC cells was estimated by qRT-PCR. As shown in Figure 1C, a significant reduction in MEG3 expression was observed in A549-DDP and H1299-DDP cells. To evaluate the effect of MEG3 on the development of DDP resistance, we performed overexpression or knockdown for MEG3 in A549-DDP and H1299-DDP cells. As shown in Figure 1D, MEG3 expression was upregulated after transfection with pcDNA-MEG3; conversely, MEG3 expression was downregulated after introduction with si-MEG3. As demonstrated by MTT analysis, MEG3-treated A549-DDP cells exhibited significantly lower IC50 value with respect to the control group, while si-MEG3-introduced H1299-DDP cells showed higher IC50 value in comparison with the si-NC group (Figure 1E). All these data indicated that the overexpression of MEG3 induced DDP sensitivity of DDP-resistant NSCLC cells.
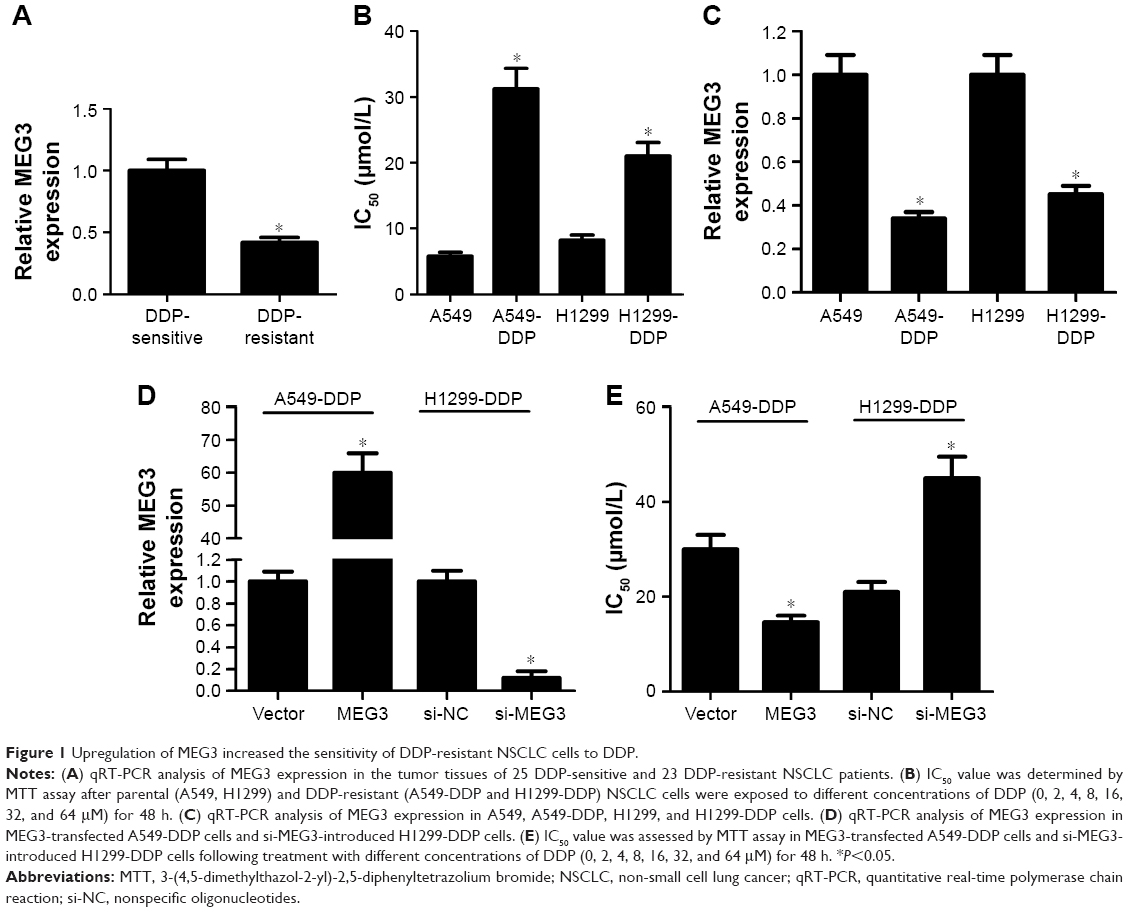 | Figure 1 Upregulation of MEG3 increased the sensitivity of DDP-resistant NSCLC cells to DDP. |
MEG3 functioned as a molecular sponge of miR-21-5p in DDP-resistant NSCLC cells
As stated in the “Introduction” section, it was previously proposed that lncRNAs function as a ceRNA by competitively binding to miRNAs. To explore whether MEG3 had the similar function to regulate some miRNAs, Starbase v.2.0 was used to predict potential miRNAs that directly interacted with MEG3. Bioinformatics analysis demonstrated the presence of sequences complementary to the seed region of miR-21-5p in MEG3 (Figure 2A). To verify the direct binding between MEG3 and miR-21-5p, luciferase reporter plasmids containing the WT or MUT in MEG3 were constructed and cotransfected with miR-21-5p or miR-NC into 293T cells. The results of luciferase reporter assay showed that miR-21-5p overexpression led to a marked decrease in luciferase activity in WT-MEG3 reporter compared with the miR-NC group, but had no obvious effect on the luciferase activity in MUT-MEG3 reporter (Figure 2B). It is well known that miRNAs control target mRNA expression by binding to Ago, a key protein in RNA-induced silencing complex (RISC), causing RNA degradation or translational repression.18 To explore whether both MEG3 and miR-21-5p are in the RISC, anti-Ago2 RIP assay was conducted in A549-DDP cell extracts, followed by qRT-PCR analysis for immunoprecipitated RNA. As shown in Figure 2C, MEG3 and miR-21-5p were significantly enriched in Ago2-containing beads compared with IgG control, indicating the endogenous interaction between MEG3 and miR-21-5p. The actual regulatory role of MEG3 on miR-21-5p expression was investigated by qRT-PCR in A549-DDP and H1299-DDP cells transfected with MEG3, si-MEG3, or matched controls. As expected, ectopic expression of MEG3 apparently inhibited miR-21-5p expression in A549-DDP cells, while MEG3 knockdown markedly promoted miR-21-5p expression in H1299-DDP cells (Figure 2D). Overall, these results elucidated that MEG3 could act as a molecular sponge, interacting with miR-21-5p.
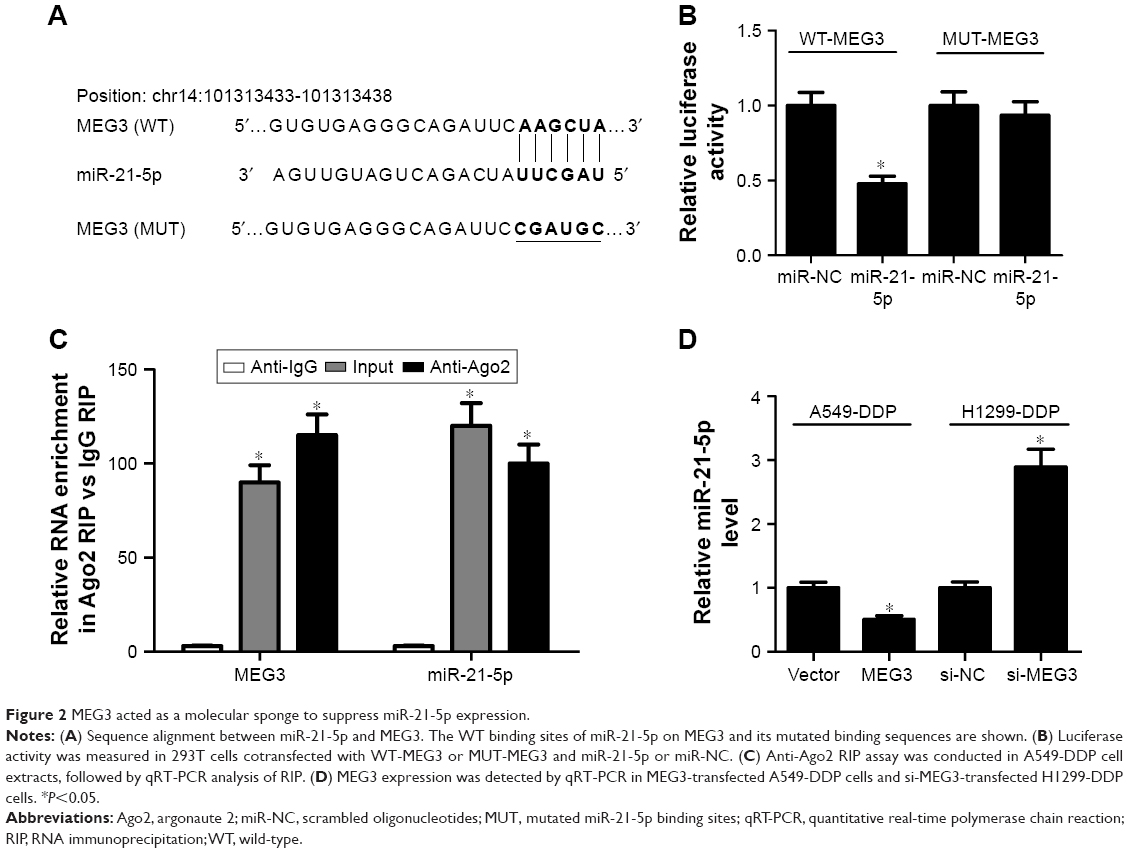 | Figure 2 MEG3 acted as a molecular sponge to suppress miR-21-5p expression. |
MEG3 overexpression increased the sensitivity of DDP-resistant NSCLC cells to DDP by sponging miR-21-5p
To gain insight into the mechanism by which MEG3 inhibits DDP resistance of NSCLC cells, we performed rescue experiments in MEG3-transfected A549-DDP cells and si-MEG3-introduced H1299-DDP cells, through miR-21-5p overexpression or suppression. MTT assay was performed to evaluate cell growth after DDP administration. The results demonstrated that ectopic expression of MEG3 remarkably blocked cell proliferation in A549-DDP cells, while miR-21-5p overexpression attenuated this inhibitory effect (Figure 3A). In contrast, MEG3 knockdown dramatically promoted cell proliferation in H1299-DDP cells, which was strikingly reversed after co-transfection with miR-21-5p inhibitor (Figure 3B). Then, we performed flow cytometry and caspase-3 activity analysis to examine cell apoptosis after DDP treatment. Flow cytometry analysis demonstrated that A549-DDP cells with upregulated MEG3 expression showed a higher apoptotic rate than that in the vector control group (Figure 3C), whereas reintroduction of miR-21-5p mimic greatly abated these effects. On the contrary, the decreased apoptotic rate of H1299-DDP cells induced by MEG3 knockdown were obviously abated after suppressing miR-21-5p expression (Figure 3D). The caspase-3 activity analysis indicated that MEG3 overexpression greatly enhanced caspase-3 activity, however, reintroduction of miR-21-5p partly lowered the caspase-3 activity in A549-DDP cells (Figure 3E). In contrast, MEG3 knockdown resulted in caspase-3 activity decrease in H1299-DDP cells, whereas suppressing miR-21-5p expression reversed this effect (Figure 3F). Collectively, these results demonstrated that MEG3 enhanced DDP sensitivity of DDP-resistant NSCLC cells by repressing proliferation and inducing apoptosis via miR-21-5p.
 | Figure 3 miR-21-5p counteracted MEG3-induced DDP sensitivity of DDP-resistant NSCLC cells. |
SOX7 was a direct target of miR-21-5p
Considering that miRNAs exert their functions via binding to the 3′UTR of specific target genes, we further predicted the candidate targets of miR-21-5p using TargetScan tool (www.targetscan.org). As shown in Figure 4A, SOX7 contained miR-21-5p binding sequence in the 3′UTR. To confirm whether SOX7 was a direct target of miR-21-5p, the luciferase reporter plasmids harboring WT or mutated miR-21-5p recognition sequences in the 3′UTR of SOX7 were cotransfected with miR-21-5p or miR-NC into A549 or H1299 cells. Luciferase reporter assay revealed that co-transfection with miR-21-5p and WT-SOX7-3′UTR significantly reduced the luciferase activity of A549 and H1299 cells compared with the control group (Figure 4B). However, the luciferase activity of A549 and H1299 cells cotransfected with miR-21-5p and MUT-SOX-3′UTR was almost unchanged when compared with the control group. Next, we evaluated the effect of miR-21-5p on the protein level of SOX7 by Western blot analysis. The results disclosed that the protein level of SOX7 was markedly decreased in miR-21-5p-treated A549 cells, while SOX7 protein expression was effectively increased following miR-21-5p inhibition in H1299 cells (Figure 4C). Moreover, the expressions of miR-21-5p and SOX7 in 25 DDP-sensitive and 23 DDP-resistant NSCLC patients were detected by qRT-PCR. As shown in Figure 4D and E, upregulated miR-21-5p expression and downregulated SOX7 expression were observed in DDP-resistant NSCLC patients compared with those in DDP-sensitive NSCLC patients. Overall, these data suggested that miR-21-5p repressed SOX7 expression by targeting 3′UTR of SOX7.
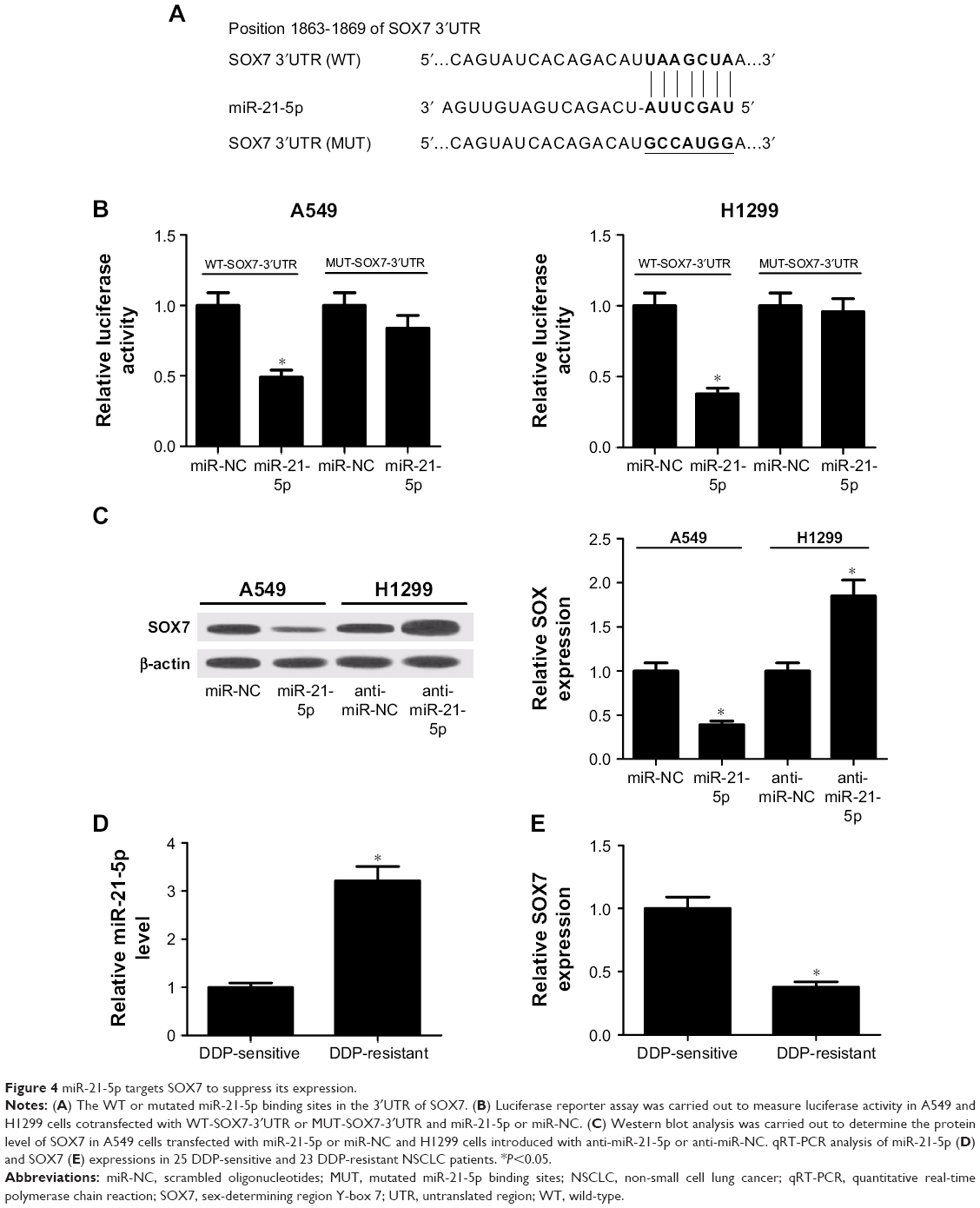 | Figure 4 miR-21-5p targets SOX7 to suppress its expression. |
MEG3 positively regulated SOX7 expression by suppressing miR-21-5p
We further investigated the regulatory role of MEG3 and miR-21-5p in SOX7 expression by performing rescue experiments in MEG3-tramsfected A549-DDP cells and si-MEG3-transfected H1299-DDP cells, via reintroduction of miR-21-5p mimic or inhibitor. The results indicated that enforced expression of miR-21-5p prominently repressed MEG3 overexpression-induced increase in SOX7 protein level in A549-DDP cells (Figure 5A). Conversely, MEG3 knockdown dramatically reduced the protein level of SOX7 in H1299-DDP cells, which was conspicuously recuperated by miR-21-5p inhibition (Figure 5B). Therefore, we concluded that MEG3 upregulated SOX7 expression by inhibiting miR-21-5p.
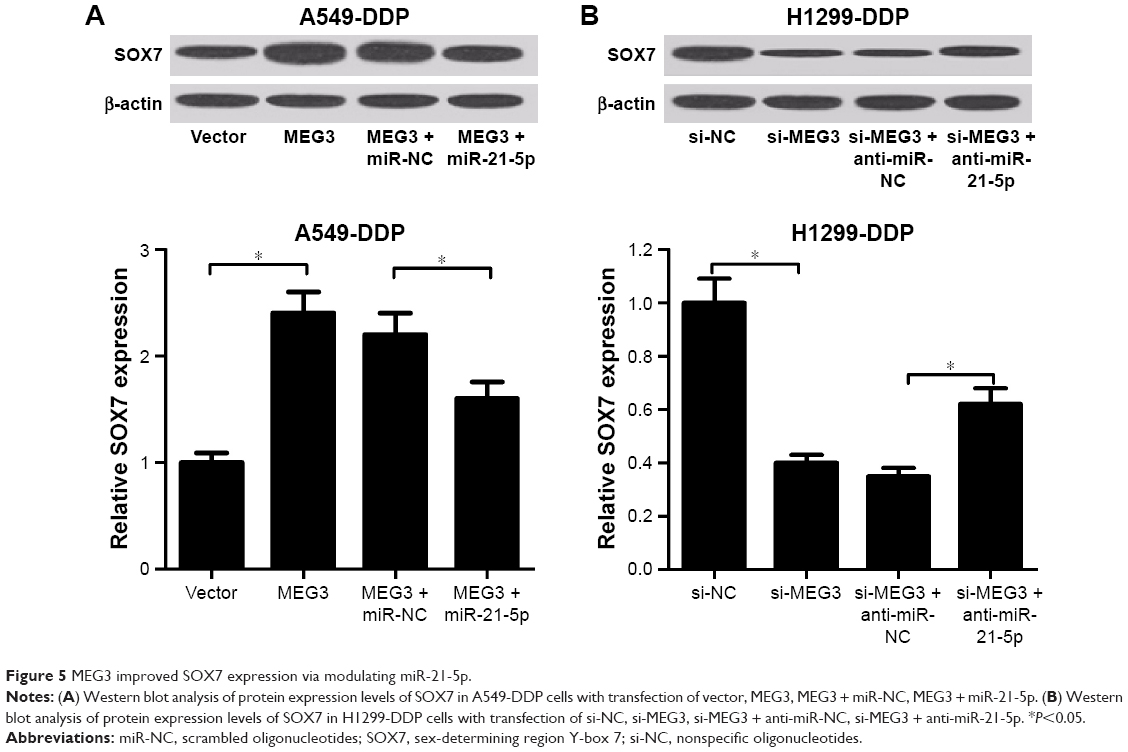 | Figure 5 MEG3 improved SOX7 expression via modulating miR-21-5p. |
MEG3 knockdown increased DDP resistance of DDP-resistant NSCLC cells by inhibiting SOX7 expression.
To further explore whether MEG3 regulated cell proliferation and apoptosis of DDP-resistant NSCLC cells through SOX7, A549-DDP and H1299-DDP cells were transfected with si-MEG3, or combined with SOX7, followed by DDP treatment. MTT assay demonstrated that MEG3 knockdown markedly promoted cell proliferation in A549-DDP (Figure 6A) and H1299-DDP (Figure 6B) cells, while SOX7 overexpression greatly reversed the effect of MEG3 silencing on cell proliferation. In addition, flow cytometry analysis revealed that si-MEG3-induced decline in apoptotic rate was evidently overturned after overexpressing SOX7 in A549-DDP (Figure 6C) and H1299-DDP (Figure 6D) cells. These data suggested that MEG3 knockdown enhanced DDP resistance of DDP-resistant NSCLC cells by repressing SOX7 expression.
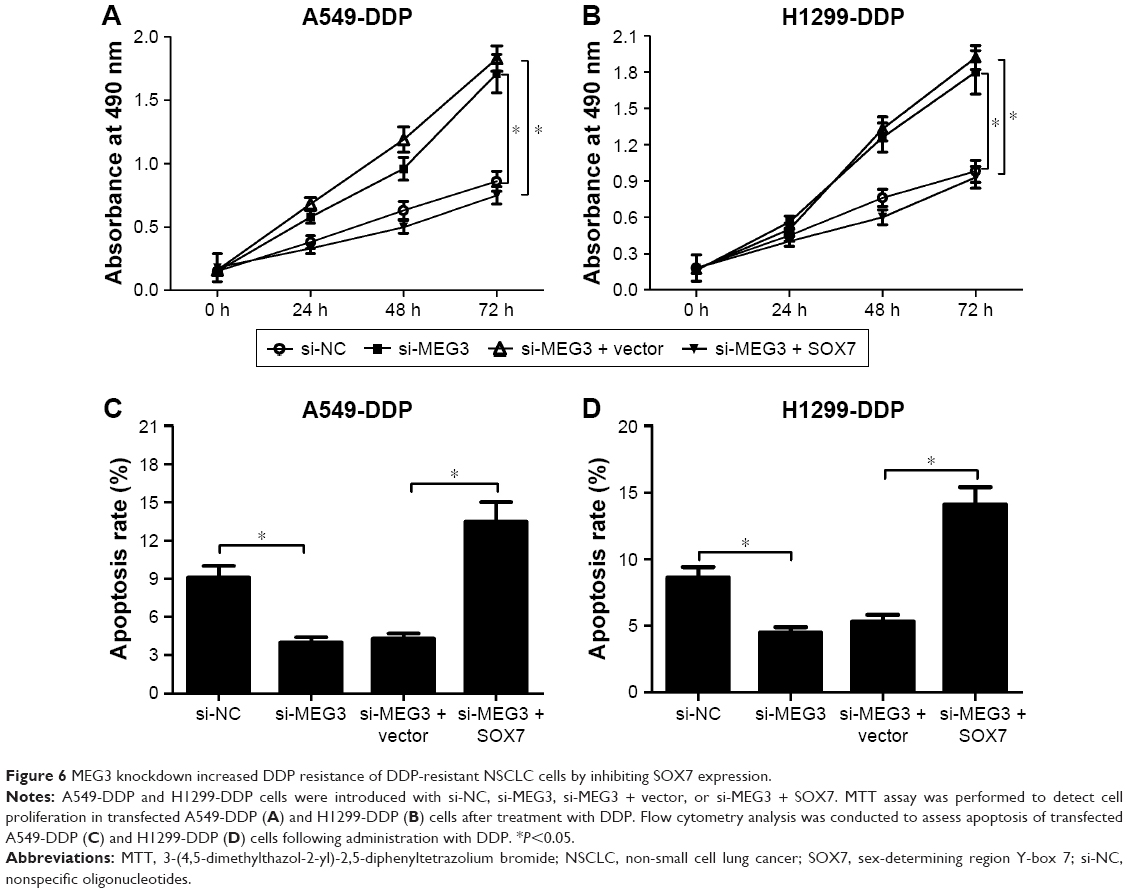 | Figure 6 MEG3 knockdown increased DDP resistance of DDP-resistant NSCLC cells by inhibiting SOX7 expression. |
MEG3 overexpression enhanced DDP sensitivity of DDP-resistant NSCLC cells in vivo
To confirm the biological role of MEG3 in the development of DDP resistance of NSCLC cells, a nude mouse xenograft model was established by subcutaneously injecting A549-DDP cells transfected with MEG3 or vector into the right flank, followed by administration with an intraperitoneal injection of 4 mg/kg DDP every 4 days. As shown in Figure 7A and B, with or without DDP treatment, MEG3 overexpression significantly restrained tumor growth, demonstrated by decreased tumor volume and depressed tumor weight. Moreover, tumor derived from MEG3-treated A549-DDP cells showed upregulated MEG3 and SOX7 expression, as well as downregulated miR-21-5p (Figure 7C). These findings indicated that MEG3 overexpression induced DDP sensitivity of NSCLC cells in vivo.
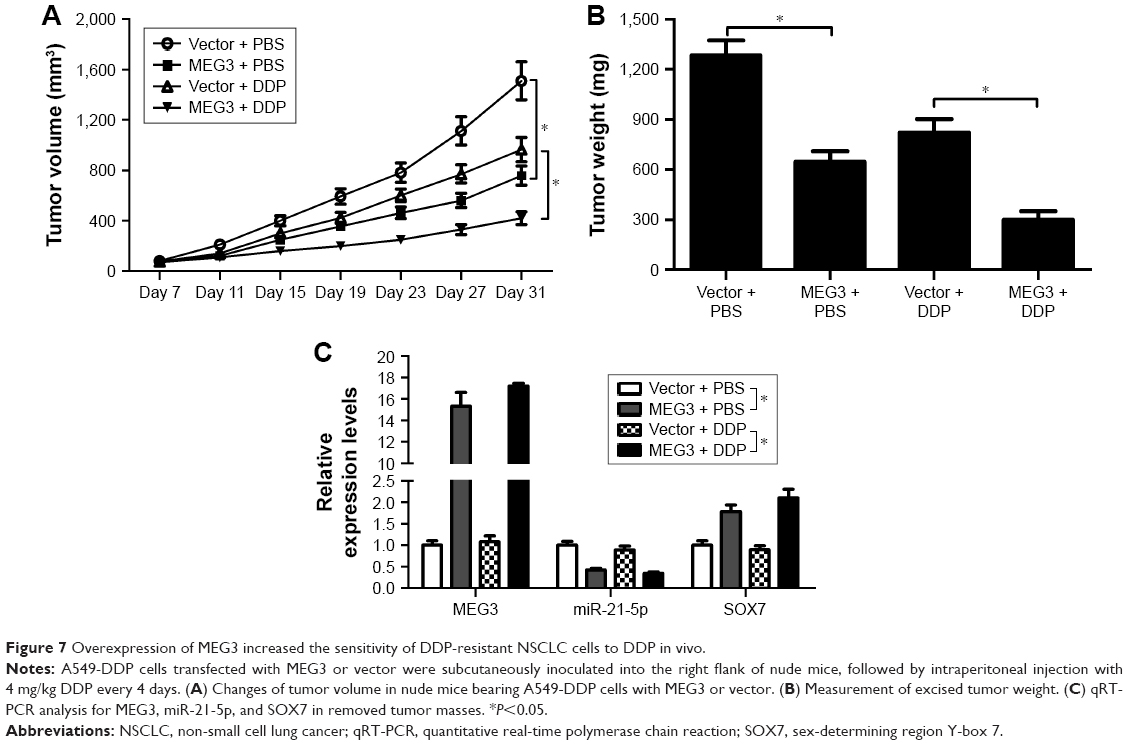 | Figure 7 Overexpression of MEG3 increased the sensitivity of DDP-resistant NSCLC cells to DDP in vivo. |
Discussion
DDP has been considered as a potential chemotherapeutic choice for NSCLC. Unfortunately, DDP resistance is becoming increasingly severe and remains one of the major impediments in the clinical therapy of lung cancer patients.19 An increasing number of lncRNAs have been demonstrated to participate in the regulation of chemoresistance of various malignancies.20 For example, ectopic expression of lncRNA urothelial carcinoma-associated 1 (UCA1) improved cell survival and enhanced resistance to tamoxifen treatment in breast cancer cells through activation of Wnt/β-catenin pathway.21 LncRNA growth arrest-specific 5 (GAS5) was reported to act as a tumor-suppressing gene to inhibit cell proliferation and reduce the chemotherapy resistance to doxorubicin in bladder transitional cell carcinoma.22 LncRNA regulator of reprogramming (ROR) silencing was revealed to enhance the sensitivity of lung adenocarcinoma cells to DDP by targeting PI3K/Akt/mTOR signaling pathway.23 In this study, we focused on the involvement of MEG3 in DDP resistance in NSCLC. Our study demonstrated that MEG3 was downregulated in DDP-resistant NSCLC tissues and cells. Besides, MEG3 overexpression improved sensitivity of DDP-resistant NSCLC cells to DDP treatment by suppressing cell proliferation and inducing apoptosis, while MEG3 knockdown showed the opposite effect. Also, forced expression of MEG3 was previously reported to decrease NSCLC cell proliferation and induce apoptosis.13,24 Meanwhile, mouse xenograft model assay uncovered that MEG3 overexpression also enhanced sensitivity to DDP in DDP-resistant NSCLC cells in vivo. Consistently, Liu et al14 found that MEG3 overexpression increased the chemosensitivity to DDP both in vitro and in vivo by hindering cell proliferation and promoting apoptosis. Xia et al15 reported that downregulation of MEG3 enhanced the DDP resistance of lung cancer cells by decreasing DDP-induced apoptosis and altering cell cycle distribution through activation of the WNT/β-catenin signaling pathway. However, the underlying molecular basis of MEG3 in acquired resistance to DDP remains to be further elaborated.
The mechanisms by which lncRNAs exert their functions were diverse in different kinds of cancers. Interestingly, increasing evidence has suggested that lncRNA may serve as ceRNAs or miRNA sponges to influence miRNAs, leading to a change in the expression of miRNA target genes.25 For example, lncRNA taurine-upregulated gene 1 (TUG1) acted as a ceRNA to sponge miR-9-5p, triggering downregulation of its target POU class 2 homeobox 1 (POU2F1) expression and facilitating the tumorigenesis of osteosarcoma.26 UCA1 functioned as a ceRNA of multidrug resistance protein-1 (MDR1) through completely binding to miR-16 in chronic myeloid leukemia cells, thereby contributing to imatinib resistance.27 Upregulated lncRNA colon cancer-associated transcript-1 (CCAT1) has been shown to enhance paclitaxel resistance in nasopharyngeal cancer cells via miR-181a/cytoplasmic polyadenylation element-binding protein 2 (CPEB2) axis.28 In our study, we demonstrated that MEG3 acted as a ceRNA to sponge miR-21-5p, repressing miR-21-5p expression. Further rescue experiments were performed after transfected cells were treated with DDP. miR-21-5p overexpression dramatically restored MEG3-induced cell dysfunction in A549-DDP cells, including growth blockage and apoptosis enhancement. In addition, miR-21-5p inhibition markedly attenuated MEG3 deficiency-elicited pro-proliferative and anti-apoptotic effect in H1299-DDP cells. These results suggested that MEG3 reduced DDP resistance of DDP-resistant lung cancer cells by sponging miR-21-5p. Similarly, in chronic myeloid leukemia, MEG3 reversed imatinib resistance through regulating miR-21, and subsequent proliferation and apoptosis.29 It is well documented that miRNAs play important roles in regulating resistance to chemotherapeutic agents in various cancers.30 miR-21-5p, an important oncogenic miRNA, has been demonstrated to confer chemoresistance in many cancers, including ovarian cancer,31 pancreatic cancer,32 and tongue cancer.33 It was documented that miR-21 silencing reversed DDP resistance of lung cancer cells by inhibiting cell growth and cell cycle, increasing apoptosis and modulating multidrug resistance (MDR)-related gene expression.34 Moreover, locked nucleic acid (LNA)-based knockdown of miR-21 enhanced sensitivity of lung cancer cells to DDP in vitro and in vivo by inhibiting growth and inducing death.35 The current study further demonstrated that SOX7 was a direct target of miR-21-5p in lung cancer cells and MEG3 positively regulated SOX7 expression by inhibiting miR-21-5p. Moreover, MEG3 knockdown-induced pro-proliferative and anti-apoptotic effects were substantially reversed in DDP-resistant NSCLC cells following SOX7 overexpression. SOX7, a member of the SOX family of transcription factors, has been proposed to act as a tumor suppressor in multiple cancers, such as oral squamous cell carcinoma,36 acute myeloid leukemia,37 glioma,38 and lung cancer.39 Overall, our data demonstrated that MEG3 upregulation enhanced DDP sensitivity in NSCLC through inhibiting cell proliferation and inducing apoptosis by regulating miR-21-5p/SOX7 axis. However, the detailed downstream pathways need to be elaborated in future studies.
Conclusion
We demonstrated that MEG3 was downregulated and the upregulation of MEG3 enhanced DDP sensitivity of DDP-resistant NSCLC cells in vitro and in vivo. Further mechanistic analyses revealed that MEG3 overexpression enhanced DDP sensitivity in NSCLC through inhibiting cell proliferation and inducing apoptosis by regulating miR-21-5p/SOX7 axis, shedding light on the molecular mechanism of MEG3 involved in DDP resistance of lung cancer cells. Therefore, MEG3 may be a potential efficacious target for reversing DDP resistance in the chemotherapy of lung cancer.
Acknowledgments
The authors thank all participants involved in this study. There was no funding for this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. | ||
Pirker R. Adjuvant chemotherapy in patients with completely resected non-small cell lung cancer. Transl Lung Cancer Res. 2014;3(5):305–310. | ||
Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016;73(13):2491–2509. | ||
Schmitt A, Chang H. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29(4):452–463. | ||
Xia H, Hui KM. Mechanism of cancer drug resistance and the involvement of noncoding RNAs. Curr Med Chem. 2014;21(26):3029–3041. | ||
Liu Y, Xu N, Liu B, et al. Long noncoding RNA RP11-838N2. 4 enhances the cytotoxic effects of temozolomide by inhibiting the functions of miR-10a in glioblastoma cell lines. Oncotarget. 2016;7(28):43835–43851. | ||
Fang S, Gao H, Tong Y, et al. Long noncoding RNA-HOTAIR affects chemoresistance by regulating HOXA1 methylation in small cell lung cancer cells. Lab Invest. 2016;96(1):60–68. | ||
Miyoshi N, Wagatsuma H, Wakana S, et al. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells. 2000;5(3):211–220. | ||
Ying L, Huang Y, Chen H, et al. Downregulated MEG3 activates autophagy and increases cell proliferation in bladder cancer. Mol Biosyst. 2013;9(3):407–411. | ||
Wang P, Ren Z, Sun P. Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J Cell Biochem. 2012;113(6):1868–1874. | ||
Yin DD, Liu ZJ, Zhang E, Kong R, Zhang ZH, Guo RH. Decreased expression of long noncoding RNA MEG3 affects cell proliferation and predicts a poor prognosis in patients with colorectal cancer. Tumour Biol. 2015;36(6):4851–4859. | ||
Lu KH, Li W, Liu XH, et al. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013;13(1):461. | ||
Liu J, Wan L, Lu K, et al. The long noncoding RNA MEG3 contributes to cisplatin resistance of human lung adenocarcinoma. PLoS One. 2015;10(5):e0114586. | ||
Xia Y, He Z, Liu B, Wang P, Chen Y. Downregulation of Meg3 enhances cisplatin resistance of lung cancer cells through activation of the WNT/β-catenin signaling pathway. Mol Med Rep. 2015;12(3):4530–4537. | ||
Liz J, Esteller M. lncRNAs and microRNAs with a role in cancer development. Biochim Biophys Acta. 2016;1859(1):169–176. | ||
Jiang Z, Yin J, Fu W, et al. MiRNA 17 family regulates cisplatin-resistant and metastasis by targeting TGFbetaR2 in NSCLC. PLoS One. 2014;9(4):e94639. | ||
Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17(5):272–283. | ||
Hou Z, Xu C, Xie H, et al. Long noncoding RNAs expression patterns associated with chemo response to cisplatin based chemotherapy in lung squamous cell carcinoma patients. PLoS One. 2014;9(9):e108133. | ||
Malek E, Jagannathan S, Driscoll JJ. Correlation of long non-coding RNA expression with metastasis, drug resistance and clinical outcome in cancer. Oncotarget. 2014;5(18):8027–8038. | ||
Liu H, Wang G, Yang L, Qu J, Yang Z, Zhou X. Knockdown of long non-coding RNA UCA1 increases the tamoxifen sensitivity of breast cancer cells through inhibition of Wnt/beta-catenin pathway. PLoS One. 2016;11(12):e0168406. | ||
Zhang H, Guo Y, Song Y, Shang C. Long noncoding RNA GAS5 inhibits malignant proliferation and chemotherapy resistance to doxorubicin in bladder transitional cell carcinoma. Cancer Chemother Pharmacol. 2017;79(1):49–55. | ||
Shi H, Pu J, Zhou XL, Ning YY, Bai C. Silencing long non-coding RNA ROR improves sensitivity of non-small-cell lung cancer to cisplatin resistance by inhibiting PI3K/Akt/mTOR signaling pathway. Tumour Biol. 2017;39(5):1010428317697568. | ||
Yan-Hua L, Xiang-Lei L, Hong L, Jian-Jun W. Long noncoding ribonucleic acids maternally expressed gene 3 inhibits lung cancer tumor progression through downregulation of MYC. Indian J Cancer. 2015;52(suppl 3):E190–E193. | ||
Wang K, Long B, Zhou LY, et al. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat Commun. 2014;5:3596. | ||
Xie CH, Cao YM, Huang Y, et al. Long non-coding RNA TUG1 contributes to tumorigenesis of human osteosarcoma by sponging miR-9-5p and regulating POU2F1 expression. Tumour Biol. 2016;37(11):15031–15041. | ||
Xiao Y, Jiao C, Lin Y, et al. LncRNA UCA1 contributes to imatinib resistance by acting as a ceRNA Against miR-16 in chronic myeloid leukemia cells. DNA Cell Biol. 2017;36(1):18–25. | ||
Wang Q, Zhang W, Hao S. LncRNA CCAT1 modulates the sensitivity of paclitaxel in nasopharynx cancers cells via miR-181a/CPEB2 axis. Cell Cycle. 2017;16(8):795–801. | ||
Zhou X, Yuan P, Liu Q, Liu Z. LncRNA MEG3 regulates imatinib resistance in chronic myeloid leukemia via suppressing microRNA-21. Biomol Ther (Seoul). 2017;25(5):490–496. | ||
Zhang W, Lei P, Dong X, Xu C. The new concepts on overcoming drug resistance in lung cancer. Drug Des Devil Ther. 2014;8(23):735–744. | ||
Lam AYC, Ngai-Na C, Tetsushi T, et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun. 2016;7:11150. | ||
Wei X, Wang W, Wang L, et al. MicroRNA-21 induces 5-fluorouracil resistance in human pancreatic cancer cells by regulating PTEN and PDCD4. Cancer Med. 2016;5(4):693–702. | ||
Zheng G, Li N, Jia X, et al. MYCN-mediated miR-21 overexpression enhances chemo-resistance via targeting CADM1 in tongue cancer. J Mol Med. 2016;94(10):1129–1141. | ||
Xu L, Huang Y, Chen D, et al. Downregulation of miR-21 increases cisplatin sensitivity of non-small-cell lung cancer. Cancer Genet. 2014;207(5):214–220. | ||
Dong Z, Ren L, Lin L, Li J, Huang Y, Li J. Effect of microRNA-21 on multidrug resistance reversal in A549/DDP human lung cancer cells. Mol Med Rep. 2015;11(1):682–690. | ||
Oh KY, Hong KO, Huh YS, Lee JI, Hong SD. Decreased expression of SOX7 induces cell proliferation and invasion and correlates with poor prognosis in oral squamous cell carcinoma. J Oral Pathol Med. Epub 2017 Mar 7. | ||
Man CH, Fung TK, Wan H, et al. Suppression of SOX7 by DNA methylation and its tumor suppressor function in acute myeloid leukemia. Blood. 2015;125(25):3928–3936. | ||
Zhao T, Yang H, Tian Y, et al. SOX7 is associated with the suppression of human glioma by HMG-box dependent regulation of Wnt/β-catenin signaling. Cancer Lett. 2016;375(1):100–107. | ||
Han L, Wei W, Wei D, Zhang L. MiR-9 is involved in TGF-β1-induced lung cancer cell invasion and adhesion by targeting SOX7. J Cell Mol Med. 2017;21(9):2000–2008. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.