")
Back to Journals » OncoTargets and Therapy » Volume 13
lncRNA MALAT1 Promotes EMT Process and Cisplatin Resistance of Oral Squamous Cell Carcinoma via PI3K/AKT/m-TOR Signal Pathway
Received 27 February 2020
Accepted for publication 22 April 2020
Published 12 May 2020 Volume 2020:13 Pages 4049—4061
DOI https://doi.org/10.2147/OTT.S251518
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Gaetano Romano
Ran Wang,1,* Xinxing Lu,2,* Riyue Yu1
1Department of Stomatology, Beijing Shijitan Hospital, Capital Medical University, Beijing, People’s Republic of China; 2Department of Urology, Beijing Chao-Yang Hospital, Capital Medical University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Riyue Yu Email [email protected]
Background: Cisplatin (DDP) is the first-line chemotherapy agent for the treatment of oral squamous cell carcinoma (OSCC). The emergence of DDP resistance leads to diminished drug efficacy and survival benefit. lncRNA MALAT1 has been considered as one of the most important factors in OSCC. It has also been reported to enhance chemo-resistance in other kinds of carcinomas. However, little is known about the role of lncRNA MALAT1 in DDP resistance of OSCC.
Materials and Methods: Two kinds of human DDP-resistant cell lines (CAL-27R and SCC-9R) were developed from cisplatin-naïve cell lines (CAL-27 and SCC-9, respectively) as in vitro cell models. Cell transfection was performed to overexpress or knockdown MALAT1 in these cells. Mouse xenograft models were also established. The following measurements were performed: cell proliferation, colony formation, wound healing, transwell, and TUNEL assays, as well as Western blot and immunofluorescence staining.
Results: DDP-resistant cells showed higher expression level of MALAT1 compared to cisplatin-naïve cells. The overexpression of MALAT1 in cisplatin-naïve cells enhanced DDP resistance and suppressed apoptosis in OSCC cells. However, the knockdown of MALAT1 in DDP-resistance cells induced apoptotic cell death and restored the sensitivity to DDP. Further analyses suggested that MALAT1 might promote DDP resistance via regulating P-glycoprotein expression, epithelial–mesenchymal transition process, and the activation of PI3K/AKT/m-TOR signaling pathway.
Conclusion: MALAT1 might be a potential therapeutic target for the treatment of DDP-resistant OSCC.
Keywords: oral squamous cell carcinoma, cisplatin resistance, lncRNA MALAT1, P-glycoprotein
Introduction
Oral squamous cell carcinoma (OSCC) is one of the most common carcinomas of the oral cavity.1,2 Despite the substantial progress in cancer management, there has been little improvement in the survival rate of OSCC over the past few decades.3 Cisplatin (DDP)-based chemotherapy is the standard first-line therapy for the treatment of locally advanced or metastatic OSCC.4 DDP is an alkylating chemotherapeutic agent that is able to form DNA adducts and cross-links, leading to mitotic stasis at the G2/M checkpoint.5 However, acquired drug resistance greatly hampers the therapeutic efficacy of DDP.6 It has been widely demonstrated that cell proliferation, apoptosis, angiogenesis, and EMT (epithelial–mesenchymal transition) are involved in DDP resistance, but overcoming drug resistance to DDP remains a challenge worldwide.7–9 Thus, it is of great significance to better understand the molecular mechanisms underlying DDP resistance and search for novel therapeutic targets for OSCC.
lncRNA is a class of non-coding RNAs with more than 200 nucleotides in length and play pivotal roles in tumorigenesis and chemo-resistance.10 Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is located on chromosome 11q13 with a length of over 8000 nucleotides.11 It was first identified as an oncogene in metastasis-associated lung adenocarcinoma as a result of its role in promoting the migration and metastasis of lung cancer cells.11 Previous data also revealed that MALAT1 was involved in a variety of pathological processes, such as carcinogenesis,12 retinal neurodegeneration,13 and vascular growth.14 Moreover, MALAT1 has been reported to promote proliferation, metastasis, and EMT through multiple signaling pathways in OSCC.15–18 However, the regulatory function of MALAT1 in DDP resistance remains unclear.
In the study, we investigated the role of MALAT1 in chemosensitivity of OSCC cells to DDP both in vitro and in vivo. Our data showed that MALAT1 overexpression induced DDP resistance in OSCC cells and MALAT1 knockdown restored the sensitivity of DDP-resistant cells by regulating P-glycoprotein (P-gp) expression, EMT process, and the activation of PI3K/AKT/m-TOR signaling pathway. Our study reported the regulatory effects of MALAT1 in DDP-resistant OSCC for the first time, which provided novel insights for the treatment of DDP-resistant OSCC.
Materials and Methods
Ethics Statement
The study protocols were approved by the Committee of Animal Experimentation and the Ethics Committee of Capital Medical University and Beijing Shijitan Hospital. All experiments were performed in accordance with the NIH guidelines for animal care and use.19
Antibodies and Reagents
All antibodies were purchased from Abcam (Cambridge, USA), including anti-GAPDH, anti-PI3K, anti-p-PI3K, anti-Akt, anti-p-Akt, anti-m-TOR, anti-p-m-TOR, anti-Bax, anti-bcl-2, anti-E-cadherin, anti-N-cadherin, anti-P-glycoprotein (P-gp) antibody (at 1:1000 dilution, respectively) and HRP-labelled goat anti-mouse IgGs (at 1:2000 dilution). Cisplatin (DDP) was purchased from Selleck Chemicals (Houston, USA). DMSO was obtained from Sigma (St. Louis, USA).
Cell Culture and Establishment of DDP-Resistant Cell Lines
Human OSCC cell lines (CAL-27 and SCC-9) were provided by the Cell Bank of Peking Union Medical College and cultured in 1640 medium (Hyclone, UT) supplemented with 10% fetal bovine serum (Hyclone, UT). DDP-resistant OSCC cells (CAL-27 and SCC-9) were established by stepwise exposure to increasing concentrations of DDP.20 The exposure was terminated when cells were able to divide normally in the medium containing 10 µM DDP. These cells were considered as DDP-resistant cells and named as CAL-27R and SCC-9R. CAL-27 and SCC-9 cells at similar passage numbers were used as ageing controls. DDP-resistant cells were maintained in complete culture medium containing 10 μM DDP. Before further experiments, DDP-resistant cells were cultured without DDP for 3 days. The degree of DDP resistance of each cell line was evaluated before each experiment.
Cell Transfection
The plasmids overexpressing MALAT1 (pcDNA3.1-MALAT1) and the negative control (Vector) were provided by Fenhui Biotechnologies (Hunan, China). The small interfering RNAs (siRNAs) targeting MALAT1 were provided by Fenhui Biotechnologies (Hunan, China) and the sequences were as follow: si-MALAT1-1, 5′ GCCCGAGACTTCTGTAAAGGA-3′, si-MALAT1-2, 5′-AGCCCGAGACTTCTGTAAAGG-3′, si-MALAT1-3, 5′-GCAGCCCGAGACTTCTGTAAA-3′, si-MALAT1-4, 5′-GCTCTAAATTGTTGTGGTTCT-3′. Cells were transfected with designated plasmids or siRNAs using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol.
RNA Isolation and qPCR
To measure the expression level of MALAT1 in OSCC cells, total RNAs were extracted from cells using TRIzol agent (Invitrogen, Carlsbad, USA) and reverse transcribed to cDNA by PrimeScript™ RT reagent Kit with gDNA Eraser (Takara). Subsequently, qPCR was performed by using Power SYBR Green PCR Master Mix (Applied Biosystems, USA) with the following primers: MALAT1, forward: 5ʹ-GCTCTGTGGTGTGGGATTGA-3ʹ and reverse: 5ʹ-GTGGCAAAATGGCGGACTTT-3ʹ; GAPDH, forward: 5ʹ- TCCTGGGCTACACTGAGCAC-3ʹ and reverse: 5ʹ- CTGTTGCTGTAGCCAAATTCGTTG-3ʹ. The relative mRNA expression of MALAT1 was calculated using the 2−ΔΔCt method and normalized to GAPDH.
Cell Proliferation Assay
The cell proliferation ability of OSCC cells was measured by using a CCK-8 kit (KeyGEN BioTECH, China). Cells were seeded into 96-well plates at a density of 5×103 cells per 200 µL in triplicate. After 24 h, culture medium was replaced by the medium containing different concentrations of DDP and cells were cultured for further 48 h. Two hours before measurement, 10 µL CCK-8 was added to each well and cells were cultured for another 2 h. The absorbance was recorded at 450 nm by an immunosorbent assay plate reader (Bio-Rad Laboratories Inc, USA). The inhibition curves of DDP for each group of cells were achieved. DDP at a dosage of 10µM inhibited approximately 40–80% cell growth, thus was selected for subsequent analyses.
Colony Formation Assay
Cells were incubated with 10 μM DDP or vehicle control for 48 h. Then they were trypsinized, seeded into 6-well culture dishes at a density of 200 cells per well, and cultured for 10 days without changing the medium. After fixation with 10% formaldehyde for 10 minutes, cells were stained by Giemsa Stain solution (Solarbio Inc, Beijing, China) for 15 minutes. Then, colonies with > 50 cells were counted under a dissecting microscope. The relative percentage of cell survival was calculated using Image J software.
Cell Migration Assay
Cell migration capacity was evaluated by wound healing assay. Cells at 90% confluency were plated in 6-well plates and incubated with vehicle control or 10 μM DDP for 24 h. Then a 200-μL pipette tip was used to scratch the cell monolayer to create a linear wound. Cells were washed twice with PBS and then treated with serum-free 1640 medium for 48 h. Images were taken under an inverted microscope at 0 h (baseline images) and 24 h (final images). The wound healing area was calculated using Image J software.
Cell Invasion Assay
Cell invasion capability was determined by transwell assay. The 24-well culture inserts (BD Biosciences, USA) with 8-μm pores coated with Matrigel (1 mg/mL; BD Biosciences) were used. The membranes of upper chamber were coated with Matrigel (BD) and then incubated for 6 h at 37 °C. Cells were treated with vehicle control or 10μM DDP for 48 h, and then transferred into the upper chamber filled with serum-free media. A volume of 0.6 mL complete medium was added to the lower chamber. After 24 h, cells were washed twice with PBS, fixed with paraformaldehyde, and stained with Giemsa Stain solution (Solarbio Inc, Beijing, China). Cells in the upper chamber were removed by cotton swabs. Cells on the bottom surface of the membranes were photographed.
Apoptosis Analysis by TUNEL Assay
The rate of apoptosis was measured by TUNEL assay. Cells were cultured in 12-well plates for 24 h followed by the exposure to designated stimuli for 48 h. The apoptosis rate was determined by a TUNEL apoptosis in situ detection kit (KeyGEN, China) according to the manufacturer’s protocol. The number of positively stained cells was counted using a microscope.
Western Blot
Protein lysates were prepared on ice using ice-cold RIPA buffer (Beyotime, Beijing, China) containing protease inhibitor cocktail (Roche, Switzerland). Total protein isolated from cells was quantified by the Bradford Protein Assay (Bio-Rad Laboratories, USA). After boiled at 100°C for 10 min, protein samples were separated by SDS-PAGE and then transferred to PVDF membrane. The membrane was blocked with 5% (w/v) fetal bovine serum at room temperature for 2 h and then incubated with mouse monoclonal primary antibody at 4°C overnight. Then, the membrane was washed three times with TBST followed by the incubation with HRP-labelled secondary antibody (1:2000; Abcam) at room temperature for 1 h. The chemo-luminescent signal was detected using Clarity™ ECL Western Substrate and quantified by Quantity One Software (Bio-Rad Laboratories). GAPDH was used as an internal control.
Animal Study
BALB/c nude mice were obtained from the experimental animal ministry of Capital Medical University. Mice were subcutaneously injected with 1×106 CAL-27 (n = 15) or CAL-27R (n = 15) cells suspended in a mixture (100 μL) of matrigel and 1640 medium (1:1) into the right axillary fossa. Mice inoculated with CAL-27 cells were randomly assigned into 3 groups (n = 5 per group). Animals injected with CAL-27R were also randomly divided into 3 groups (n = 5 per group). The intervention treatment was initiated when tumors reached a volume of 200 mm3. DDP (5mg/kg) or vehicle control was intraperitoneally injected every 7 days (Day 1, 8, 15, 22). Tumor sizes were measured every 3 days by caliper during the study. Tumor volume was calculated by the formula: L×S2×0.5, in which L represented the longest diameter and S represented the shortest diameter. Mice were anesthetized and sacrificed at the end of the study, and tumors were isolated and weighted.
Immunofluorescence Staining
Xenograft tumors were fixed in 10% formalin and paraffin-embedded for immunofluorescence staining. The 5-µm-thick sections were deparaffinized with xylene, rehydrated in an alcohol gradient, and immersed in 3% H2O2. Then samples were incubated with anti-PCNA (1:1000 diluted, Abcam) antibody at 4 °C for 12 h and then with a secondary antibody (Invitrogen). After staining with DAPI, the images were captured under a fluorescence microscope.
Statistical Analysis
All statistical analyses were performed using SPSS (22.0) and the data were shown as mean ± standard deviation (SD). Student’s t-test was used to analyze the differences between two groups and one-way ANOVA was used for multiple comparisons. Differences were considered significant when p < 0.05.
Results
Upregulated MALAT1 Expression in DDP-Resistant OSCC Cell Lines
The expression level of MALAT1 in DDP-resistant cells (CAL-27R and SCC-9R) was measured by qPCR. Data showed that the expression MALAT1 was significantly upregulated in DDP-resistant cells as compared with DDP naïve cells (Figure 1A), suggesting a potential role of MALT1 in DDP-resistant OSCC.
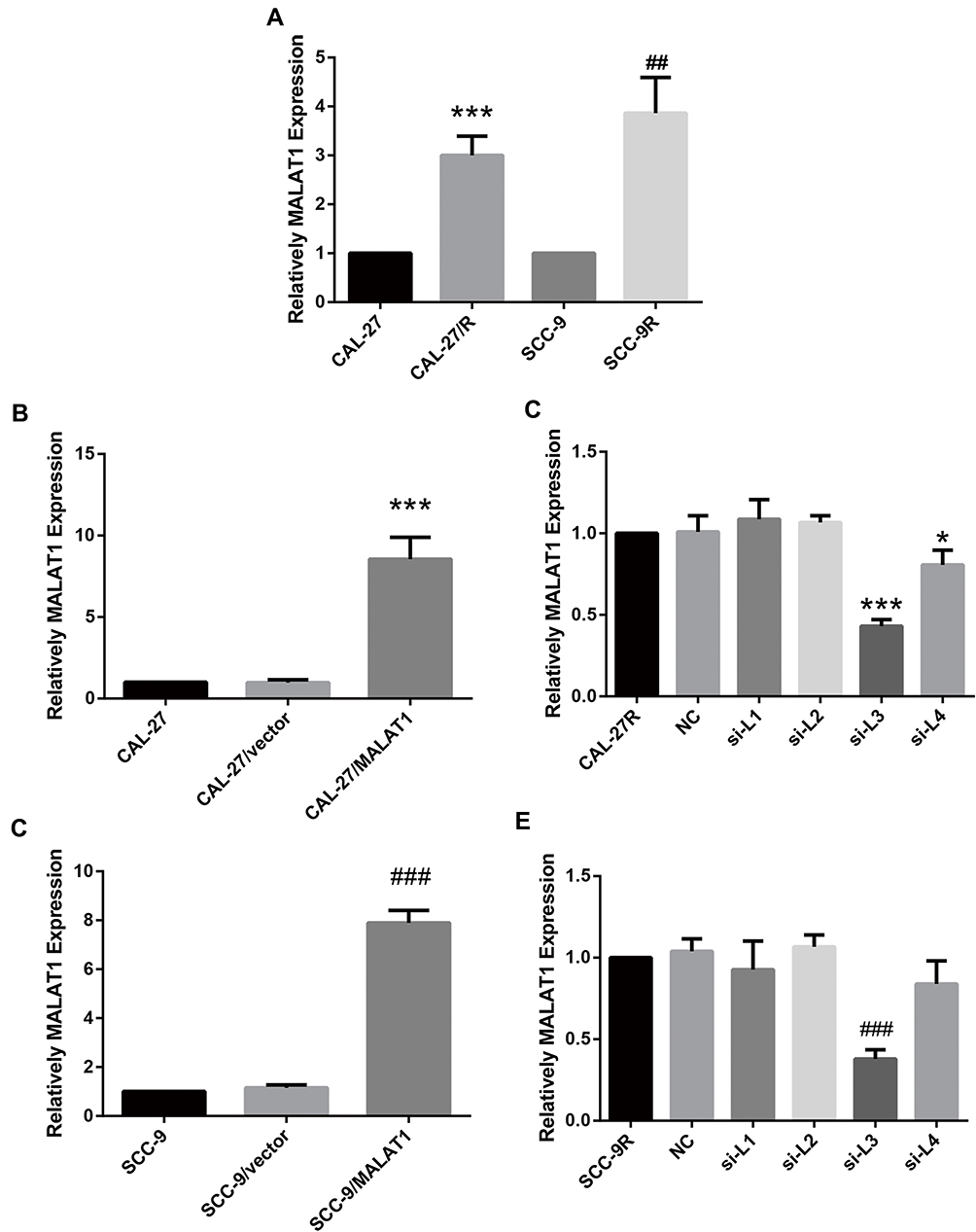 |
Figure 1 MALAT1 was upregulated in DDP-resistant OSCC cells compared with their parental DDP naïve cells. (A) qPCR: MALAT1 was up-regulated in DDP-resistant OSCC cells compared with the parental DDP naïve cells. (B) qPCR: MALAT1 overexpression validation in CAL-27. (C) qPCR: MALAT1 knockdown validation in CAL-27R. (D) qPCR: MALAT1 overexpression validation in SCC-9. (E) qPCR: MALAT1 knockdown validation in CAL-27R. *P<0.05, ***P<0.001, compared with CAL-27 or CAL-27R group. ##P<0.01, ###P<0.001, compared with SCC-9 or SCC-9R group. |
Then, DDP naïve cells were transfected with plasmids overexpressing MALAT1 or siRNAs against MALAT1. The efficiency of transfection was measured by qPCR (Figure 1B–E). As shown in Figure 1C and E, si-L3 led to the most efficient inhibition of MALAT1 expression in cells. Thus, we chose si-L3 (hereafter termed “si-MALAT1”) for subsequent experiments.
Effects of MALAT1 Expression on Cell Proliferation and Colony in DDP Naïve Cells and DDP-Resistant Cells
CCK-8 assay was performed to evaluate the viability of cells treated with DPP or vehicle. CAL-27R and SCC-9R cells showed significantly stronger resistance against DDP treatment compared with their parental DDP naïve cells (CAL-27 and SCC-9) (Figure 2A and B). CAL-27 and SCC-9 cells overexpressing MALAT1 exhibited significantly enhanced resistance to DDP when compared with control cells or cells transfected with control vector. On the contrary, the transfection of DDP-resistant cells with si-MALAT1 significantly suppressed the resistance of cells to DDP as compared to control cells or cells delivered with si-NC.
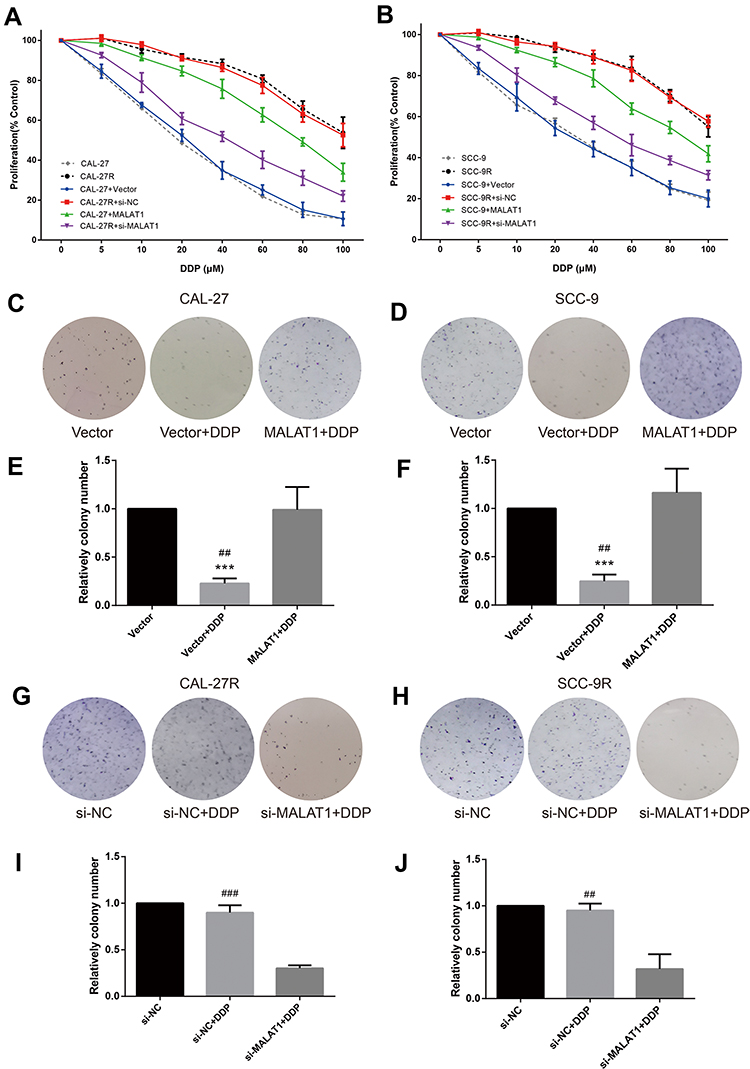 |
Figure 2 MALAT1 up-regulation enhanced DDP resistance and MALAT1 down-regulation reversed DDP resistance on cell proliferation and colony formation in human OSCC cell lines. (A, B) CCK-8 assay: indicated cells were treated with different concentrations of DDP as indicated. Cell viability was determined in 48h. (B) qPCR: MALAT1 overexpression validation in CAL-27. (C–J) colony formation assay was utilized to evaluate the capability of indicated cells with indicated treatment on colony formation. ***P<0.001, compared with Vector group. ##P<0.01, ###P<0.001, compared with MALAT1+DDP or si-MALAT1+DDP group. |
The effect of MALAT1 expression on DDP resistance of OSCC cells were also investigated by colony formation assays. Consistent with the results of CCK-8 assay, the number of colonies in DDP naïve cells (CAL-27 and SCC-9) transfected with control vectors was significantly decreased after the treatment with 10μM DDP (Figure 2C–F), while MALAT1 overexpression significantly increased the number of colonies in DDP-treated cells. DDP-resistant cells (CAL-27R and SCC-9R) transfected with si-NC showed almost complete resistance to 10μM DDP, whereas the knockdown of MALAT1 by si-MALAT1 restored the sensitivity of CAL-27R and SCC-9R to DDP, as evidenced by reduced colony number (Figure 2G–J).
Effects of MALAT1 Expression on Cell Migration and Invasion in DDP Naïve Cells and DDP-Resistant Cells
Wound healing assay was performed to evaluate cell migration ability in OSCC cells incubated with 10μM DDP or vehicle control. Results revealed that the migration of DDP naïve cells (CAL-27 and SCC-9) was significantly inhibited by DDP treatment compared with the Vector group (Figures 3A and B and 4A and B). However, the overexpression of MALAT1 significantly induced the resistance of DDP naïve cells to DDP treatment, as shown by increased migration ability compared to the Vector + DDP group. The DDP treatment did not inhibited the migration of both DDP-resistant cell lines (CAL-27R and SCC-9R) (Figures 3C and D and 4C and D). The knockdown of MALAT1, however, significantly suppressed the migration of DDP-resistant cells.
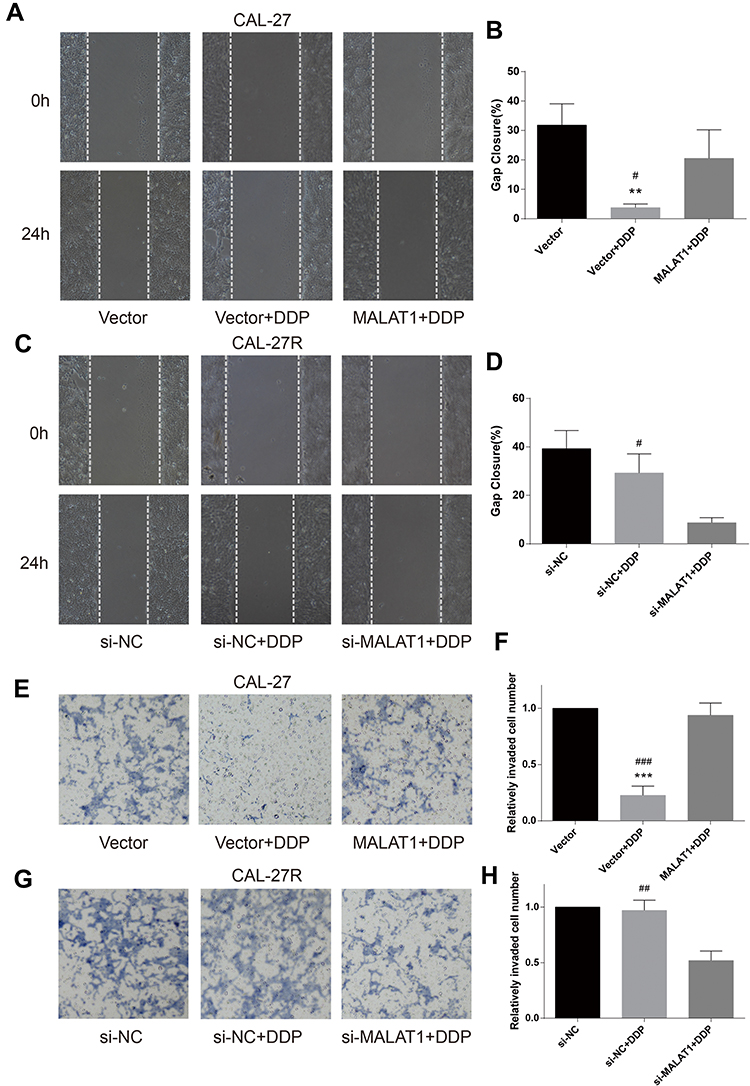 |
Figure 3 MALAT1 up-regulation enhanced DDP resistance and MALAT1 down-regulation reversed DDP resistance on migration and invasion in CAL-27 cell lines. (A–D) wound healing assay were carried out to measure the migratory ability of indicated cells. (E–H) Transwell assays were carried out to measure the invasive ability of indicated cells. **P<0.01, ***P<0.001, compared with Vector group. #P<0.05, ##P<0.01, ###P<0.001, compared with MALAT1+DDP or si-MALAT1+DDP group. |
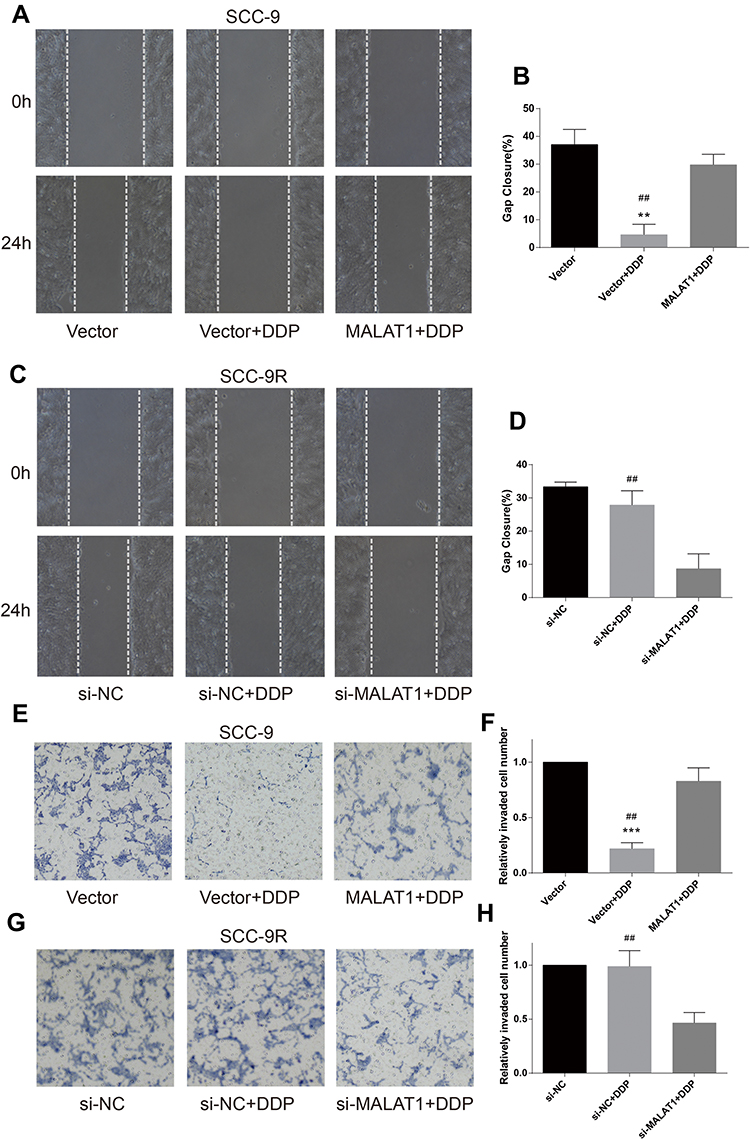 |
Figure 4 MALAT1 up-regulation enhanced DDP resistance and MALAT1 down-regulation reversed DDP resistance on migration and invasion in SCC-9 cell lines. (A–D) wound healing assay were carried out to measure the migratory ability of indicated cells. (E–H) Transwell assays were carried out to measure the invasive ability of indicated cells. **P<0.01, ***P<0.001, compared with Vector group. ##P<0.01, compared with MALAT1+DDP or si-MALAT1+DDP group. |
The invasion capacity of cells was detected by Transwell assay, and the results were consistent with those of wound healing assay (Figures 3E–H and 4E–H). Taken together, the upregulation of MALAT1 enhanced DDP resistance in DDP naïve cells and MALAT1 knockdown restored the sensitivity of CAL-27R and SCC-9R to DDP in terms of cell migration and invasion.
Effects of MALAT1 Expression on the Apoptosis of CAL-27 and CAL-27R Cell Lines
The TUNEL assay showed that the apoptosis of CAL-27 cells was significantly induced by DDP treatment, but suppressed by the overexpression of MALAT1 (Figure 5A and B). Although the exposure to DDP did not alter the apoptosis rate in CAL-27R cells (Figure 5C and D), the downregulation of MALAT1 significantly induced apoptotic cell death in DDP-resistant cells.
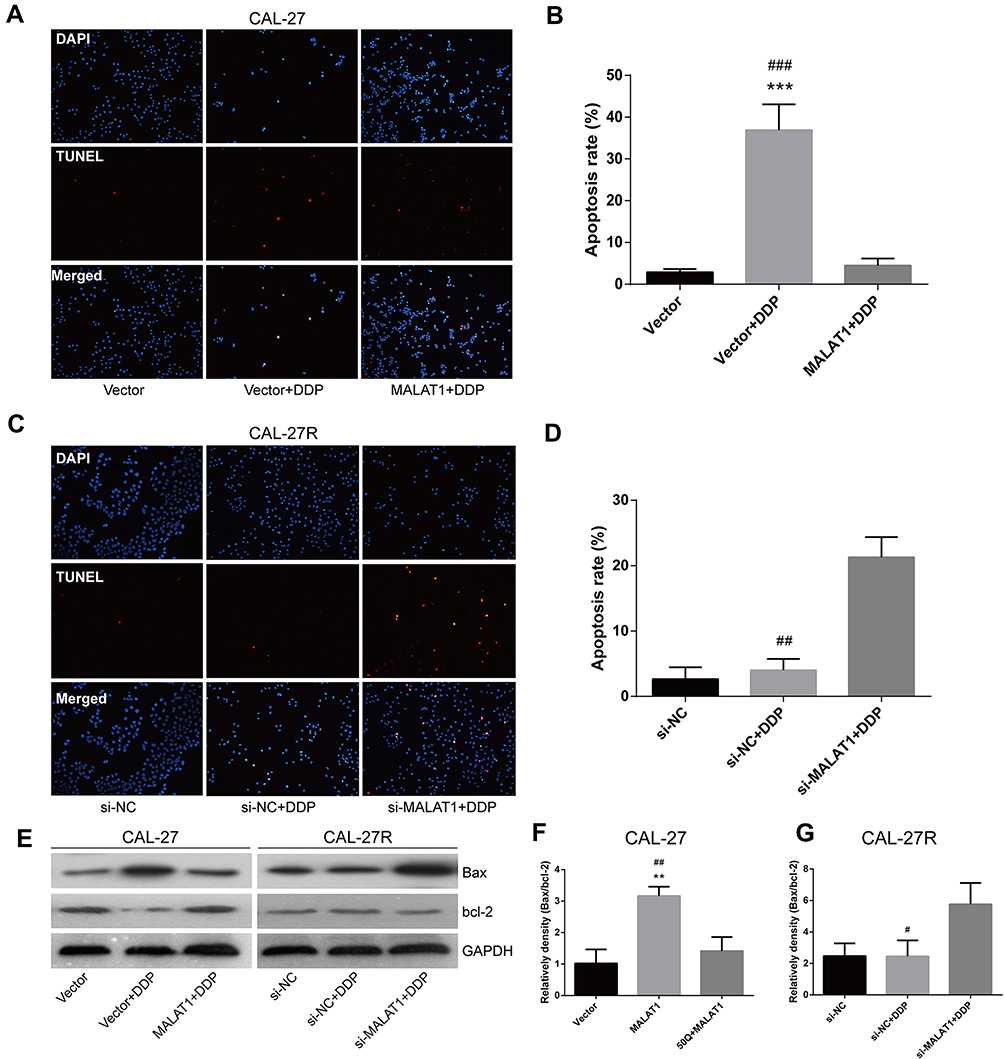 |
Figure 5 MALAT1 up-regulation enhanced DDP resistance and MALAT1 down-regulation reversed DDP resistance via cell apoptosis in CAL-27 cell lines. (A–D) TUNEL assay was used to assess the apoptosis rate of cells under indicated treatment. (E–G) Associated proteins that participated in the apoptosis were examined by the Western blot analysis. **P<0.01, ***P<0.001, compared with Vector group. #P<0.05, ##P<0.01, ###P<0.001, compared with MALAT1+DDP or si-MALAT1+DDP group. |
The expressions of apoptosis-related proteins (Bax and Bcl-2) were measured by Western blot (Figure 5E). We found that the overexpression of MALAT1 protected CAL-27 cells from apoptosis (Figure 5E and F), while MALAT1 knockdown significantly induced the apoptosis of CAL-27R cells by regulating the expressions of anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bax. Taken together, our results indicated that MALAT1 might be an important target in DDP-mediated apoptosis in OSCC cells.
MALAT1 Regulated DDP Resistance in Mouse Xenograft Models of OSCC
To explore the effect of MALAT1 on DDP resistance in vivo, CAL-27 cells, MALAT1-overexpressed CAL-27 cells, CAL-27R cells, CAL-27R cells with MALAT1 knockdown were subcutaneously injected into nude mice. All mice underwent an administration of DDP (5mg/kg) or vehicle every 7 days, and the tumor size was measured every 3 days. At 28 days after injection, mice were sacrificed and tumors were removed and weighed (Figure 6A). The tumors of the Vector + DDP group were smaller compared with the CAL-27 vector group, while mice inoculated with cells overexpressing MALAT1 and treated with DDP showed a tumor volume similar to that in the Vector group (Figure 6B). In the xenograft model of CAL-27R cells, no significant difference was observed in tumor volume between si-NC group and si-NC + DDP group, whereas MALAT1 knockdown significantly suppressed tumor growth as compared to si-NC + DDP group (Figure 6C). The results of tumor weight were consistent with that of tumor growth (Figure 6D and E).
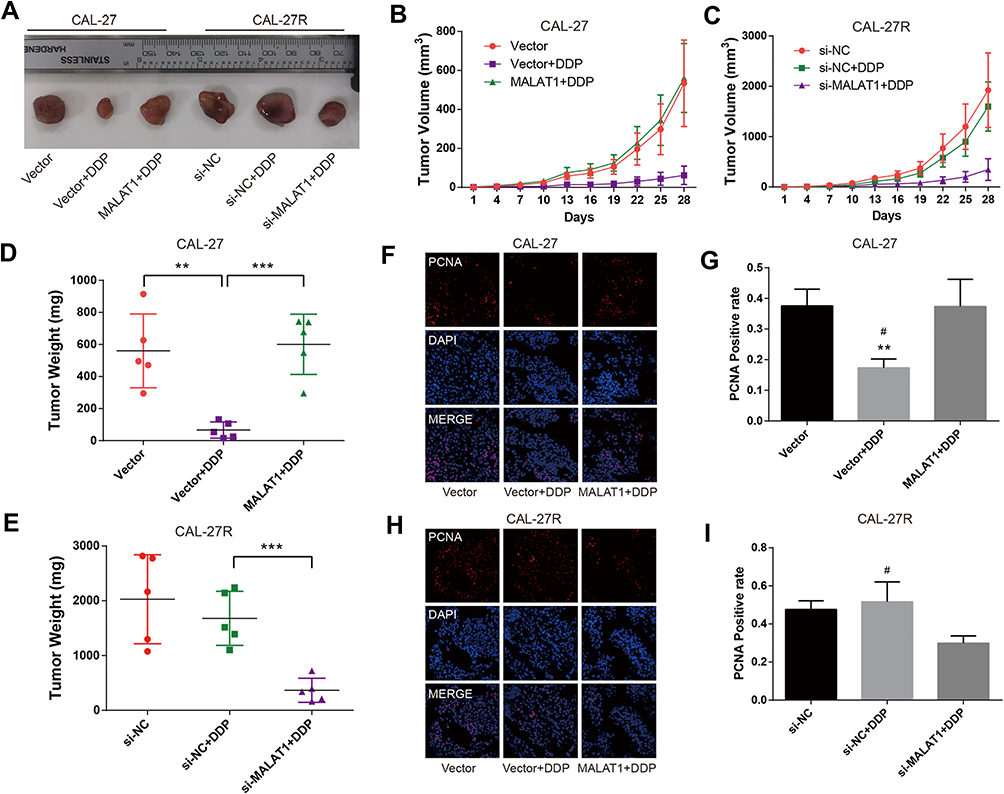 |
Figure 6 MALAT1 regulated DDP resistance in nude mice xenograft models of OSCC. (A) Photographs of dissected tumors in nude mice. (B, C) The tumor volume fluctuation of the mice was measured every three days. (D, E), The tumor weight of the mice was measured in the end. (F–I) immunofluorescence staining detected PCNA-positive cells in xenograft tumor tissue. **P<0.01, ***P<0.001, compared with Vector group. #P<0.05, compared with MALAT1+DDP or si-MALAT1+DDP group. |
In addition, immunofluorescence staining demonstrated that the expression of PCNA was suppressed by DDP treatment in mice injected with CAL-27 cells, but restored by the upregulation of MALAT1 (Figure 6F and G). In CAL-27R cell xenograft tumors, DDP treatment did not affect the expression of PCNA. However, the sensitivity to DDP was restored by the downregulation of MALAT1 via si-MALAT transfection (Figure 6H and I). These results implied that MALAT1 played an important regulatory role in DDP resistance of OSCC in vivo.
MALAT1 Regulated DDP Resistance via Inducing P-Gp Expression, EMT Process, and the Activation of PI3K/AKT/m-TOR Signaling Pathway
To explore the mechanisms involved in MALAT1-regulated DDP resistance in OSCC, the expressions of P-gp, EMT markers (E-cadherin & N-cadherin), and proteins associated with PI3K/AKT/m-TOR signaling pathway were detected by Western blot (Figure 7A). The overexpression of MALAT1 in CAL-27 cells significantly increased the expression ratio of p-PI3K/PI3K, p-AKT/AKT, p-m-TOR/m-TOR, and P-gp/GAPDH, and decreased the ratio of E-cadherin/N-cadherin (Figure 7B). In CAL-27R cells, the knockdown of MALAT1 significantly decreased the expression ratio of p-PI3K/PI3K, p-AKT/AKT, p-m-TOR/m-TOR, and P-gp/GAPDH, but elevated the levels of E-cadherin (Figure 7C). Collectively, the above findings indicated that MALAT1 might promote EMT and DDP resistance of OSCC cells via upregulating P-gp and activating the PI3K/AKT/m-TOR signaling pathway.
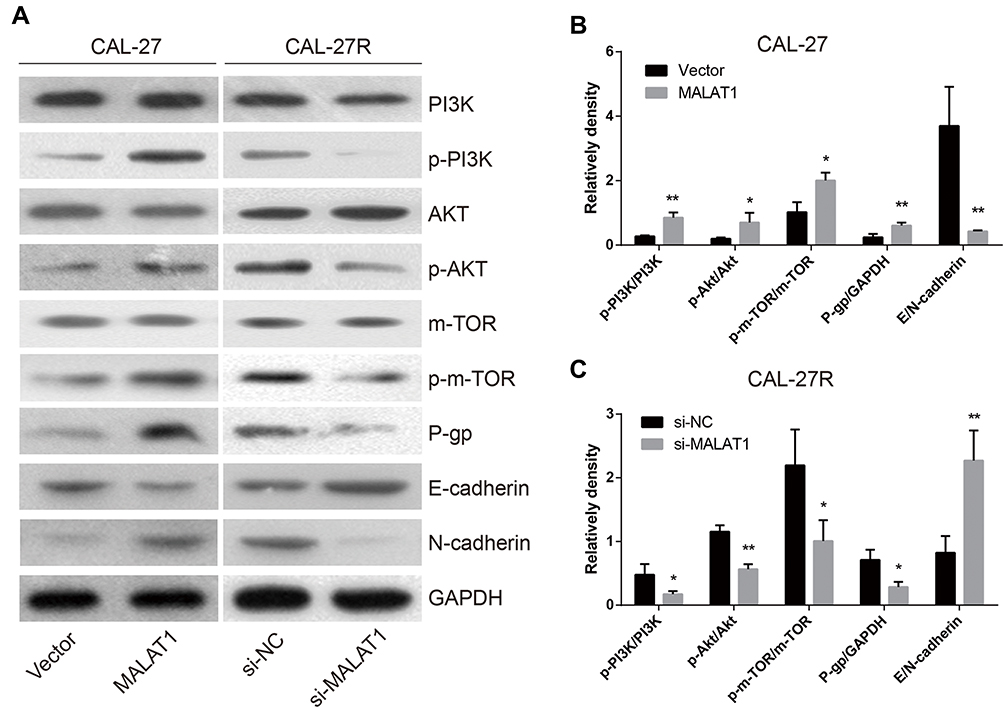 |
Figure 7 MALAT1 regulated DDP resistance via up-regulation of P-gp, activation of EMT and PI3K/AKT/m-TOR signal pathway. (A–C) The Western blot assay was applied to examine the expression levels of P-gp and the corresponding proteins in the EMT process and PI3K/AKT/m-TOR signaling pathway. *P<0.05, **P<0.01, compared with Vector or si-NC group. |
Discussion
OSCC is a common fatal cancer that contributes to a major cause of health burden around the world.21 Despite great advances in the development of chemotherapy, acquired resistance remains an obstacle for the treatment of OSCC.22–24 DDP resistance is one of the most important factors for poor prognosis in OSCC patients. Thus, there is an urgent need to investigate the mechanisms involved in DDP resistance in OSCC. Previous studies showed that the expression of MALAT1 was significantly upregulated in OSCC and positively correlated with poor prognosis.25 Moreover, MALAT1 knockdown suppressed tumorigenesis and promoted apoptosis of OSCC,26 indicating that MALAT1 might be a potential factor for the diagnosis, prognosis, and treatment of OSCC. Recently, MALAT1 has been reported as a chemotherapeutic resistant factor in various cancers, such as colorectal cancer,27 lymphoma,28 glioblastoma,29 ovarian cancer, and lung cancer.30,31 Therefore, we assumed that MALAT1 might play an important role in DDP resistance of OSCC.
In this study, two DDP-resistant OSCC cell lines (CAL-27R and SCC-9R) were developed from DDP naïve OSCC cell lines (CAL-27 and SCC-9, respectively) by stepwise exposure of parental cells with increasing concentrations of DDP. We found that the expression of lncRNA MALAT1 in DDP-resistant OSCC cells was significantly higher than that in DDP naïve cells. Then we stably transfected DDP naïve OSCC cell lines (CAL-27 and SCC-9) with plasmids overexpressing MALAT1 and delivered siRNAs against MALAT1 in DDP-resistant OSCC cell lines (CAL-27R and SCC-9R). Further analyses showed that DDP naïve cells with MALAT1 overexpression showed stronger resistance against DDP compared with control vector-transfected DDP naïve cells, while the knockdown of MALAT1 restored the sensitivity of DDP-resistant cells to DDP, as shown by decreased proliferation, colony formation, migration, invasion, and increased apoptosis. These findings were also confirmed by the data from mouse xenograft models.
The underlying mechanism of DDP resistance in OSCC cells was complicated. To further investigate the regulation of MALAT1 in this process, we examined the protein expressions of CAL-27 cells, CAL-27 cells overexpressing MALAT1, CAL-27R cells, and CAL-27R cells with MALAT1 knockdown (Figure 7A). The aberrant activation of PI3K/AKT/m-TOR signaling pathway has been considered as a leading cause of DDP resistance. The activation of this cascade leads to excessive expressions of multi-drug resistance proteins, as well as multiple oncogenes and growth factors, such as VEGF, c-myc, and survivin.32–34 Our results suggested that the activation of PI3K/AKT/m-TOR signaling pathway was significantly induced by MALAT1 overexpression, and suppressed by the knockdown of MALAT1. The above results implied that MALAT1 might promote DDP resistance in OSCC cells partially via activating the PI3K/AKT/m-TOR signaling pathway.
It has been proved that EMT is another major cause of DDP resistance.34–36 Here, we observed that the level of MALAT1 was closely correlated with the expressions of EMT markers, E-cadherin and N-cadherin. MALAT1 upregulated N-cadherin and downregulated E-cadherin, therefore promoting the EMT process. These data confirmed the regulatory effect of MALAT1 on EMT in DDP resistance.
P-gp is an important factor in chemo-resistance, especially for the development of multidrug resistance.37,38 It acts as a drug efflux pump for multiple chemotherapeutic drugs, including DDP. P-gp is encoded by the multidrug resistance-1 (MDR1) gene, which is overexpressed in DDP cells.39 In the present study, the level of P-gp in MALAT1-overexpressed OSCC cells was increased compared to cells with normal MALAT1 expression, while P-gp was downregulated by the knockdown of MALAT1 in DDP-resistant cells. These findings suggested that MALAT1 upregulation stimulated DDP resistance partially by increasing P-gp expression.
Conclusions
In summary, our results demonstrated that MALAT1 induced EMT and DDP resistance in OSCC cells via the activation of PI3K/AKT/m-TOR signaling pathway and the upregulation of P-gp, suggesting that MALAT1 overexpression might be an important factor for acquired DDP resistance in OSCC. These findings may provide evidence for the potential use of MALAT1 as a biomarker and a therapeutic target for the treatment of DDP-resistant OSCC.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Leemans CR, Tiwari R, Nauta JJ, Van der Waal I, Snow GB. Regional lymph node involvement and its significance in the development of distant metastases in head and neck carcinoma. Cancer. 1993;71(2):452–456. doi:10.1002/1097-0142(19930115)71:2<452::AID-CNCR2820710228>3.0.CO;2-B
3. Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45(4–5):309–316. doi:10.1016/j.oraloncology.2008.06.002
4. Adelstein D, Gillison ML, Pfister DG, et al. NCCN guidelines insights: head and neck cancers, version 2.2017. J Natl Compr Canc Netw. 2017;15(6):761–770. doi:10.6004/jnccn.2017.0101
5. Yuan L, Yu W-M, Qu C-K. DNA damage-induced G2/M checkpoint in SV40 large T antigen-immortalized embryonic fibroblast cells requires SHP-2 tyrosine phosphatase. J Biol Chem. 2003;278(44):42812–42820. doi:10.1074/jbc.M305075200
6. Zhang P, Zhang Z, Zhou X, Qiu W, Chen F, Chen W. Identification of genes associated with cisplatin resistance in human oral squamous cell carcinoma cell line. BMC Cancer. 2006;6(1):224. doi:10.1186/1471-2407-6-224
7. Shen D-W, Pouliot LM, Hall MD, Gottesman MM, Sibley DR. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64(3):706–721. doi:10.1124/pr.111.005637
8. Galluzzi L, Vitale I, Michels J, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5(5):e1257.
9. Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res. 2016;106:27–36. doi:10.1016/j.phrs.2016.01.001
10. Iyer MK, Niknafs YS, Malik R, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47(3):199. doi:10.1038/ng.3192
11. Ji P, Diederichs S, Wang W, et al. MALAT-1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22(39):8031. doi:10.1038/sj.onc.1206928
12. Latorre E, Carelli S, Raimondi I, et al. The ribonucleic complex HuR-MALAT1 represses CD133 expression and suppresses epithelial–mesenchymal transition in breast cancer. Cancer Res. 2016;76(9):2626–2636. doi:10.1158/0008-5472.CAN-15-2018
13. Yao J, Wang XQ, Li YJ, et al. Long non‐coding RNA MALAT1 regulates retinal neurodegeneration through CREB signaling. EMBO Mol Med. 2016;8(4):346–362. doi:10.15252/emmm.201505725
14. Michalik KM, You X, Manavski Y, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114(9):1389–1397. doi:10.1161/CIRCRESAHA.114.303265
15. Zhou X, Liu S, Cai G, et al. Long non coding RNA MALAT1 promotes tumor growth and metastasis by inducing epithelial-mesenchymal transition in oral squamous cell carcinoma. Sci Rep. 2015;5:15972. doi:10.1038/srep15972
16. Zhang T-H, Liang L-Z, Liu X-L, et al. Long non-coding RNA MALAT1 interacts with miR-124 and modulates tongue cancer growth by targeting JAG1 retraction in/10.3892/or. 2018.6688. Oncol Rep. 2017;37(4):2087–2094. doi:10.3892/or.2017.5445
17. Fang Z, Zhang S, Wang Y, et al. Long non-coding RNA MALAT-1 modulates metastatic potential of tongue squamous cell carcinomas partially through the regulation of small proline rich proteins. BMC Cancer. 2016;16(1):706. doi:10.1186/s12885-016-2735-x
18. Chang SM, Hu WW. Long non‐coding RNA MALAT1 promotes oral squamous cell carcinoma development via microRNA‐125b/STAT3 axis. J Cell Physiol. 2018;233(4):3384–3396. doi:10.1002/jcp.26185
19. Council NR. Guide for the Care and Use of Laboratory Animals. National Academies Press; 2010.
20. Lin W, Li D, Lin Y, Du C, Chen J, Hong C. Establishment of a cisplatin-induced human nasopharyngeal carcinoma drug-resistant cell line and its biological characteristics. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2008;22(21):992–994, 997.
21. Ramqvist T, Dalianis T. An epidemic of oropharyngeal squamous cell carcinoma (OSCC) due to human papillomavirus (HPV) infection and aspects of treatment and prevention. Anticancer Res. 2011;31(5):1515–1519.
22. Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869. doi:10.1038/onc.2011.384
23. Perez R. Cellular and molecular determinants of cisplatin resistance. Eur J Cancer. 1998;34(10):1535–1542. doi:10.1016/S0959-8049(98)00227-5
24. Xiong P, Li Y-X, Tang Y-T, Chen H-G. Proteomic analyses of Sirt1-mediated cisplatin resistance in OSCC cell line. Protein J. 2011;30(7):499–508. doi:10.1007/s10930-011-9354-9
25. Liu S, Zhou X, Wang X, et al. Metastasis-associated lung adenocarcinoma transcript 1 modulates oral squamous cell carcinoma invasion in vitro and in vivo. Chin J Clin Oncolo. 2015;9:460–465.
26. Guo Y, Ma Y, Hu X, Song R, Zhu L, Zhong M. Long non-coding RNA CEBPA-AS1 correlates with poor prognosis and promotes tumorigenesis via CEBPA/Bcl2 in oral squamous cell carcinoma. Cancer Biol Ther. 2018;19(3):205–213. doi:10.1080/15384047.2017.1416276
27. Li P, Zhang X, Wang H, et al. MALAT1 is associated with poor response to oxaliplatin-based chemotherapy in colorectal cancer patients and promotes chemoresistance through EZH2. Mol Cancer Ther. 2017;16(4):739–751.
28. Li L-J, Chai Y, Guo X-J, Chu S-L, Zhang L-S. The effects of the long non-coding RNA MALAT-1 regulated autophagy-related signaling pathway on chemotherapy resistance in diffuse large B-cell lymphoma. Biomed Pharmacother. 2017;89:939–948. doi:10.1016/j.biopha.2017.02.011
29. Chen W, Xu X-K, Li J-L, et al. MALAT1 is a prognostic factor in glioblastoma multiforme and induces chemoresistance to temozolomide through suppressing miR-203 and promoting thymidylate synthase expression. Oncotarget. 2017;8(14):22783.
30. Bai L, Wang A, Zhang Y, Xu X, Zhang X. Knockdown of MALAT1 enhances chemosensitivity of ovarian cancer cells to cisplatin through inhibiting the Notch1 signaling pathway. Exp Cell Res. 2018;366(2):161–171. doi:10.1016/j.yexcr.2018.03.014
31. Cui Y, Li G, Zhang X, Dai F, Zhang R. Increased MALAT1 expression contributes to cisplatin resistance in non‑small cell lung cancer. Oncol Lett. 2018;16(4):4821–4828. doi:10.3892/ol.2018.9293
32. Lee S, Choi E-J, Jin C, Kim D-H. Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA amplification contributes to cisplatin resistance in an ovarian cancer cell line. Gynecol Oncol. 2005;97(1):26–34. doi:10.1016/j.ygyno.2004.11.051
33. Johnson-Holiday C, Singh R, Johnson EL, Grizzle WE, Lillard JW, Singh S. CCR9-CCL25 interactions promote cisplatin resistance in breast cancer cell through Akt activation in a PI3K-dependent and FAK-independent fashion. World J Surg Oncol. 2011;9(1):46. doi:10.1186/1477-7819-9-46
34. Tan J, You Y, Xu T, et al. Par-4 downregulation confers cisplatin resistance in pancreatic cancer cells via PI3K/Akt pathway-dependent EMT. Toxicol Lett. 2014;224(1):7–15. doi:10.1016/j.toxlet.2013.10.008
35. Bingjing Z, Yuliang W, Hu L, Xiangdong Z. CA916798 gene is involved in cisplatin resistance in human lung cancer through PI3K/AKT pathway. J Third Mil Med Univ. 2013;7:11.
36. Haslehurst AM, Koti M, Dharsee M, et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer. 2012;12(1):91. doi:10.1186/1471-2407-12-91
37. Lu C, Shan Z, Li C, Yang L. MiR-129 regulates cisplatin-resistance in human gastric cancer cells by targeting P-gp. Biomed Pharmacother. 2017;86:450–456. doi:10.1016/j.biopha.2016.11.139
38. Zhang Y, Xu W, Ni P, Li A, Zhou J, Xu S. MiR-99a and MiR-491 regulate cisplatin resistance in human gastric cancer cells by targeting CAPNS1. Int J Biol Sci. 2016;12(12):1437. doi:10.7150/ijbs.16529
39. Veneroni S, Zaffaroni N, Daidone M, Benini E, Villa R, Silvestrini R. Expression of P-glycoprotein and in vitro or in vivo resistance to doxorubicin and cisplatin in breast and ovarian cancers. Eur J Cancer. 1994;30(7):1002–1007. doi:10.1016/0959-8049(94)90132-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.