")
Back to Journals » OncoTargets and Therapy » Volume 14
LncRNA ILF3-AS1 Promotes the Progression of Colon Adenocarcinoma Cells Through the miR-619-5p/CAMK1D Axis
Received 8 December 2020
Accepted for publication 24 February 2021
Published 12 March 2021 Volume 2021:14 Pages 1861—1872
DOI https://doi.org/10.2147/OTT.S296441
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Wei Liu,1 Shan Li2
1Department of Gastrointestinal Surgery, Xiantao First People’s Hospital Affiliated to Yangtze University, Xiantao, 433000, People’s Republic of China; 2Department of Endocrinology, Xiantao First People’s Hospital Affiliated to Yangtze University, Xiantao, 433000, People’s Republic of China
Correspondence: Shan Li
Xiantao First People’s Hospital Affiliated to Yangtze University, Xiantao, 433000, People’s Republic of China
Email [email protected]
Introduction: Colon adenocarcinoma (COAD) is the third most common tumor of the digestive tract. Recent studies reported that lncRNA’s abnormal expression might play a vital role in the occurrence and development of COAD.
Methods: In the present study, we investigated the expression of ILF3-AS1 in COAD cell lines, human normal colon epithelial cell line, patient tumor tissues and adjacent normal tissues by real-time quantitative PCR (RT-qPCR). Small interfering RNAs (siRNAs) were transfected into COAD cells to inhibit the expression of ILF3-AS1. The effects of ILF3-AS1 on cell proliferation, migration, invasion and apoptosis were measured by CCK-8 assay, transwell migration and invasion assay, and flow cytometry apoptosis assay, respectively. The direct binding of ILF3-AS1 and miR-619-5p/CAMK1D was validated by the luciferase reporter assay. The expression of CAMK1D and epithelial-mesenchymal transformation (EMT)-related proteins was detected by Western Blot analysis. Besides, in vivo experiments were conducted to demonstrate the oncogenic role of ILF3-AS1 in COAD.
Results: The results showed that the expression of ILF3-AS1 was significantly higher in COAD tissue than in normal adjacent samples, and this conclusion was confirmed in the available public datasets. After ILF3-AS1 knockdown, the proliferation of COAD cell lines SW480 and HT29 was significantly inhibited. At the same time, the apoptosis was increased, and the invasion and migration abilities were decreased. After further exploring the mechanisms, we found that ILF3-AS1 serves as a competitive endogenous RNA of mir-619-5p. It can bind to mir-619-5p and reduce its expression, thus regulating the target gene CAMK1D. In addition, we found that high expression of ILF3-AS1 was significantly associated with tumor grade, tumor size, and distant metastasis in COAD samples. In vivo experiments confirmed that ILF3-AS1 promotes tumor growth in COAD models.
Conclusion: The present study demonstrated that ILF3-AS1 plays an oncogenic role in COAD through regulating the miR-619-5p/CAMK1D axis, and inhibition of ILF3-AS1 may pave the way for COAD treatment.
Keywords: lncRNA ILF3-AS1, miR-619-5p, CAMK1D, COAD
Introduction
Colon cancer is the second leading cause of cancer-related death in western countries, and its occurrence is closely associated with a high-fat, low-fiber diet.1 Colon adenocarcinoma (COAD) is the most common type of colon cancer. Early COAD (Stage I) confined to the mucous membranes has a good prognosis, with a five-year survival rate of over 90% after treatment.2 However, because early COAD tends to be asymptomatic, many patients are not detected until the cancer is in the late stages. Metastasis occurs in COAD patients with advanced stages. Patients with lymph node metastasis but no distant metastasis had a 5-year survival rate of 33% to 76%. Despite advances in screening and treatment in recent years, less than 5% of patients with distant metastases, such as liver and lung, survive five years.2
The first step in tumor cell metastasis is EMT, in which tumor cells change epithelial phenotype to mesenchymal-like. During EMT, intercellular interaction and apicobasal polarity disappeared, and mesenchymal markers were obtained on the surface of tumor cells. As a result, tumor cells got the ability to invade and metastasize.3 The EMT process occurs in a variety of epithelial-cell derived tumors and promotes tumor invasion and metastasis. COAD is one of these tumors.4,5
In recent years, with the development of high-throughput sequencing technology, long non-coding RNA (lncRNA) has gradually come into researchers’ sight. LncRNAs are RNAs that are longer than 200 nucleotides and cannot be translated into protein. It has been found to play essential roles in various physiological and pathological processes. It is of vital importance in the development of tumors. The same lncRNA acts differently in different tumors and even in different stages of the same tumor. It may play as an oncogene or a tumor suppressor. Various activities of lncRNAs were reported in previous studies. LncRNAs also play critical roles in COAD. For example, RAMS11 was associated with unfavorable disease-free survival and COAD metastasis. It transcriptionally activated Topoisomerase II alpha (TOP2α) through recruiting Chromobox protein 4, and it was considered a potential biomarker for metastatic COAD patients.6 LNAPPCC formed a positive feedback loop with PCDH7 and was associated with a poor prognosis for COAD patients.7 Some studies have found that lncRNA ILF3-AS1 was related to COAD recurrence and can be used as a marker for COAD relapse.8 But its functional role in COAD has not been studied yet. Previous studies have reported that ILF3-AS1 was dysregulated in various human cancers, and it played critical roles in the development, progression and metastasis of tumors. In retinoblastoma, upregulation of ILF3-AS1 promotes tumor cell proliferation and invasion through the miR-132-5p/SMAD2 axis.9 In melanoma, ILF3-AS1 and ILF3 formed a positive feedback loop to promote proliferation, migration and invasion of tumor cells.10
In the present study, we explored the role of ILF3-AS1 in COAD. Our study demonstrated that ILF3-AS1 was highly expressed in COAD samples and cell lines. It could promote the EMT process of COAD cell line HT-29 and SW480 through the miR-619-5p/CAMK1D pathway, thus promoting the proliferation and invasion of tumor cells.
Materials and Methods
Clinical Samples
Tumor tissues and adjacent normal tissues from 34 COAD samples (21 males and 13 females, 57.92±8.23y) were enrolled in the present. The samples were collected from Xiantao first people’s Hospital Affiliated to Yangtze University from 2011–2018. The patients were diagnosed with COAD pathologically. Patients who received radiotherapy, chemotherapy, immunotherapy, or biotherapy before surgery were excluded from the study. All samples were frozen in the liquid nitrogen tank immediately after surgical removal and were stored at −80°C. The study was approved by the Research Ethics Committee of Xiantao first people’s Hospital Affiliated to Yangtze University, and the informed consent was signed by all participants. The present study was conducted in accordance with the Declaration of Helsinki.
GEO Database
Expression data of ILF3-AS1 in COAD tissues and normal controls were obtained from GSE9348 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE9348) and GSE37364 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37364) datasets. There are 70 tumor samples and 12 normal controls in GSE9348 and 27 tumor samples and 38 normal controls in GSE37364 datasets.
Cell Culture
Human normal colon epithelial cell line NCM460 and colorectal cancer cell line SW620, SW480, HT-29 and HCT116 were purchased from American Type Culture Collection (ATCC, US). Cells were cultured in RPMI-1640 medium (Gibco, US) supplemented with 10% Fetal Bovine Serum (FBS, Gemini, US), 100U/mL penicillin and 100mg/mL streptomycin (Gibco, US). Cells were kept in the incubator at 37°C with 5% CO2.
Cell Transfection
To change the expression of target genes, cell transfection was carried out using Lipofectamine 2000 (Thermo, US) according to the manufacturer’s instruction. Briefly, 1*10^5 cells/well were seeded into a 24-well plate before transfection. On the next day, cells were ready for transfection when the confluence reaches 90%. 0.8ug/50ul DNA and 2ul/50ul Lipofectamine 2000 were added to Opti-MEM (Thermo, US) culture medium, respectively. Next, DNA and Lipofectamine were gently mixed and let stand for 20min at room temperature. Finally, the mixture of DNA and Lipofectamine 2000 was added to the cells. Cells were checked for gene expression by RT-qPCR or Western blot after 48h transfection. siRNA sequence for ILF3-AS1: sense 5ʹ- ACGAAAGAACCCAAGAACCCG-3ʹ, antisense 5ʹ-GGUUCUUGGGUUCUUUCGUCC-3ʹ(si-ILF3-AS1-1); sense 5ʹ- AAUUUUUCCAGGAAAUGGCGG-3ʹ, antisense 5ʹ- GCCAUUUCCUGGAAAAAUUGG-3ʹ (si-ILF3-AS1-2). Sequence for siRNA negative control (si-NC): sense 5ʹ- UUCUCCGAACGUGUCACGU-3ʹ, antisense 5ʹ- ACGUGACACGUUCGGAGAA-3ʹ. Sequence for si-CAMK1D: sense 5ʹ- UAAAACCACUUCGGAAAAGGC’, antisense 5ʹ- CUUUUCCGAAGUGGUUUUAGC-3ʹ. siRNA sequences were synthesized by RiboBio (China). miR-619-5p mimics (miR mimics) and miRNA mimics negative control (mimics NC) were purchased from GenePharma (China).
Luciferase Reporter Assay
The binding sites of miR-619-5p and ILF3-AS1 or CAMK1D were predicted by miRDB (http://mirdb.org/). The mutated binding sites were generated by site-directed mutagenesis (InFusion HD Cloning Plus, Takara, Japan). The fragments containing the binding sites (WT) or the mutant sites (MUT) were inserted into the pGL3 basic vector after synthesis (referred to as ILF3-AS1 WT, CAMK1D WT, ILF3-AS1 MUT, and CAMK1D MUT). HT-29 and SW480 cells were seeded into a 24-well plate and co-transfected with reporter plasmids and miR mimics/mimics-NC. 48h after transfection, cells were collected, and the luciferase activity was detected by a Dual-Luciferase Reporter Assay System (Promega, US).
Transwell Migration and Invasion Assay
2*10^4 cells were seeded into a 24-well transwell chamber (8mm, Corning, US) for migration assay or into a Matrigel-precoated (BD Biosciences) chamber for invasion assay. The lower chamber was filled with 0.5mL RPMI 1640 medium supplemented with 10% FBS. After incubation at 37°C for 48h, cells in the upper chamber were removed. Cells on the surface of the lower chamber were fixed with 4% formaldehyde for 15min and were stained with 0.5% crystal violet for 20min. The results were observed using a microscope (X200, Olympus, JP). ImageJ software (NIH, US, version 1.6.2) was used for data analysis.
Cell Counting Kit-8 (CCK-8) Assay
Cell proliferation was measured through the CCK-8 assay. Briefly, 2*10^4 cells were seeded into a 96-well plate and were cultured for 24, 48 and 72h. Next, 10ul/well CCK-8 solution (Dojindo, Japan) was added to each well for 3h at 37°C. The absorbance was measured at 450nm using a microplate reader (Bio-Rad, US).
Flow Cytometry
Cell apoptosis was measured by flow cytometry apoptotic assay. Briefly, 4*10^5 cells were collected and stained with Annexin V and 7-AAD antibodies (BD Biosciences, US) for 15min. Cell apoptosis was detected by BD FACSCalibur (BD Biosciences), and the results were analyzed through FlowJo software (FlowJo LLC, version 10.0.7).
Western Blot
Protein was extracted using RIPA buffer (10X, Cell Signaling Technology, US). Protein samples (30ug/well) were loaded onto the 10% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide) gel and then were transferred to the PVDF (polyvinylidene fluoride) membrane. The membranes were blocked using 5% nonfat milk followed by the primary antibody incubation at 4°C overnight. The membranes were incubated with HRP-conjugated secondary antibody (1:1000) for 1h at room temperature on the next day. The protein bands were visualized using an ECL substrate kit (Abcam, US). ImageJ software (NIH, US, version 1.6.2) was used for densitometry. Primary antibody used in the present study: anti-CAMK1D (Abcam, ab172618, 1:1000), anti-E-cadherin (Abcam, ab40772, 1:1000), anti-N-cadherin (Abcam, ab76011, 1:1000), anti-Vimentin (Abcam, ab92547, 1:1000), anti-β-Actin (Abcam, ab8226, 1:10000).
Quantitative Reverse Transcription PCR (RT-qPCR) Assay
Total RNA was extracted using TRIzol reagent (Invitrogen, US). Reverse transcription was carried out using Prime ScriptTM RT Master Mix (Takara, JP) according to the manufacturer’s instruction. The quantitative PCR was subsequently performed using SYBR premix Ex Taq II (Takara, JP). Primer sequences were as follows: ILF3-AS1 forward 5ʹ- GCTTTGACGCATGTGTACCT-3ʹ, reverse 5ʹ- CGAAAGAACCCAAGAACCCG-3ʹ; miR-619-5p forward 5ʹ-ACACTCCAGCTGGGCGGGGTTTTGAGGGCG-3ʹ, reverse 5ʹ-CTCAACTGGTGTCGTGGAGTCGG −3ʹ; CAMK1D forward 5ʹ-GACCGGATAGTGGAGAAGGG-3ʹ, reverse 5ʹ- ACATCTCCTTTGCCCTCCAT-3ʹ. GAPDH forward 5ʹ-CACATCGCTCAGACACCATG-3ʹ, reverse 5ʹ-TTGAGGTCAATGAAGGGGTC-3ʹ.
In vivo Experiment
5*105 HT-29 cells/mouse were injected into athymic nude mice (BALB/c, 6-week-old, male, n=4/group) subcutaneously. Mice were divided into the si-NC group or si-ILF3-AS1 treatment group. Mice were treated with 20nM si-NC or si-ILF3-AS1 (tumor mass injection) for 21 days. Tumor volume was calculated according to the formula: Volume=(Length*width)2/2. All mice were sacrificed on day 28. The experiment was conducted under the approval of the Committee on the Ethics of Animal Experiments of Yangtze University. Guidelines for the welfare and treatment of the laboratory animals were followed by the Ministry of Science and Technology of the People’s Republic of China.
Statistical Analysis
Statistical analysis was conducted using GraphPad Prism 8.0 (GraphPad Software, US). All experiments were performed in triplicate. The data was presented as mean±SD. Pairwise comparisons were conducted using the Student’s t-test. Multigroup comparisons were conducted using the two-way ANOVA, followed by Bonferroni’s post hoc test. Survival analysis was carried out using a Kaplan–Meier curve by SPSS Statistics 21.2 software (IBM, US). Univariate and multivariate Cox regression analysis were also conducted using SPSS Statistics 21.2 software. Statistical significance was considered when p<0.05.
Results
ILF3-AS1 Was Upregulated in COAD Tissues and Cell Lines
First, we compared the expression levels of ILF3-AS1 in clinical samples by RT-qPCR. We found that the expression of ILF3-AS1 was significantly upregulated in COAD tissues compared to adjacent normal tissues (5.25±0.93 vs 3.53±0.96, P<0.0001) (Figure 1A). We also validated our findings using a public database. We obtained COAD patient data with ILF3-AS1 expression from the GEO database. Similarly, the results showed that ILF3-AS1 expression was significantly increased in COAD tumor tissues compared to normal tissues (GSE37364: 6.53±2.38 vs 6.24±1.23, P=0.0195, GSE9348: 5.72±3.24 vs 5.09±1.21, P=0.0007) (Figure 1C–D). The expression of ILF3-AS1 was also detected in COAD cell lines and a normal colon cancer epithelial cell line NCM460. The results revealed that the expression of ILF3-AS1 in COAD cell lines (SW460, SW480, HT-29 and HCT116) was 3.30–5.67 folds higher than that in the NCM460 cell line (Figure 1B). Besides, we investigated the impact of the expression of ILF3-AS1 on the survival of COAD patients. As shown in Figure 1E, patients with high ILF3-AS1 expression had a worse prognosis than those with low ILF3-AS1 expression (P=0.0042). Univariate and multivariate cox regression analysis also demonstrated that ILF3-AS1 was an independent prognostic factor for COAD (Table 1).
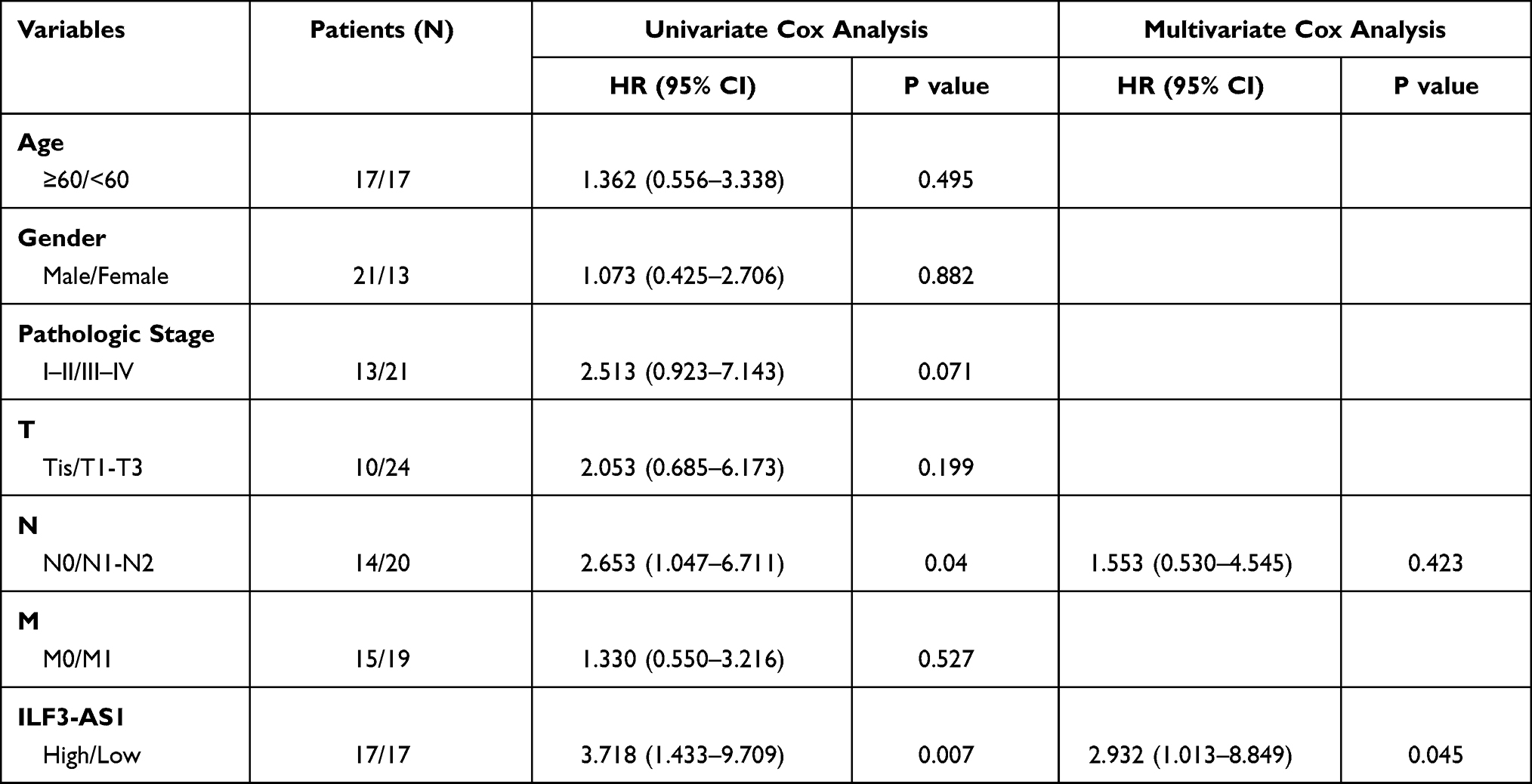 |
Table 1 Univariate and Multivariate Cox Regression Analysis for Clinical Factors and ILF3-AS1 |
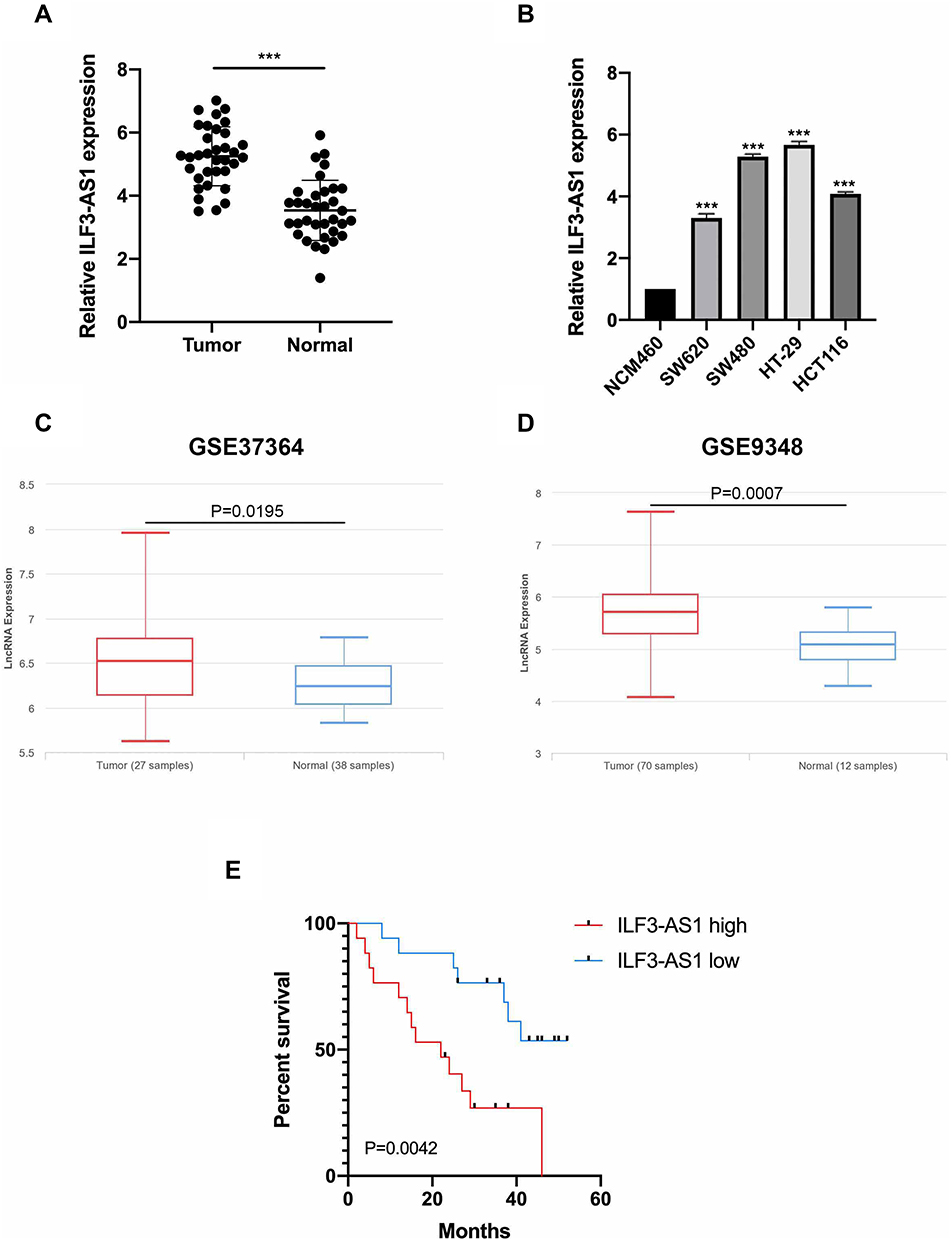 |
Figure 1 ILF3-AS1 was upregulated in COAD tissues and cell lines. The expression of ILF3-AS1 in (A) COAD tissues and adjacent normal tissues, (B) normal colon cancer epithelial cell line NCM460 and COAD cell lines (SW620, SW480, HT-29, HCT116). (C and D) The expression of ILF3-AS1 in COAD samples and normal controls from GSE37364 and GSE9348 datasets. (E) Kaplan–Meier curve for COAD patients with different levels of ILF3-AS1 expression. ***P<0.001. |
ILF3-AS1 Knockdown Inhibited Cell Growth, Migration and Invasion in COAD Cell Lines
Since ILF3-AS1 was highly expressed in COAD samples and cell lines and was associated with poor prognosis, we hypothesized that it might be an oncogene in COAD. Therefore, ILF3-AS1 knockdown was performed in COAD cell line HT-29 and SW480 by siRNA transfection. The knockdown efficiency was validated by RT-qPCR (Figure 2A). Cell proliferation, apoptosis, invasion and migration analysis were carried out using ILF3-AS1 knockdown cells. CCK-8 results showed that the proliferation of ILF3-AS1 knockdown cells was significantly inhibited compared with the negative control and the blank group in both HT-29 and SW480 cell lines (Figure 2B). Apoptosis assay showed that after the expression of ILF3-AS1 was reduced, the apoptosis of HT-29 and SW480 cells was significantly increased (Figure 2C). Transwell migration and invasion assays also revealed that the migration and invasion ability of HT-29 cells was reduced considerably after ILF3-AS1 knockdown (Figure 2D). Statistical results of apoptosis and invasion/migration assays were presented in Figure 2E and F.
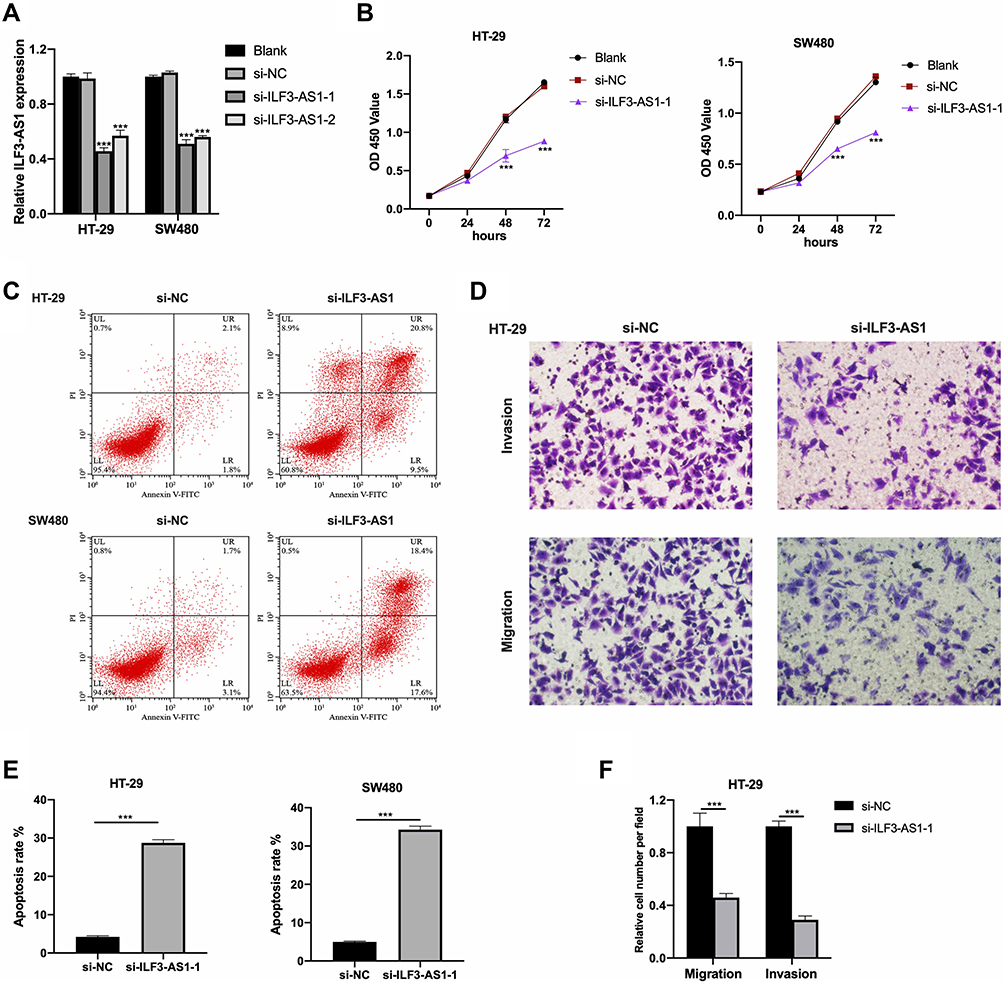 |
Figure 2 ILF3-AS1 knockdown inhibited cell growth, migration and invasion in COAD cell lines. (A) Transfection efficiency of si-ILF3-AS1s in HT-29 and SW480 cells validated by RT-qPCR. The results of (B) CCK-8 assay and (C) flow cytometry apoptosis assay in HT-29 and SW480 cells, and the results of (D) transwell invasion/migration assay in HT-29 cells after ILF3-AS1 knockdown. (E and F) The statistical results of apoptosis assay and transwell invasion/migration assay were shown as bar plots. ***P<0.001. |
ILF3-AS1 Was a Molecular Sponge for miR-619-5p
It has been reported that lncRNAs bind to miRNA and prevent the latter from binding to target genes, thus playing a role in promoting the function of target genes. MiRDB website tool was used to predict miRNAs that could bind to ILF3-AS1, and mir-619-5p was one of them. The predicted binding sites of miR-619-5p and ILF3-AS1 were shown in Figure 3A. We first investigated the expression of miR-619-5p clinical samples. As shown in Figure 3B, miR-619-5p was significantly decreased in COAD tumor samples compared to normal controls. Besides, we also found that mir-619-5p was negatively correlated with the expression of ILF3-AS1 (Figure 3C). Further studies confirmed that the expression level of miR-619-5p in COAD cells transfected with ILF3-AS1 was significantly decreased (Figure 3D). These results demonstrated that ILF3-AS1 regulated the expression of miR-619-5p. To verify the binding between miR-619-5p and ILF3-AS1, the luciferase reporter assay was used. Before luciferase reporter assay, miR-619-5p mimics were transfected into HT-29 and SW480 cells. The transfection efficiency was validated through RT-qPCR (Figure 3E). Luciferase reporter assay showed that luciferase activity was significantly lower in cells co-transfected with ILF3-AS1 WT and miR-619-5p mimics than in cells co-transfected with ILF3-AS1 and mimics-NC. In contrast, in ILF3-AS1 WT cell lines, Luciferase activity showed no significant difference between the miR mimics group and the mimics-NC group (Figure 3F).
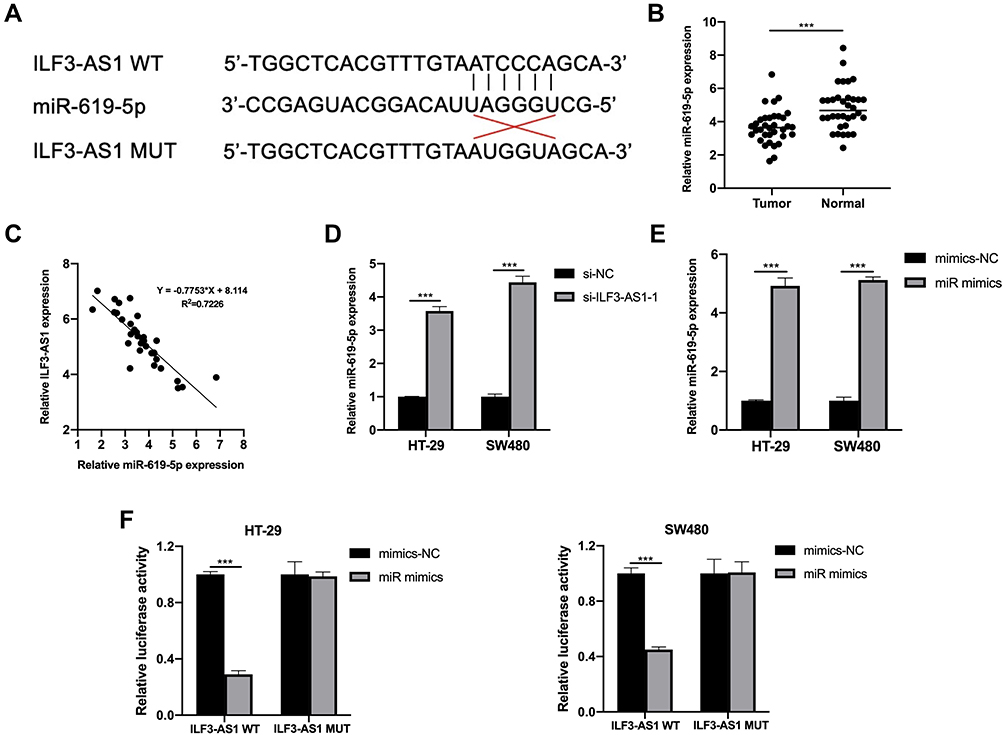 |
Figure 3 ILF3-AS1 was a molecular sponge for miR-619-5p. (A) Predicted binding sites of ILF3-AS1 and miR-619-5p. (B) The expression of miR-619-5p in COAD tissues and adjacent normal tissues. (C) The relationship between the expression of ILF3-AS1 and miR-619-5p. (D) The expression of miR-619-5p after ILF3-AS1 knockdown in HT-29 and SW480 cells. (E) Transfection efficiency of miR mimics validated by RT-qPCR in HT-29 and SW480 cells. (F) Luciferase activities of HT-29 and SW480 cells co-transfected with miR mimics/mimics-NC and luciferase reporters harboring ILF3-AS1 MUT or ILF3-AS1 WT. ***P<0.001. |
CAMK1D Was a Direct Target of miR-619-5p
Next, we explored the downstream target genes of miR-619-5p to find the mechanism by which ILF3-AS1 promoted COAD development. Among the many miR-619-5p target genes predicted by the miRDB web tool, CAMK1D was selected for further validation. This is because CAMK1D has been reported to promote tumor invasion in breast cancer.11 We first examined CAMK1D expression in tumor tissues and found that its expression in COAD samples was significantly higher than in normal controls (Figure 4C). Then we conducted the luciferase reporter assay. The predicted binding sites of CAMK1D and miR-619-5p were shown in Figure 4A. Luciferase reporter assay showed that luciferase activity was significantly lower in the CAMK1D MUT cells transfected with miR-mimics than the control group transfected with mimics-NC. No significant difference between the miR mimics group and the mimics-NC group was observed in luciferase activity in CAMK1D WT cells (Figure 4B). Subsequently, CAMK1D expression was detected in cells transfected with miR-mimics and ILF3-AS1. The transfection efficiency of the ILF3-AS1 plasmid was displayed in Figure 4E. The results showed that the expression of CAMK1D in the miR-mimics group was significantly lower than that in the control group. In comparison, the expression of CAMK1D in the ILF3-AS1 group was considerably higher. For cells co-transfected with miR-mimics and ILF3-AS1, CAMK1D expression showed no significant difference than the control group (Figure 4D–G).
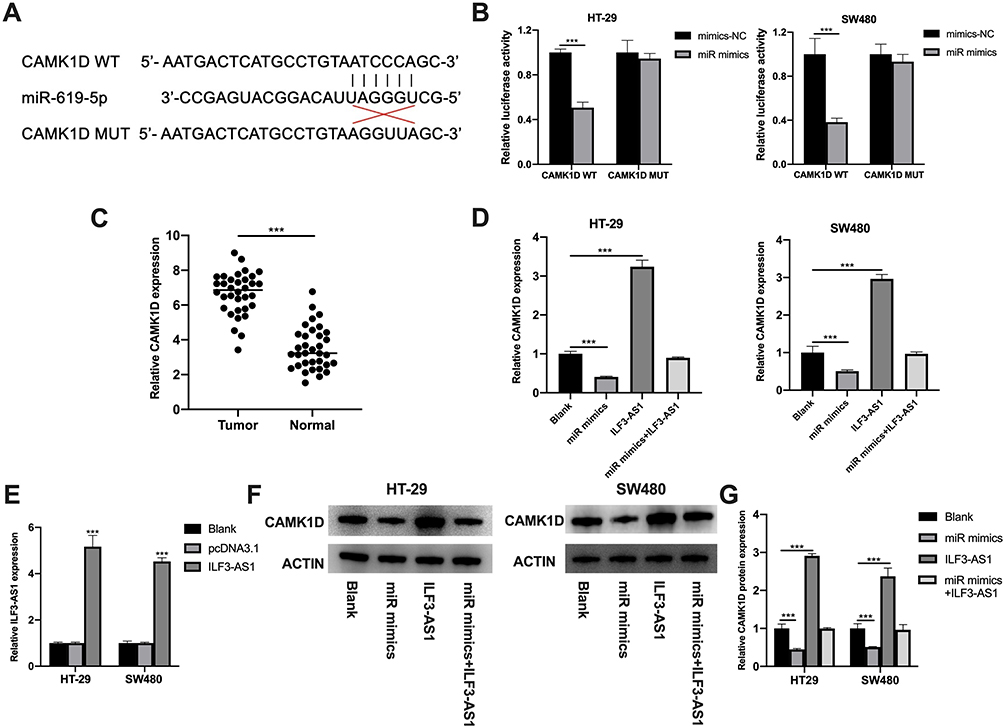 |
Figure 4 CAMK1D was a direct target of miR-619-5p. (A) Predicted binding sites of CAMK1D and miR-619-5p. (B) Luciferase activities of HT-29 and SW480 co-transfected with miR mimics/mimics-NC and luciferase reporters harboring CAMK1D MUT or CAMK1D WT. (C) The expression of CAMK1D in COAD tissues and adjacent normal controls by RT-qPCR. (D) The expression of CAMK1D in the blank, miR mimics, ILF3-AS1 overexpression (ILF3-AS1) and miR mimics+ILF3-AS1 overexpression (miR mimics+ILF3-AS1) group tested by RT-qPCR in HT-29 and SW480 cells. (E) The expression of ILF3-AS1 after ILF3-AS1 plasmid transfection in HT-29 and SW480 cells tested by RT-qPCR. (F) The protein expression of CAMK1D in the blank, miR mimics, ILF3-AS1, and miR mimics+ILF3-AS1 group in HT29 and SW480 cells. (G) The statistical results of the protein expression. ***P<0.001. |
ILF3-AS1 Exerted Its Oncogenic Role Through CAMK1D by Promoting the EMT Process
Next, we tried to verify whether ILF3-AS1 promotes COAD cell growth and invasion through CAMK1D. We transfected siRNA targeting CAMK1D to reduce its expression in HT-29 and SW480 cells. qPCR results showed that the expression of CAMK1D was significantly decreased after si-CAMK1D transfection (Figure 5A). CCK-8, Transwell invasion/migration assay, and apoptosis were subsequently performed. The results showed that in CAMK1D knockdown cells, cell proliferation, cell invasion/migration were significantly decreased compared with the control group, while apoptosis was increased considerably. In contrast, the cells overexpressing ILF3-AS1 showed increased cell proliferation, cell invasion/migration, and significantly reduced apoptosis. In cells co-transfected with ILF3-AS1 and si-CAMK1D, no significant difference in cell proliferation and invasion/migration was observed compared with the control group (Figure 5B–D). A previous study stated that CAMK1D was closely related to the EMT process. Meanwhile, EMT was closely associated with the metastasis of COAD. Therefore, we speculated that ILF3-AS1 promoted the EMT process through CAMK1D in COAD cells. We tested the expression of EMT-related proteins and found that E-cadherin expression was significantly reduced in CAMK1D knockdown cells. In contrast, the expression of N-cadherin and Vimentin increased significantly. The opposite trend appeared in ILF3-AS1 overexpressing cells. Besides, when the cells were co-transfected with si-CAMK1D and ILF3-AS1, there was no significant difference in the expression of the proteins mentioned above compared with the control group (Figure 5E). These results suggested that ILF3-AS1 promoted the EMT process of COAD cells through CAMK1D.
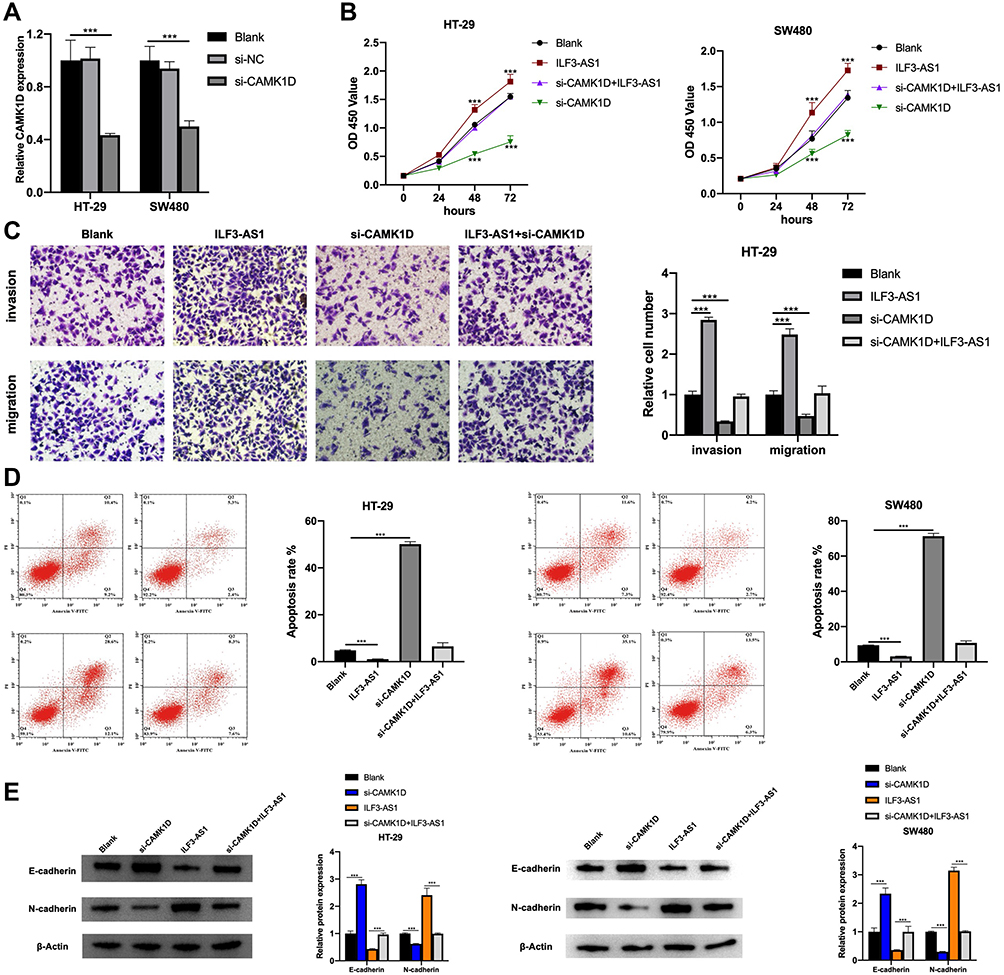 |
Figure 5 ILF3-AS1 exerted its oncogenic role through CAMK1D by promoting the EMT process. (A) The expression of CAMK1D in HT-29 and SW480 cells after si-CAMK1D transfection. The results of (B) CCK-8 assay, (C) The results of the transwell invasion/migration assay in the blank, ILF3-AS1, si-CAMK1D and si-CAMK1D+ILF3-AS1 group in HT-29 cells. (D) flow cytometry apoptosis assay and(E) the expression of EMT-related proteins (E-cadherin, N-cadherin and vimentin) in the blank, ILF3-AS1, si-CAMK1D and si-CAMK1D+ILF3-AS1 group in HT-29 and SW480 cells.***P<0.001. |
ILF3-AS1 Promoted COAD Cell Growth in vivo
To further validate effects of ILF3-AS1 on COAD progression, we conducted in vivo experiments. The volumes of the subcutaneous tumors were measured weekly. It was found that the tumor volumes in the si-ILF3-AS1 group were significantly smaller than those in the si-NC group from the 7th day after the subcutaneous injection of HT-29 (Figure 6B). On the 28th day after HT-29 injection, all the mice were euthanized, and the tumor mass was examined (Figure 6A and C). It was found that the tumor mass in the si-ILF3-AS1 group was significantly lower than that in the si-NC group. These results confirmed that ILF3-AS1 could promote the growth of COAD cells in vivo.
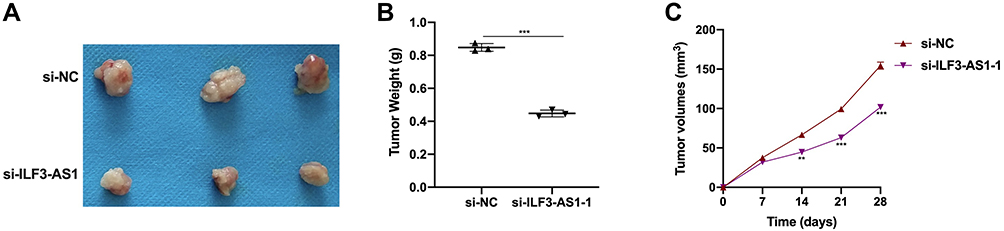 |
Figure 6 ILF3-AS1 promoted COAD cell growth in vivo. (A) Tumor size, (B) tumor weight and (C) tumor volume in HT-29 injected mice treated with si-NC and si-ILF3-AS1. **P<0.01; ***P<0.001. |
Discussion
LncRNAs play essential roles in the development of COAD. For example, LncRNA USP2-AS1 could promote colon cancer development through Hippo/YAP1 pathway.12 SNHG17 regulated the expression of CBX3 by binding to miR-375, thus promoting the progression of colon cancer.13 LINC01578 promoted colon cancer metastasis by forming positive feedback with the NF-κB/YY1 axis.14 In this study, we found that ILF3-AS1 expression was significantly higher in COAD samples and cell lines than in the control group. After siRNA was transfected to reduce the expression of ILF3-AS1, the proliferation, invasion and migration of HT-29 and SW480 cells were significantly decreased, while the apoptosis was increased considerably. These results suggested that ILF3-AS1 played an oncogenic role in COAD. ILF3-AS1 has been reported to play an important role in the development of a variety of tumors. For example, ILF3-AS1 promoted cell metastasis in nasopharyngeal carcinoma, and its high expression indicated poor prognosis.15 In gastric cancer, ILF3-AS1 promoted gastric cancer progression through the miR-29a/PTBP1 pathway.16 In osteosarcoma, ILF3-AS1 regulated SOX5 expression through miR-212.17 But the exact mechanism of action in colon cancer is not clear.
In this study, it was found that ILF3-AS1 could bind to miR-619-5p, thus weakening the latter’s regulatory effect on the expression of target genes. At the same time, we found that the expression of miR-619-5p was significantly decreased in COAD samples and cells, which was negatively correlated with the expression of ILF3-AS1. MiR-619-5p was reported to play an anti-tumor role in COAD. The low expression of miR-619-5p was significantly associated with poor prognosis.18 LncRNA SBF2-AS1 promoted the development of COAD by inhibiting the activity of miR-619-5p.19 These findings are consistent with our results. Interestingly, downregulation of ILF3-AS1 has been reported to be associated with colon cancer recurrence,8 which is contrary to our conclusion. We hypothesized that this might be because ILF3-AS1 acts differently at different stages of colon cancer. The dual roles of the same gene in the same tumor have previously been reported. TGF-β, for example, acts as a tumor suppressor gene in normal and early-stage cancerous cells but serves as an oncogene during tumor progression and is associated with poor prognosis.20–23 This is due to mutations or epigenetic modifications in the downstream signaling pathways of TGF-β, resulting in resistance to the tumor suppressor signals emitted by TGF-β. In addition, other signaling pathways of TGF-β are prevalent, and these signaling pathways have no tumor suppressor effect and promote tumor genesis and development.24 ILF3-AS1 may also have dual roles in colon cancer, and we may continue to investigate its function in patients with recurrent COAD in the future.
After exploring the downstream genes of miR-619-5p, we selected CAMK1D as the target for subsequent study. CAMK1D is a member of the calcium/calmodulin-dependent protein kinase 1 family. CAMK1D is involved in various physiological processes, including activation of CREB-dependent gene transcription,25 differentiation and activation of neutrophils,26 and regulation of apoptosis of erythrocyte leukemia cells.25 Recent studies have shown that overexpression of CAMK1D could promote the proliferation of breast cancer cells, promote the EMT process, and increase cell invasion and migration.11 In this study, we also found that when CAMK1D was knocked down in HT-29 and SW480 cells, cell proliferation, invasion/migration abilities were significantly reduced, and apoptosis was significantly increased. WB results showed that after overexpression of ILF3-AS1, E-cadherin switched to Vimentin and N-cadherin, while CAMK1D knockdown showed opposite results. These results suggested that ILF3-AS1 promoted the EMT process in COAD cells through CAMK1D, enabling the cells to acquire invasion and migration abilities. In addition, it was also reported that CAMK1D was one of the causes of drug resistance and treatment failure in the treatment of COAD with anti-PD-L1 drugs.27 These results suggested that CAMK1D may promote the occurrence and development of COAD through various mechanisms.
In summary, our study demonstrated that ILF3-AS1 regulates CAMK1D expression by binding with miR-619-5p, thus promoting the development of COAD. ILF3-AS1 may be a potential target for the treatment of colon cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
1. O’Keefe SJ. Diet, microorganisms and their metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol. 2016;13(12):691–706. doi:10.1038/nrgastro.2016.165
2. Link KH, Sagban TA, Mörschel M, et al. Colon cancer: survival after curative surgery. Langenbecks Arch Surg. 2005;390(2):83–93. doi:10.1007/s00423-004-0508-5
3. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi:10.1016/j.cell.2009.11.007
4. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. doi:10.1038/nrc3447
5. Bates RC, Pursell BM, Mercurio AM. Epithelial-mesenchymal transition and colorectal cancer: gaining insights into tumor progression using LIM 1863 cells. Cells Tissues Organs. 2007;185(1–3):29–39. doi:10.1159/000101300
6. Silva-Fisher JM, Dang HX, White NM, et al. Long non-coding RNA RAMS11 promotes metastatic colorectal cancer progression. Nat Commun. 2020;11(1):2156. doi:10.1038/s41467-020-15547-8
7. Li T, Li Z, Wan H, et al. Recurrence-associated long non-coding RNA LNAPPCC facilitates colon cancer progression via forming a positive feedback loop with PCDH7. Mol Ther Nucleic Acids. 2020;20:545–557. doi:10.1016/j.omtn.2020.03.017
8. Zhou M, Hu L, Zhang Z, Wu N, Sun J, Su J. Recurrence-associated long non-coding RNA Signature for determining the risk of recurrence in patients with colon cancer. Mol Ther Nucleic Acids. 2018;12:518–529. doi:10.1016/j.omtn.2018.06.007
9. Han S, Song L, Chen Y, Hou M, Wei X, Fan D. The long non-coding RNA ILF3-AS1 increases the proliferation and invasion of retinoblastoma through the miR-132-3p/SMAD2 axis. Exp Cell Res. 2020;393(2):112087. doi:10.1016/j.yexcr.2020.112087
10. Gao G, Li W, Liu S, et al. The positive feedback loop between ILF3 and lncRNA ILF3-AS1 promotes melanoma proliferation, migration, and invasion. Cancer Manag Res. 2018;10:6791–6802. doi:10.2147/CMAR.S186777
11. Bergamaschi A, Kim YH, Kwei KA, et al. CAMK1D amplification implicated in epithelial-mesenchymal transition in basal-like breast cancer. Mol Oncol. 2008;2(4):327–339. doi:10.1016/j.molonc.2008.09.004
12. Li D, Bao J, Yao J, Li J. lncRNA USP2-AS1 promotes colon cancer progression by modulating Hippo/YAP1 signaling. Am J Transl Res. 2020;12(9):5670–5682.
13. Liu J, Zhan Y, Wang J, Wang J, Guo J, Kong D. lncRNA-SNHG17 promotes colon adenocarcinoma progression and serves as a sponge for miR-375 to regulate CBX3 expression. Am J Transl Res. 2020;12(9):5283–5295.
14. Liu J, Zhan Y, Wang J, Wang J, Guo J, Kong D. Long noncoding RNA LINC01578 drives colon cancer metastasis through a positive feedback loop with the NF-κB/YY1 axis. Mol Oncol. 2020;14(12):3211–3233. doi:10.1002/1878-0261.12819
15. Yang X, Lin F, Gao F. Up-regulated long non-coding RNA ILF3-AS1 indicates poor prognosis of nasopharyngeal carcinoma and promoted cell metastasis. Int J Biol Markers. 2020;1724600820955199.
16. Ren ZH, Shang GP, Wu K, Hu CY, Ji T. WGCNA co-expression network analysis reveals ILF3-AS1 functions as a CeRNA to regulate PTBP1 expression by sponging miR-29a in gastric cancer. Front Genet. 2020;11:39. doi:10.3389/fgene.2020.00039
17. Hu XH, Dai J, Shang HL, Zhao ZX, Hao YD. SP1-mediated upregulation of lncRNA ILF3-AS1 functions a ceRNA for miR-212 to contribute to osteosarcoma progression via modulation of SOX5. Biochem Biophys Res Commun. 2019;511(3):510–517. doi:10.1016/j.bbrc.2019.02.110
18. Qiu G, Zhang XB, Zhang SQ, et al. Dysregulation of MALAT1 and miR-619-5p as a prognostic indicator in advanced colorectal carcinoma. Oncol Lett. 2016;12(6):5036–5042. doi:10.3892/ol.2016.5312
19. Chen G, Gu Y, Han P, Li Z, Zhao JL, Gao MZ. Long noncoding RNA SBF2-AS1 promotes colorectal cancer proliferation and invasion by inhibiting miR-619-5p activity and facilitating HDAC3 expression. J Cell Physiol. 2019;234(10):18688–18696. doi:10.1002/jcp.28509
20. Colak S, Ten Dijke P. Targeting TGF-β signaling in cancer. Trends Cancer. 2017;3(1):56–71. doi:10.1016/j.trecan.2016.11.008
21. Massagué J. TGFbeta in Cancer. Cell. 2008;134(2):215–230. doi:10.1016/j.cell.2008.07.001
22. Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350–1356. doi:10.1038/nm.3967
23. Calon A, Lonardo E, Berenguer-Llergo A, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 2015;47(4):320–329. doi:10.1038/ng.3225
24. Neel J-C, Humbert L, Lebrun -J-J. The dual role of TGFβ in human cancer: from tumor suppression to cancer metastasis. ISRN Mol Biol. 2012;2012:381428. doi:10.5402/2012/381428
25. Yamada T, Suzuki M, Satoh H, Kihara-Negishi F, Nakano H, Oikawa T. Effects of PU.1-induced mouse calcium-calmodulin-dependent kinase I-like kinase (CKLiK) on apoptosis of murine erythroleukemia cells. Exp Cell Res. 2004;294(1):39–50. doi:10.1016/j.yexcr.2003.10.023
26. Gaines P, Lamoureux J, Marisetty A, Chi J, Berliner N. A cascade of Ca(2+)/calmodulin-dependent protein kinases regulates the differentiation and functional activation of murine neutrophils. Exp Hematol. 2008;36(7):832–844. doi:10.1016/j.exphem.2008.02.009
27. Volpin V, Michels T, Sorrentino A, et al. CAMK1D triggers immune resistance of human tumor cells refractory to anti-PD-L1 treatment. Cancer Immunol Res. 2020;8(9):1163–1179. doi:10.1158/2326-6066.CIR-19-0608
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.