")
Back to Journals » OncoTargets and Therapy » Volume 12
lncRNA FOXD2-AS1 confers cisplatin resistance of non-small-cell lung cancer via regulation of miR185-5p–SIX1 axis
Authors Ge P, Cao L, Yao YJ, Jing RJ, Wang W, Li HJ
Received 7 December 2018
Accepted for publication 29 April 2019
Published 30 July 2019 Volume 2019:12 Pages 6105—6117
DOI https://doi.org/10.2147/OTT.S197454
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Peng Ge,1 Lei Cao,2 Yue-Juan Yao,1 Rui-Jun Jing,1 Wei Wang,1 Han-Jie Li1
1Department of Cardiothoracic Surgery, Second Affiliated Hospital of Xi’an Medical University, Xi’an, Shaanxi, People’s Republic of China; 2Department of Gynecology, Second Affiliated Hospital of Xi’an Medical University, Xi’an, Shaanxi, People’s Republic of China
Background: Chemoresistance is a major obstacle for chemotherapy failure in non-small-cell lung cancer (NSCLC). lncRNAs are a class of pivotal regulators in various cancers, and the lncRNA FOXD2-AS1 is implicated in the progression of NSCLC. However, it is still unclear whether it regulates chemosensitivity.
Methods: Expression levels of FOXD2-AS1, miR185-5p, and SIX1 mRNA were identified by reverse-transcription qPCR. CCK8 assay was performed to assess cell proliferation and chemosensitivity of cisplatin-resistant A549/DDP and H1299/DDP cells. Colony-forming assay was utilized to detect colony numbers. Cell migration and invasion ability were measured by transwell assay. The protein levels of LRP, Pgp, MRP1, and SIX1 were examined by Western blot assay. The correlation between FOXD2-AS1 and miR185-5p or miR185-5p and SIX1 were validated by bioinformatic, dual-luciferase, and RNA immunoprecipitation assays. Tumor xenografts were constructed to confirm the function and mechanism of FOXD2-AS1 in chemosensitivity of DDP-resistant NSCLC.
Results: FOXD2-AS1 and SIX1 were upregulated and miR185-5p downregulated in DDP-resistant NSCLC. Absence of FOXD2-AS1 enhanced drug sensitivity of A549/DDP and H1299/DDP cells, reflected by the reduced colony formation, cell proliferation, migration, invasion, and drug resistance–associated protein expression. FOXD2-AS1 acted as a molecular sponge for miR185-5p and relieved the binding of miR185-5p and its target gene SIX1, leading to the derepression of SIX1 in A549/DDP and H1299/DDP cells. Rescue experiments validated the functional interaction among FOXD2-AS1, miR185-5p, and SIX1. Moreover, FOXD2-AS1 interference receded the growth of DDP-resistant NSCLC tumors in vivo.
Conclusion: FOXD2-AS1/miR185-5p/SIX1 regulates the progression and chemosensitivity of DDP-resistant NSCLC, suggesting a potential therapeutic target for cisplatin-resistant NSCLC patients.
Keywords: FOXD2-AS1, miR185-5p–SIX1, DDP resistance, NSCLC
Background
Lung cancer is the most common malignancy and remains the leading cause of cancer-related deaths around the world,1 in which non-small-cell lung cancer (NSCLC) accounts for almost 80%–85%.2 Although great advances have been achieved in the treatment of NSCLC, the 5-year relative survival rate is still poor, mainly due to delays in diagnosis.3 Currently, cisplatin-based chemotherapy is applied as a first-line adjuvant-treatment strategy for NSCLC patients after surgical resection.4 However, intrinsic or acquired chemoresistance has become a major obstacle for cancer treatment.5 As such, it is an urgent matter to understand the molecular basis underlying NSCLC progression and cisplatin resistance.
lncRNAs are defined as eukaryote transcripts >200 nucleotides, acting as pivotal regulators of gene expression through chromatin modification, transcription, and posttranscriptional mechanisms.6 Growing evidence has demonstrated the regulatory roles of lncRNAs in various diseases.7 Ectopic expression of lncRNAs is associated with a variety of cancer progression through modulation of cellular processes, including cell proliferation, apoptosis, migration, and invasion.8 Moreover, great efforts have been made to elucidate the function and mechanism of lncRNAs in drug resistance of a wide range of tumors, including lung cancer. For instance, silencing of lncRNA HOTAIR sensitizes SCLC to antitumor drugs via inactivation of HOXA1 methylation by decreasing DNMT1 and DNMT3β expression.9 Subsequent study further confirmed the sensitization of HOTAIR knockdown to temozolomide through the blockade of autophagy by inhibiting ULK1 phosphorylation.10 Recently, a novel regulatory loopback suggests that the mainstream regulative mode of lncRNA on cancer phenotype is to serve as a ceRNA to suppress miRNA expression, thus resulting in the derepression of miRNA target genes.11,12 Hu et al, reported that lncRNA CCTA1 acted as a molecular sponge for miR130a-3p, triggering the upregulation of SOX4 and enhancing the cisplatin resistance of NSCLC.13 Wang et al pointed that lncRNA MEG3 resulted in the reduction of cisplatin resistance of NSCLC by regulation of the miR21-5p–SOX7 pathway.14
FOXD2-AS1 is a widely known lncRNA identified in various cancers and plays a central role in the regulation of cancer progression and drug resistance. Upregulation of FOXD2-AS1 is associated with poor prognosis of patients with esophageal squamous-cell carcinoma.15 By sponging miR185-5p, FOXD2-AS1 contributes to the proliferation of colorectal cancer cells.16 FOXD2-AS1 functions as an oncogene in bladder cancer through an FOXD2-AS1–Akt–E2F1 feedback loop.17 In addition, FOXD2-AS1 reduces the gemcitabine sensitivity of bladder cancer by acting as a sponge for miR143.18 In NSCLC, patients with high FOXD2-AS1 suffer a poor prognosis, and elevated expression of FOXD2-AS1 drives tumor formation via regulation of Wnt–β-catenin signaling.19 Although the role of FOXD2-AS1 in NSCLC progression has been validated, its function and mechanism in drug resistance are still unclarified.
In the present study, we proved the upregulation of FOXD2-AS1 in DDP-resistant NSCLC tissue and cells. Silencing of FOXD2-AS1 contributed to the sensitivity of NSCLC to cisplatin. Moreover, FOXD2-AS1 was identified as a sponge for miR185-5p, leading to the derepression of the miR185-5p target SIX1. Therefore, our findings highlight the underlying mechanism of FOXD2-AS1 in DDP resistance of NSCLC.
Methods
Patients
A total of 40 fresh tumor tissue samples were collected from NSCLC patients, who underwent surgical resection at the Second Affiliated Hospital of Xi’an Medical University between 2015 and 2017. These specimens were divided into cisplatin (DDP)-sensitive (n=20) and -resistant (n=20) groups dependent on patient outcomes following cisplatin treatment. All fresh specimens were rapidly frozen in liquid nitrogen and preserved in a freezer at −80°C for following RNA extraction. Written informed consent was obtained from all participants, and approval from the Research Ethics Committee of the Second Affiliated Hospital of Xi’an Medical University was obtained.
Cell culture and transfection
The NSCLC cell lines A549 and H1299 were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The stable cisplatin-resistant cell line A549/DDP was obtained from the American Type Culture Collection. DDP-resistant H1299 cell lines (H1299/DDP) were established through continuous exposure with stepwise increasesin concentrations of cisplatin. Briefly, H1299 cells grown in the logarithmic phase were initially treated with 0.5 μg/mL DDP (Sigma-Aldrich, St Louis, MO, USA). When cells had become resistant to thatcurrent concentration, cisplatin gradually increased until a final DDP concentration of 8 μg/mL. When the induced cells had survived in 8 μg/mL of cisplatin for about 2 months with a normal morphology and activity, they were confirmed to be cisplatin-resistant. A549 and H1299 cells were respectively cultured in specific Ham’s F12K (Thermo Fisher Scientific, Waltham, MA, USA) or RPMI 1640 medium (Thermo Fisher Scientific) in presence of 10% qualified FBS (Thermo Fisher Scientific). A549/DDP and H1299/DDP were grown in RPMI 1640 medium plus 10% FBS and 2 µg/mL DDP to maintain drug resistance. All cells were maintained at 37°C in a humidified chamber with 5% CO2 until the logarithmic growth phase.
siRNA specially targeting FOXD2-AS1 (si-FOXD2-AS1) or SIX1 (si-SIX1) and scramble control (si-NC), miR185-5p mimics (miR185-5p), negative control (miRNC), miR185-5p inhibitor (anti-miR185-5p) and inhibitor control (anti-miRNC), shRNA targeting FOXD2-AS1 (sh- FOXD2-AS1) and relative control (sh-NC) were obtained from GenePharma (Shanghai, China). FOXD2-AS1 or SIX1-overexpression plasmid (pcDNA-FOXD2-AS1 or pcDNA-SIX1) was established through inserting the full-length FOXD2-AS1 or SIX1 sequences into pcDNA3.1 vector (Thermo Fisher Scientific), with pcDNA3.1 empty vector (pcDNA) as a control. Oligonucleotides (50 nM) or 0.5 µg plasmids were transiently transfected into A549/DDP and H1299/DDP using Lipofectamine 3000 (Thermo Fisher Scientific).
RT-qPCR
Total RNA was extracted from tumor tissue or cells using RNAiso Plus reagent (Takara, Dalian, China) according to the manual, and then reverse transcription (RT) of lncRNA, miRNA, and target genes was performed using an M-MLV RT Kit (Thermo Fisher Scientific) or TaqMan miRNA RT kit (Thermo Fisher Scientific). For detection of RNA levels, qPCR analyses were carried out in triplicate using SYBR Green PCR Master Mix (Thermo Fisher Scientific) and run on Thermo Fisher Scientific 7500 real-time PCR systems (Thermo Fisher Scientific). Relative expression was calculated by the 2−ΔΔCt method, with GAPDH or U6 snRNA as the endogenous control.
Sequences of the RT primers were 5ʹ-GTCGTATCC AGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCAGGA-3ʹ for miR185-5p and 5ʹ- AACGCTTCACGAATTTGCGT-3ʹ for U6 snRNA. qRT-PCR primers were FOXD2-AS1, 5ʹ- TGGACCTAGCTGCAGCTCCA3-3ʹ (forward) and 5ʹ-AGTTGAAGGTGCACACACTG-3ʹ (reverse); miR185-5p, 5ʹ-GCGCGATTGGAGAGAAAGGCAGT-3ʹ (forward) and 5ʹ- ATCCAGTGCAGGG TCCGAGG-3ʹ (reverse); SIX1, 5ʹ-AAGGAGAAGTCGAGGGGTGT-3ʹ (forward) and 5ʹ- TGCTTGTTGGAGGAGGAGTT-3ʹ (reverse); GAPDH, 5ʹ-TATGATGATATCAAGAGGGTAGT-3ʹ (forward) and 5ʹ-TGTATCCAAACTCATTGTCATAC-3ʹ (reverse); and U6, 5ʹ-CTCGCTTCGGCAGCACA-3ʹ (forward) and 5ʹ-AACGCTTCACGAATTTGCGT-3ʹ (reverse).
CCK8
Treated or transfected cells (3×103/well) in 100 μL culture medium were seeded into 96-well plates and treated with varying concentrations of DDP (total volume 50 mg; Tocris Bioscience, Ellisville, MO, USA) for 24 hours or transfected with oligonucleotides or plasmids for various periods. Afterward, CCK8 assay was performed by adding 10 µL CCK8 reagent (Beyotime, Haimen, China) into each well and incubated for another 2 hours, followed by detection of absorbance at 450 nm on a microplate reader (Thermo Fisher Scientific). Cell growth–inhibition rate was calculated — (1 – OD values of drug group/OD values of control group) ×100% — followed by assessment of IC50 values.
Colony-forming assay
A549/DDP and H1299/DDP cells in logarithmic growth phase were digested with 0.25% trypsin (Thermo Fisher Scientific), dispersed into single cells by repeated pipetting, and suspended in RPMI 1640 medium supplemented with 10% FBS. Suspensions were diluted by gradient dilutions and seeded into a 10 mL culture dish preheated to 37°C. Following incubation at 37°C, 5% CO2 for 2–3 weeks, visible colonies were fixed with 4% paraformaldehyde (Sigma-Aldrich) and stained using 0.1% crystal violet (Sigma-Aldrich). Having been washed with PBS three times and air-dried, the culture dish was inverted and overlaid onto a grid of transparency to count clones larger than ten cells on microscopy (low magnification), followed by calculation of colony-formation rate.
Transwell assay
Cell migration and invasion ability of transfected A549/DDP and H1299/DDP cells were assessed by transwell assay. For migration assay, cells resuspended in serum-free medium were seeded into the upper chamber (Corning, NNY, USA) and complete medium containing 10% FBS added into the bottom chamber. After incubation for 8 hours at 37°C, migrating cells on the basal side of the membrane were fixed with 4% paraformaldehyde and stained using 0.5% crystal violet. Finally, migrating cells in five random visual fields were observed and counted on microscopy (Olympus, Tokyo, Japan). For invasion assay, the chambers were coated with matrigel (BD Biosciences, San Jose, CA, USA) and cell-invasion ability detected using a similar approach.
Western blot
Total proteins were extracted from NSCLC cells and tumor tissue using RIPA buffer (Beyotime) mixed with 1% protease inhibitor mixture (Sigma-Aldrich), followed by the detection of protein concentration using a Pierce BCA protein assay kit (Thermo Fisher Scientific). Proteins (30 μg) denatured at 97°C for 5 minutes were divided by SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). After blockade of aspecific signals using 5% non-fat milk powder, membranes were incubated overnight at 4°C with specific primary antibodies against LRP, MDR1, MRP1, SIX1, and β-actin (Abcam, Cambridge, UK) and then further incubated with HRP- conjugated secondary antibody (Abcam) for another 1.5 hours at 37°C. Ultimately, protein blots were visualized using an enhanced-chemiluminescence reagent (Beyotime) and band densities analyzed using Image Lab software (Bio-Rad, Hercules, CA, USA).
Luciferase assay
Wild-type FOXD2-AS1 sequences containing predicted miR185-5p-binding sites or mutants of each site were cloned into psiCHECK-2 plasmid (Promega, Madison, WI, USA) and named FOXD2-AS1-WT or FOXD2-AS1-Mut reporter. Similarly, partial fragments of SIX1 3ʹUTR containing putative miR185-5p-binding sites or mutants of each site were cloned into psiCHECK-2 plasmid (Promega, Madison, WI, USA), named SIX1 3ʹUTR-WT or SIX1 3ʹUTR-MUT reporter. Then, luciferase reporter was transfected into A549/DDP cells along with miR185-5p, anti-miR185-5p, or relative controls. About 48 hours after transfection, luciferase activities were measured using a dual luciferase–reporter assay system (Promega).
RNA immunoprecipitation
RNA immunoprecipitation (RIP) assay was carried out using a Magna RIP RNA-binding protein immunoprecipitation kit (Millipore). In short, cell extracts in RIP buffer were incubated with protein A/G magnetic beads conjugated with primary antibodies against Ago2 (Cell Signaling Technology, Beverly, MA, USA) and IgG (Cell Signaling Technology). Finally, the RNA in the immunoprecipitant complex was isolated and enrichment levels of FOXD2-AS1 and miR185-5p measured by RT-qPCR.
Tumor xenograft
All animal procedures were performed following the Guidelines for Care and Use of Laboratory Animals with approval of the Ethics Committee of the Second Affiliated Hospital of Xi’an Medical University. Six-week-old male BALB/c nude mice were purchased from Vital River Laboratory Animal Technology (Beijing, China). A549/DDP cells (6×106) transfected with sh-FOXD2-AS1 or sh-NC were subcutaneously injected into nude mice. When tumor volume had reached almost 60 mm3, nude mice wre intraperitoneally administered 3 mg/kg DDP every 3 days. At 35 days after inoculation, tumors were measured every week with slide calipers and volume calculated as (length × width2)/2. About 35 days after implantation, mice were killed and tumor specimens weighed and collected for molecular analyses.
Statistical analysis
All experiments performed in this study were repeated three times. Statistical analysis was performed using SPSS 20.0, and results were shown as means ± SD. All comparisons between groups were assessed using paired-samples t-test and one-way ANOVA, with P<0.05 representing statistical significance.
Results
FOXD2-AS1 was upregulated in cisplatin-resistant NSCLC tissue and cells
Firstly, we observed that the abundance of FOXD2-AS1 was higher in cisplatin-resistant NSCLC tissue than cisplatin-sensitive NSCLC tissue, which was detected by RT-qPCR (Figure 1A). To explore the role of FOXD2-AS1 in drug sensitivity of NSCLC cells, cisplatin-resistant NSCLC cell lines A549/DDP and H1299/DDP were employed. CCK8 assay showed that increasing cisplatin concentrations resulted in the inhibition of cell viability (Figure 1, B and C). Moreover, within the mass concentration range of 0–10 µg/mL DDP, cell viability and IC50 in A549/DDP and H1299/DDP cells were markedly higher than in A549 and H1299 cells (Figure 1, B and C). However, cell-growth inhibition in DDP-resistant cells was lowerthan in DDP-sensitive cells (Figure S1), indicating the successful establishment of cisplatin-resistant NSCLC cells. Next, RT-qPCR assay revealed that FOXD2-AS1 expression in A549/DDP and H1299/DDP cells presented a significant increase compared to their counterparts (Figure 1D).
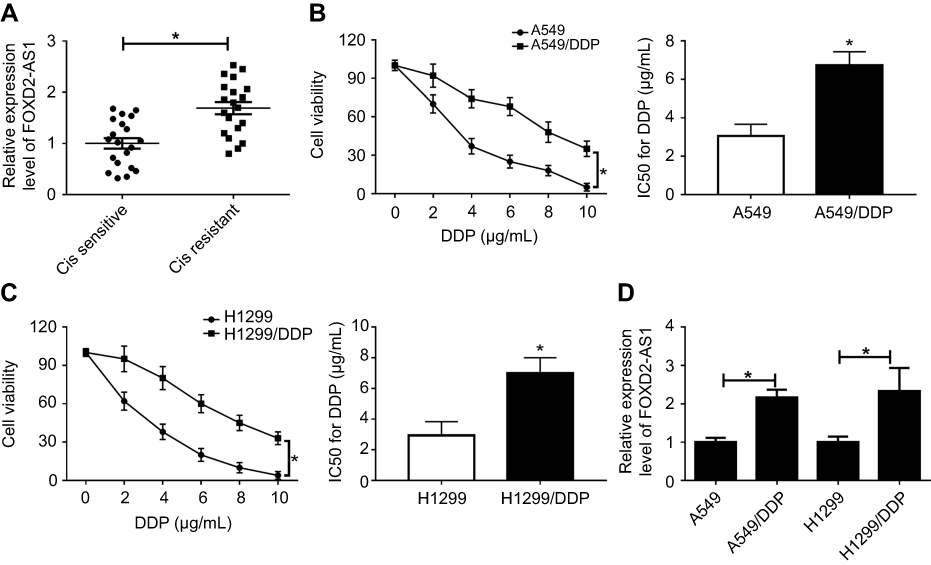 |
Figure 1 FOXD2-AS1 was highly expressed in cisplatin-resistant non-small-cell lung cancer (NSCLC) and cells. Notes: (A) Expression of FOXD2-AS1 in cisplatin-sensitive or -resistant NSCLC tissue was detected by reverse-transcription (RT)-qPCR. (B, C) CCK8 assay assessed viability and IC50 of DDP-sensitive or -resistant NSCLC. (D) Expression of FOXD2-AS1 in DDP-sensitive or -resistant NSCLC cells was measured by RT-qPCR. *P<0.05. |
FOXD2-AS1 depletion enhanced chemosensitivity of cisplatin-resistant NSCLC cells
si-FOXD2-AS1 was transfected into A549/DDP and H1299/DDP cells to suppress FOXD2-AS1 expression (Figure 2A). Next, loss-of-function experiments were performed to probe the regulatory role of FOXD2-AS1 in DDP-tolerant NSCLC cells. Results of colony-forming assays revealed that colony numbers of A549/DDP and H1299/DDP cells decreased remarkably following si-FOXD2-AS1 transfection (Figure 2B). In addition, CCK8 and transwell analyses suggested that FOXD2-AS1 interference significantly repressed the proliferation (Figure 2, C and D), migration (Figure 2E), and invasion ability (Figure 2F) of A549/DDP and H1299/DDP cells compared to respective controls. Moreover, expression levels of MRP1, Pgp, and LRP detected by Western blot assay had obviously declined in si-FOXD2-AS1–transfected A549/DDP and H1299/DDP cells relative to the si-NC–transfected group (Figure 2, G and H).
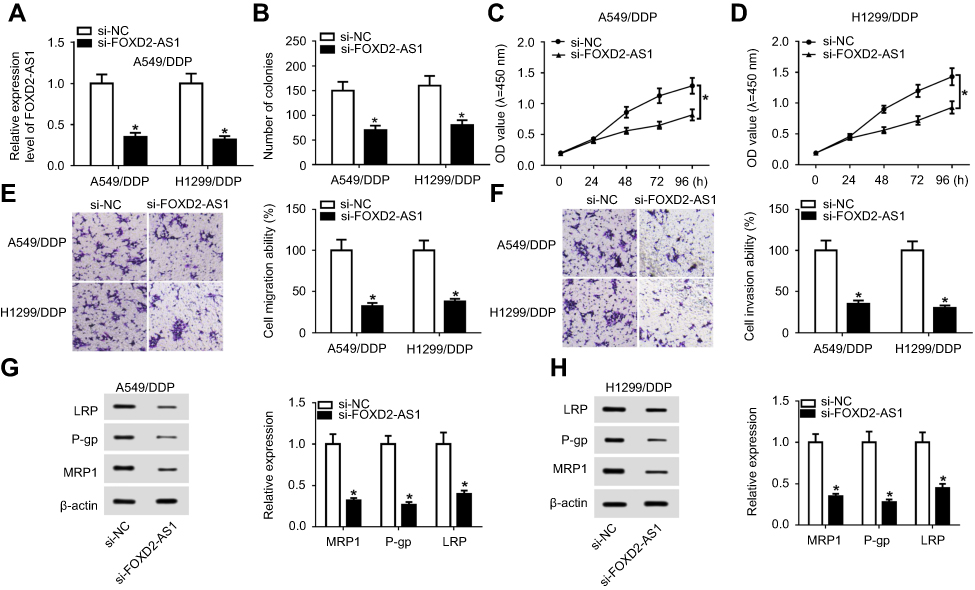 |
Figure 2 Lack of FOXD2-AS1 enhanced drug sensitivity of DDP-resistant NSCLC cells. A549/DDP and H1299/DDP cells were transfected with si-NC or si-FOXD2-AS1. |
FOXD2-AS1 served as a molecule sponge for miR185-5p
Growing evidence is appearing on the correlation between lncRNAs and microRNAs in various cancers by complementary base pairing.20 Here, using the bioinformatic tool miRTarBase, we verified the existence of complementary binding sites between FOXD2-AS1 and miR185-5p (Figure 3A). Afterward, luciferase and RIP analyses were carried out to confirm the true interaction between the two RNAs. Results showed that transfection of miR185-5p suppressed and anti-miR185-5p induced the luciferase activity of FOXD2-AS1-WT reporter in A549/DDP cells, whereas efficacy was lost in response to FOXD2-AS1-Mut (Figure 3, B and C). Moreover, FOXD2-AS1 and miR185-5p were highly enriched by Ago2, while IgG showed little efficacy of enrichment (Figure 3D). Seeing that miR185-5p was targeted by FOXD2-AS1, levels of miR185-5p in DDP-sensitive and -resistant NSCLC tissue and cells were determined by RT-qPCR. Results displayed a remarkable decrease in miR185-5p in DDP-tolerated NSCLC tissue and cell lines compared to their counterparts (Figure 3, E and F). To verify whether FOXD2-AS1 could modulate the expression of miR185-5p, pcDNA-FOXD2-AS1 or si-FOXD2-AS1 was transfected into A549/DDP and H1299/DDP cells to overexpress or inhibit FOXD2-AS1 (Figure 3G), followed by the detection of miR185-5p expression. Results showed that addition of FOXD2-AS1 impeded miR185-5p expression and knockdown of FOXD2-AS1 conferred miR185-5p expression (Figure 3H).
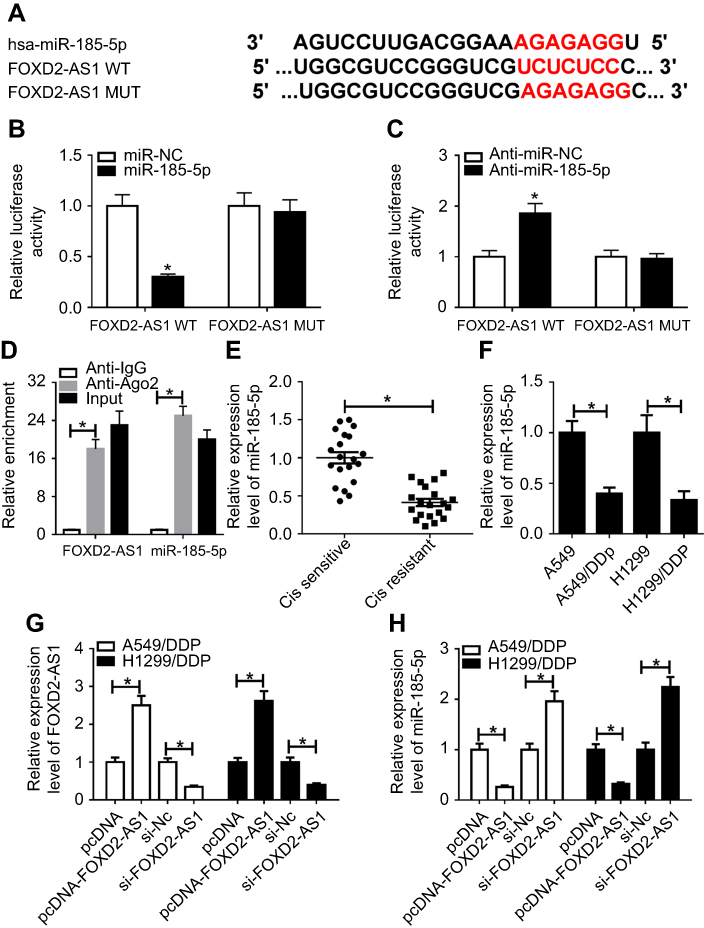 |
Figure 3 FOXD2-AS1 bound to miR185-5p and negatively regulated its expression. Notes: (A) Putative binding regions of FOXD2-AS1 in miR185-5p predicted with biological software. (B, C) Luciferase activity of wild-type or mutant FOXD2-AS1 in A549/DDP cells following miRNC, miR185-5p, anti-miRNC, or anti-miR185-5p transfection determined by luciferase reporter assay. (D) Correlations between FOXD2-AS1 and miR185-5p detected by RIP assay. (E, F) Expression of miR185-5p in DDP-resistant non-small-cell lung cancer (NSCLC) tissue and cell lines detected by RT-qPCR assay. (G, H) Expression of FOXD2-AS1 and miR185-5p in A549/DDP and H1299/DDP cells, initially transfected with pcDNA, pcDNA-FOXD2-AS1, si-NC, or si-FOXD2-AS1, determined by reverse-transcription (RT) qPCR assay. *P<0.05. |
miR185-5p was required for FOXD2-AS1-mediated drug sensitivity of cisplatin-tolerated NSCLC cells
To validate whether miR185-5p was responsible for FOXD2-AS1-mediated drug sensitivity of cisplatin-tolerated NSCLC cells, miR185-5p mimics were transfected into DDP-resistant NSCLC cells to overexpress miR185-5p, with miRNC as a negative control (Figure 4A). After that, rescue experiments were performed by transfection with miRNC, miR185-5p, miR185-5p + pcDNA, and miR185-5p + pcDNA-FOXD2-AS1 in A549/DDP and H1299/DDP cells. Colony-forming analysis showed that supplementation of miR185-5p decreased colony numbers of DDP-tolerated NSCLC cells, which was reversed following pcDNA-FOXD2-AS1 transfection (Figure 4B). Moreover, CCK8 and transwell analyses revealed that restoration of FOXD2-AS1 abolished the inhibitory effects of miR185-5p on cell proliferation (Figure 4, C and D), migration (Figure 4E), and invasion (Figure 4F) in A549/DDP and H1299/DDP cells. Western blot assay further validated that reexpression of FOXD2-AS1 overturned and miR185-5p-suppressed expression of LRP, Pgp, and MRP1 proteins in A549/DDP and H1299/DDP cells (Figure 4, G and H).
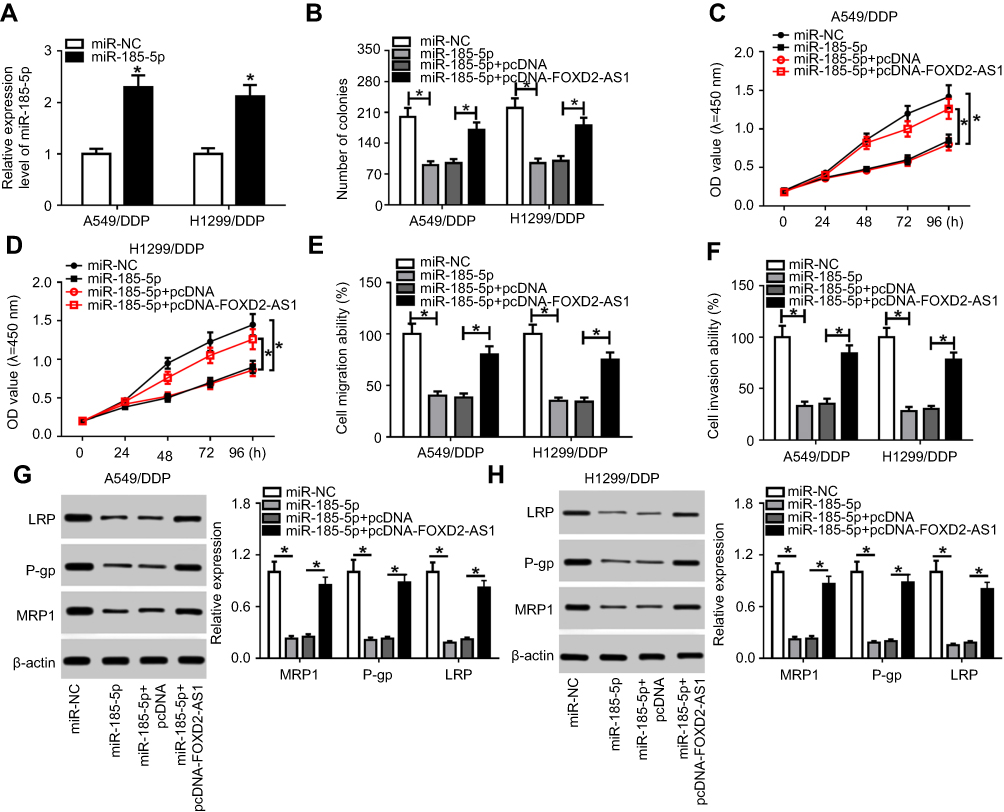 |
Figure 4 FOXD2-AS1 reversed miR185-5p-enhanced drug sensitivity in DDP-toleratant non-small-cell lung cancer (NSCLC) cells.Notes: (A) miR185-5p expression in A549/DDP and H1299/DDP cells after transfection of miRNC or miR185-5p mimics. A549/DDP and H1299/DDP cells were transfected with miRNC, miR185-5p, miR185-5p + pcDNA, or miR185-5p + pcDNA-FOXD2-AS1, followed by evaluation of colony numbers (B), cell proliferation (C, D), migration (E), invasion (F), drug resistance–related LRP, Pgp, and MRP1 expression (G, H). *P<0.05. |
SIX1 was directly targeted by miR185-5p
Here, bioinformatic analysis validated the existence of putative binding sites between miR185-5p and SIX1 3ʹUTR using TargetScan software (Figure 5A). To elucidate whether miR185-5p directly targeted SIX1, luciferase-reporter plasmids containing wild -ype or mutant SIX1 3ʹUTR–binding sites were constructed. The results of luciferase-activity assays showed that overexpression of miR185-5p decreased and knockdown of miR185-5p increased the luciferase activity of SIX1 3ʹUTR-WT reporter in A549/DDP cells (Figure 5, B and C). Next, we determined levels of SIX1 in DDP-sensitive and -resistant NSCLC tissue and cell lines. Results displayed that SIX1 expression at mRNA and protein levels were remarkably increased in DDP-tolerated NSCLC tissue and cell lines compared to their counterparts (Figure 5, D–F). Moreover, supplementation of miR185-5p suppressed and absence of miR185-5p conferred SIX1 protein expression (Figure 5G).
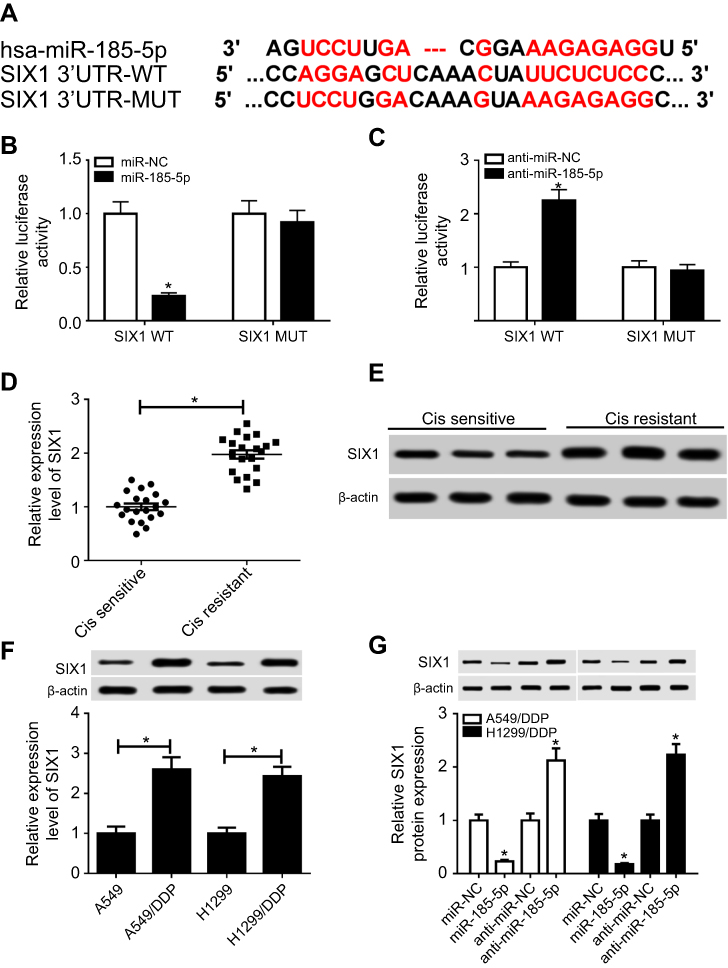 |
Figure 5 SIX1 was a target of miR185-5p. Notes: (A) Putative binding regions of miR185-5p in SIX1 3ʹUTR. (B, C) Luciferase activity of wild-type or mutant SIX1 in A549/DDP cells after transfection of miRNC, miR185-5p, anti-miRNC, or anti-miR185-5p. (D–F) mRNA and protein levels of SIX1 in DDP-resistant non-small-cell lung cancer (NSCLC) tissue and cell lines detected by reverse-transcription (RT) qPCR and Western blot assays. (G) Protein levels of SIX1 in miR185-5p or anti-miR185-5p–transfected A549/DDP and H1299/DDP cells. *P<0.05. |
FOXD2-AS1 attenuated chemosensitivity of cisplatin-resistant NSCLC cells via miR185-5p–SIX1 axis
Ee initially observed decreased levels of miR185-5p and SIX1 in A549/DDP and H1299/DDP cells following anti-miR185-5p or si-SIX1 transfection (Figure 6, A–C), hinting that anti-miR185-5p and si-SIX1 could be used to inhibit miR185-5p and SIX1 in DDP-tolerant NSCLC cells. To further investigate the potential mechanism underlying miR185-5p-mediated drug sensitivity of DDP-tolerant NSCLC cells, si-SIX1 was transfected into A549/DDP and H1299/DDP cells together with anti-miRNC or anti-miR185-5p, while pcDNA-SIX1 and its control pcDNA-NC were transfected into A549/DDP and H1299/DDP cells. The results showed that overexpressed SIX1 significantly increased cell proliferation, migration, and invasion (Figure S2). Following rescue experiments revealed that interference of SIX1 resulted in reductions in colony number (Figure 6D), cell proliferation (Figure 6, E and F), migration (Figure 6G), and invasion (Figure 6H), as well as the downregulation of LRP, Pgp, and MRP1 (Figure 6, I and J) in A549/DDP and H1299/DDP cells. Further introduction of anti-miR185-5p reversed the inhibitory effects of si-SIX1 on colony forming, cell proliferation, migration, invasion, and drug resistance (Figure 6, D–J). To explore whether FOXD2-AS1 was required for the drug sensitivity of DDP-resistant NSCLC cells through regulation of the miR185-5p–SIX1 axis, anti-miRNC or anti-miR185-5p was transfected into FOXD2-AS1–inhibited A549/DDP and H1299/DDP cells. After that, the abundance of SIX1 was determined, and results displayed that miR185-5p deficiency weakened si-FOXD2-AS1–inhibited SIX1 expression (Figure 6K). Altogether, these data indicated the implication of the FOXD2-AS1–miR185-5p–SIX1 pathway in the regulation of DDP resistance in NSCLC cells.
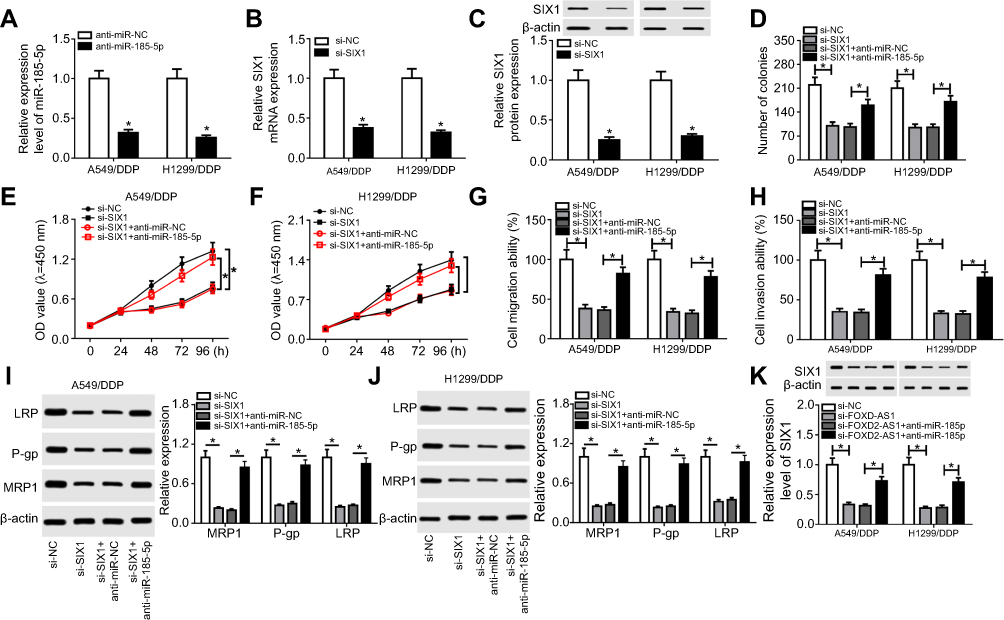 |
Figure 6 FOXD2-AS1 reduced the chemosensitivity of DDP-resistant non-small-cell lung cancer (NSCLC) cells via the miR185-5p–SIX1 axis. Notes: (A) miR185-5p expression in anti-miRNC or anti-miR185-5p–transfected A549/DDP and H1299/DDP cells. (B and C) SIX1 expression at mRNA and protein levels in si-NC or si-SIX1–transfected A549/DDP and H1299/DDP cells. (C) A549/DDP and H1299/DDP cells were transfected with si-NC, si-SIX1, si-SIX1 + anti-miRNC, or si-SIX1 + anti-miR185-5p, followed by evaluation of colony numbers (D), cell proliferation (E and F), migration (G), invasion (H), and drug resistance–related proteins LRP, Pgp, and MRP1 expression (I and J). (K) SIX1 expression in A549/DDP and H1299/DDP following transfection of si-NC, si-FOXD2-AS1, si-FOXD2-AS1 + anti-miRNC, or si-FOXD2-AS1 + anti-miR185-5p. *P<0.05. |
Knockdown of FOXD2-AS1 attenuated DDP resistance of NSCLC in vivo
To further probe the effect of FOXD2-AS1 on chemosensitivity of NSCLC in vivo, sh-NC or sh-FOXD2-AS1–transfected A549/DDP cells were inoculated into BABL/c nude mice. As a result, tumor volume progressively increased with time (Figure 7A). Knockdown of FOXD2-AS1 reduced tumor growth compared with the control group (Figure 7A). Tumor weight was obviously inhibited by FOXD2-AS1 interference (Figure 7B). Furthermore, expression levels of FOXD2-AS1 (Figure 7C) and SIX1 (Figure 7, E and F) were evidently decreased, while miR185-5p (Figure 7D) was remarkably enhanced in tumor tissue in the sh-FOXD2-AS1 group relative to the control group. These findings uncovered that lack of FOXD2-AS1 inhibited the growth of DDP-resistant tumors through the miR185-5p–SIX1 axis in vivo.
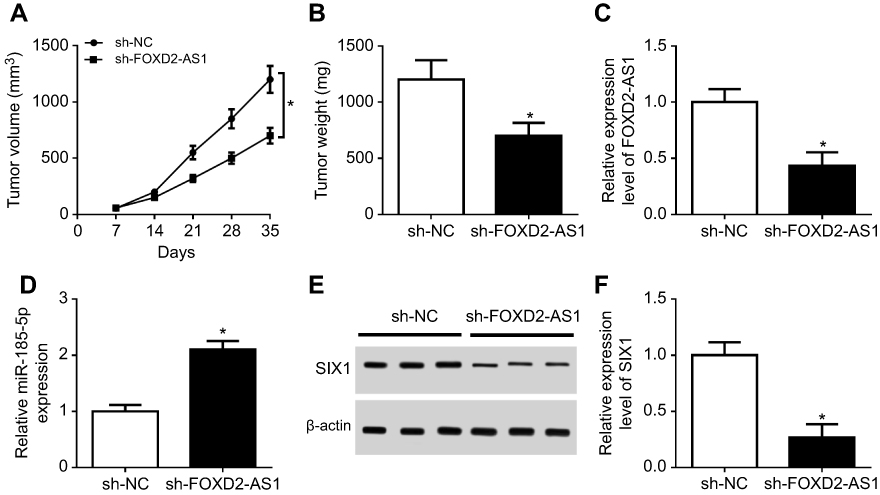 |
Figure 7 Knockdown of FOXD2-AS1 reduced DDP resistance of non-small-cell lung cancer (NSCLC) in vivo. Notes: (A) Tumor volume detected every week after injection. (B) Tumor weight investigated at the end point of 35 days. Expression of FOXD2-AS1 (C), miR185-5p (D), and SIX1 protein (E, F) in tumor tissue examined. *P<0.05. |
Discussion
A growing body of evidence has revealed that aberrant expression of lncRNAs participates in the chemoresistance of multiple malignancies, including NSCLC. Here, we showed that knockdown of FOXD2-AS1 retarded the progression and enhanced the sensitivity of DDP-resistant NSCLC cells via sponging miR185-5p by downregulating SIX1. We initially provided evidence that the FOXD2-AS1–miR185-5p–SIX1 axis was involved in the regulation of drug resistance in NSCLC.
FOXD2-AS1 has been identified as an oncogene in different types of cancer, such as colorectal cancer,16 nasopharyngeal carcinoma,21 and gastric cancer.22 Also, FOXD2-AS1 has been demonstrated to be associated with the chemoresistance of bladder cancer.18 In this study, we focused on the function and mechanism of FOXD2-AS1–mediated DDP resistance in NSCLC. Results showed that FOXD2-AS1 was upregulated in DDP-resistant NSCLC tissue and cells. Functionally, silencing of FOXD2-AS1 reduced tumor progression in DDP-resistant NSCLC in vitro and in vivo. Moreover, the sensitivity of DDP-resistant NSCLC cells was enhanced following FOXD2-AS1 knockdown, as reflected by the decreased levels of LRP, Pgp, and MRP1 proteins.
An increasing amount of evidence shows that lncRNAs can function as molecular sponges for miRNAs to impede the binding of miRNAs and their target mRNAs, leading to the release of target mRNAs. Here, we confirmed that the true interaction between FOXD2-AS1 and miR185-5p was through complementary binding. mir185-5p is a cancer-related miRNA, which is usually found to be dysregulated in various cancers, leading to the inhibition of cancer progression. For example, miR185-5p is negatively correlated with malignant clinical features of breast cancer, and enforced expression of this miRNA reverses epithelial–mesenchymal transition by suppressing RAGE.23 Supplementation of miR185-5p inhibits oral squamous-cell carcinoma progression via targeting cyclin D2.24 Also, miR185-5p inhibited by ZNF139 plays a key role in the modulation of multidrug resistance in gastric cancer.25 Via targeting ABCC1, miR185-5p contributes to cisplatin sensitivity and cell apoptosis, but restrains cell proliferation in NSCLC.26 In line with these studies, we also demonstrated that introduction of miR185-5p inhibited cell progression and improved the sensitivity of NSCLC to cisplatin, but FOXD2-AS1 overexpression abolished the effects of miR185-5p on cell growth and cisplatin sensitivity.
SIX1, located in a cluster of related genes on chromosome 14, plays instrumental functions in the regulation of cancer progression, including cervical,27 ovarian,28 and breast cancer.29 It is important to note that SIX1 significantly triggers VEGFC expression, resulting in lymphangiogenesis and distant metastasis of breast cancer cells.29 Li et al reported that elevated expression of SIX1 in pancreatic cancer obviously inhibited cell-cycle arrest and facilitated proliferation by upregulatingcyclin D1.30 Xu et al pointed that SIX1 induced stimulatory effects in tumor growth and metastasis by promoting angiogenesis and recruiting tumor-associated macrophages.31 In this study, we provide the first evidence that SIX1 is a target of miR185-5p and knockdown of SIX1 retards tumor progression and diminishes chemoresistance of DDP-resistant NSCLC cells. Similarly, a previous study showed that depletion of SIX1 conferred sensitivity of drug-resistant breast cancer cells to paclitaxel.32 Enhanced sensitivity of hepatoma cells to paclitaxel has been confirmed to be attributable to the inhibition of SIX1.33 Also, enforced abundance of SIX1 is associated with the poor prognosis of esophageal squamous-cell carcinoma patients, and should be responsible for the radioresistance of this cancer.34 Furthermore, we found that knockdown of miR185-5p substantially weakened si-SIX1 inhibition of cell growth and DDP resistance. Absence of FOXD2-AS1 resulted in a reduction SIX1, and miR185-5p knockdown abrogated the inhibitory effect of si-FOXD2-AS1 on SIX1 expression.
In conclusion, our study indicated that FOXD2-AS1 stimulated tumor progression and DDP resistance of NSCLC cells via modulation of the miR185-5p–SIX1 axis, hinting at the central role of the FOXD2-AS1–miR185-5p–SIX1 network in cisplatin resistance of NSCLC cells. These findings suggest that FOXD2-AS1 may be an effective target for overcoming cisplatin resistance during chemotherapy for NSCLC.
Ethics approval and consent to participate
Written informed consent was obtained from all participants, and approval from the Research Ethics Committee of the Second Affiliated Hospital of Xi’an Medical University was obtained. All animal procedures were performed following Guidelines for Care and Use of Laboratory Animals with approval of the ethics committee.
Acknowledgment
This work was supported by Shaanxi Provincial Research Center for the Project of Prevention and Treatment of Respiatory Diseases (Grant No: 2018GCKF04).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fitzmaurice C, Allen C, Barber RM, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. Jama Oncol. 2017;3(4):524–548. doi:10.1001/jamaoncol.2016.5688
2. Peters S, Adjei AA, Gridelli C, et al. Metastatic non-small-cell lung cancer (NSCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;24 Suppl 6(suppl_3):vi99–vi105. doi:10.1093/annonc/mdt178
3. Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Springer Int Publ. 2016;893:1–19. doi:10.1007/978-3-319-24223-1_1
4. Pirker R. Adjuvant chemotherapy in patients with completely resected non-small cell lung cancer. Transl Lung Cancer Res. 2014;3(5):305–310. doi:10.3978/j.issn.2218-6751.2014.09.13
5. Chang A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer. 2011;71(1):3–10. doi:10.1016/j.lungcan.2010.08.022
6. Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell. 2011;145(2):178–181. doi:10.1016/j.cell.2011.03.014
7. Chen G, Wang Z, Wang D, et al. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013;41(Databaseissue):D983–D986. doi:10.1093/nar/gks1099
8. Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012;9(6):703–719. doi:10.4161/rna.20481
9. Fang S, Gao H, Tong Y, et al. Long noncoding RNA-HOTAIR affects chemoresistance by regulating HOXA1 methylation in small cell lung cancer cells. Lab Invest. 2016;96(1):60–68. doi:10.1038/labinvest.2015.123
10. Yang Y, Jiang C, Yang Y, et al. Silencing of LncRNA-HOTAIR decreases drug resistance of non-small cell lung cancer cells by inactivating autophagy via suppressing the phosphorylation of ULK1. Biochem Biophys Res Commun. 2018;18(497):1003–1010. doi:10.1016/j.bbrc.2018.02.141
11. Ballantyne MD, Mcdonald RA, Baker AH. LncRNA/microRNA interactions in the vasculature. Clin Pharmacol Ther. 2016;99(5):494–501. doi:10.1002/cpt.355
12. Juan L, Wang G, Radovich M, et al. Potential roles of microRNAs in regulating long intergenic noncoding RNAs. Bmc Med Genomics. 2013;6(S1):S7. doi:10.1186/1755-8794-6-S1-S7
13. Hu B, Zhang H, Wang Z, Zhang F, Wei H, Li L. LncRNA CCAT1/miR-130a-3p axis increases cisplatin resistance in non-small-cell lung cancer cell line by targeting SOX4. Cancer Biol Ther. 2017;18(12):974–983. doi:10.1080/15384047.2017.1385679
14. Wang P, Chen D, Ma H, Li Y. LncRNA MEG3 enhances cisplatin sensitivity in non-small cell lung cancer by regulating miR-21-5p/SOX7 axis. Oncotarget Ther. 2017;10:5137–5149. doi:10.2147/OTT.S146423
15. Bao J, Zhou C, Zhang J, et al. Upregulation of the long noncoding RNA FOXD2-AS1 predicts poor prognosis in esophageal squamous cell carcinoma. Cancer Biomarkers. 2017;32(1):108–112. doi:10.5301/jbm.5000240
16. Zhu Y, Qiao L, Zhou Y, Ma N, Wang C, Zhou J. Long non-coding RNA FOXD2-AS1 contributes to colorectal cancer proliferation through its interaction with microRNA-185-5p. Cancer Sci. 2018;109(7):2235–2242. doi:10.1111/cas.13632
17. Su F, He W, Chen C, et al. The long non-coding RNA FOXD2-AS1 promotes bladder cancer progression and recurrence through a positive feedback loop with Akt and E2F1. Cell Death Dis. 2018;9(2):233. doi:10.1038/s41419-018-0275-9
18. An Q, Zhou L, Xu N. Long noncoding RNA FOXD2-AS1 accelerates the gemcitabine-resistance of bladder cancer by sponging miR-143. Biomed Pharmacother. 2018;103:415–420. doi:10.1016/j.biopha.2018.03.138
19. Rong L, Zhao R, Lu J. Highly expressed long non-coding RNA FOXD2-AS1 promotes non-small cell lung cancer progression via Wnt/Î2-catenin signaling. Biochem Biophys Res Commun. 2017;484(3):586–591. doi:10.1016/j.bbrc.2017.01.141
20. Yang G, Lu X, Yuan L. LncRNA: a link between RNA and cancer. Biochimica Et Biophysica Acta. 2014;1839(11):1097–1109. doi:10.1016/j.bbagrm.2014.08.012
21. Chen G, Sun W, Hua X, Zeng W, Yang L. Long non-coding RNA FOXD2-AS1 aggravates nasopharyngeal carcinoma carcinogenesis by modulating miR-363-5p/S100A1 pathway. Gene. 2018;645:76. doi:10.1016/j.gene.2017.12.026
22. Li CY, Liang GY, Yao WZ, et al. Integrated analysis of long non-coding RNA competing interactions reveals the potential role in progression of human gastric cancer. Int J Oncol. 2016;48(5):1965–1976. doi:10.3892/ijo.2016.3407
23. Yin C, Yin C, Zhang G, et al. miR-185-5p inhibits F-actin polymerization and reverses epithelial mesenchymal transition of human breast cancer cells by modulating RAGE. Mol Med Rep. 2018;18(3):2621–2630. doi:10.3892/mmr.2018.9294
24. Sun CC, Zhang L, Li G, et al. The lncRNA PDIA3P Interacts with miR-185-5p to modulate oral squamous cell carcinoma progression by targeting cyclin D2. Mol Ther Nucleic Acids. 2017;9(C):100–110. doi:10.1016/j.omtn.2017.08.015
25. Tan B, Li Y, Zhao Q, Fan L, Wang D. ZNF139 increases multidrug resistance in gastric cancer cells by inhibitingmiR-185. Bioscience Rep. 2018;38(5):BSR20181023. doi:10.1042/BSR20181023
26. Pei K, Zhu JJ, Wang CE, Xie QL, Guo JY. MicroRNA-185-5p modulates chemosensitivity of human non-small cell lung cancer to cisplatin via targeting ABCC1. European Rev Med Pharmacol Sci. 2016;20(22):4697–4704.
27. Tan J, Zhang C, Qian J. Expression and significance of Six1 and Ezrin in cervical cancer tissue. Tumor Biol. 2011;32(6):1241–1247. doi:10.1007/s13277-011-0228-8
28. Kearns S, Menke C, Thorburn J, Behbakht K, Thorburn A. Abstract #5141: six1 mediated TRAIL induced apoptosis resistance in ovarian cancer. Semin Cancer Biol. 2012;22(3):187–193.
29. Wang CA, Jedlicka P, Patrick AN, et al. SIX1 induces lymphangiogenesis and metastasis via upregulation of VEGF-C in mouse models of breast cancer. J Clin Invest. 2012;122(5):1895–1906. doi:10.1172/JCI59858
30. Li Z, Tian T, Lv F, et al. Six1 promotes proliferation of pancreatic cancer cells via upregulation of cyclin D1 expression. Plos One. 2013;8(3):e59203. doi:10.1371/journal.pone.0059203
31. Xu H, Zhang Y, Peña MM, Pirisi L, Creek KE. Six1 promotes colorectal cancer growth and metastasis by stimulating angiogenesis and recruiting tumor-associated macrophages. Carcinogenesis. 2017;38(3):281–292. doi:10.1093/carcin/bgw121
32. Li Z, Tian T, Hu X, et al. Six1 mediates resistance to paclitaxel in breast cancer cells. Biochem Biophys Res Commun. 2013;441(3):538–543. doi:10.1016/j.bbrc.2013.10.131
33. Li B, Zhao S, Geng R, Huo Z, Zhang H. The sineoculis homeobox homolog 1 (SIX1) gene regulates paclitaxel resistance by affecting reactive oxygen species and autophagy in human hepatocellular carcinoma cell line HepG2. Med Sci Monit. 2018;24:2271–2279.
34. He Z, Li G, Tang L, Li Y. SIX1 overexpression predicts poor prognosis and induces radioresistance through AKT signaling in esophageal squamous cell carcinoma. Oncotarget Ther. 2017;10:1071–1079. doi:10.2147/OTT.S125330
Supplementary materials
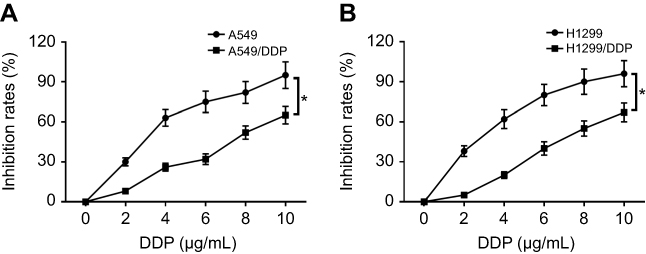 |
Figure S1 DDP-resistant NSCLC cells showed a lower growth inhibition rate compared with DDP-sensitive group. (A) The growth inhibition rate of A549 or A549/DDP was measured by CCK8 assay. (B) The growth inhibition rate of H1299 or H1299/DDP was measured by CCK8 assay. *p<0.05. |
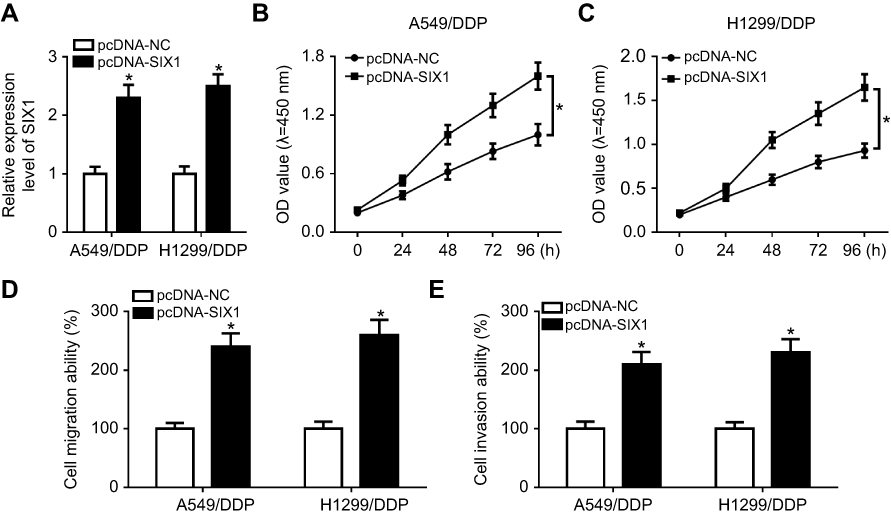 |
Figure S2 Overexpression of SIX1 attenuated drug sensitivity of DDP-resistant NSCLC cells. A549/DDP and H1299/DDP cells were transfected with pcDNA-NC or pcDNA-SIX1. (A) SIX1 expression was determined by RT-qPCR. (B and C) CCK-8 assay was performed to detect cell proliferation. (D and E) Transwell analysis was carried out for the detection of cell migration and invasion. *p<0.05. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.